
Abstract
Background
Ribosomal RNA (rRNA) comprises at least 90% of total RNA extracted from mammalian tissue or cell line samples. Informative transcriptional profiling using massively parallel sequencing technologies requires either enrichment of mature poly-adenylated transcripts or targeted depletion of the rRNA fraction. The latter method is of particular interest because it is compatible with degraded samples such as those extracted from FFPE and also captures transcripts that are not poly-adenylated such as some non-coding RNAs. Here we provide a cross-site study that evaluates the performance of ribosomal RNA removal kits from Illumina, Takara/Clontech, Kapa Biosystems, Lexogen, New England Biolabs and Qiagen on intact and degraded RNA samples.
Results
We find that all of the kits are capable of performing significant ribosomal depletion, though there are differences in their ease of use. All kits were able to remove ribosomal RNA to below 20% with intact RNA and identify ~ 14,000 protein coding genes from the Universal Human Reference RNA sample at >1FPKM. Analysis of differentially detected genes between kits suggests that transcript length may be a key factor in library production efficiency.
Conclusions
These results provide a roadmap for labs on the strengths of each of these methods and how best to utilize them.
Similar content being viewed by others

Background
Ribosomal depletion is a critical method in transcriptomics that allows for efficient detection of functionally relevant coding as well as non-coding transcripts through removal of highly abundant rRNA species. Use of oligo dT primer to capture the polyadenylated 3′ end of the transcripts and isolate mRNA is routine in many RNA sequencing preparations; however this method lacks the ability to handle degraded samples where most of the RNA is separated from the 3′ tail, or to isolate non-polyadenylated transcripts such as lncRNAs. Ribosomal removal methods address these issues by directly depleting the rRNA while leaving other transcripts intact. This technique is widely utilized and is a basic component of many large datasets [Full size image
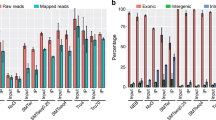
By comparison, the kits that degraded the rRNAs by either RNase H treatment or using ZapR showed more consistent results. Excluding single sites that failed with the NEBNext Ultra rRNA and SMARTer Pico kits, those two kits as well as the Takara/Clontech RiboGone and Kapa RiboErase kits performed very well with no differences observed between intact and degraded RNA. The RNaseH methods all showed very low rRNA fractions overall with the noted exception to the NEB kit. The SMARTer Pico kit had a slightly higher rRNA level, similar to that observed with Illumina Ribozero Gold degraded samples.
For those samples with successful rRNA depletion, we next ascertained the quality of the RNA sequencing data. Samples with greater than 50% rRNA were excluded from further analysis to eliminate artifacts that may be caused by improper implementation of the protocol. All the kits showed strong strand bias as expected by the protocols (Fig. 2b). Notably, SMARTer Pico reads mapped to the sense strand, which is the opposite strand from the other methods. While this is expected, care should be taken in adapting existing informatics pipelines to this kit. Differences were observed among the kits in how they handled mtRNAs. These were a major contaminant in the Clontech kits, particularly the RiboGone method that only targets the 12 s mtRNA and not the 16 s mtRNA. The other methods that addressed all mtRNA reads, such as Illumina Ribozero Gold, significantly reduced the fraction of reads from mtRNAs (Fig. 2c). This is especially noticeable in the RiboZero samples where site3 utilized a standard Human/Mouse/Rat kit instead of the RiboZero Gold.
Looking at the non-rRNA reads in each sample, the vast majority align to protein coding genes based on the ENSEMBL annotation. All samples show > 60% protein coding with most over 80% and the Clontech RiboGone kit having the largest fraction map** (Fig. 3a). Most of the samples identified ~ 14,000 genes expressed at greater than one RPKM and ~ 16,000 at over 0.1 RPKM (Fig. 3b). A single site using the Takara/Clontech SMARTer Pico kit did show a somewhat reduced number of genes, which was associated with a lower library complexity observed from that site. Antisense mapped reads and reads map** to the signal recognition particle RNAs (SRPs) were the most variable aspect of each sample though the source of these differences were unclear as they varied widely from site to site.

Protein Coding Gene Detection in rRNA Depleted Libraries: a) Non-mtRNA reads were mapped to the ENSENBL annotation and grouped by transcript type. Fraction of reads associated with transcript types > 1% shown. Data sets ordered by site then intact/degraded status within each kit top to bottom. b) Number of genes detected at >1RPKM (dark blue) and > 0.1RPKM (light blue) shown for each replicate. Genes ordered the same as in A but left to right. c) Changes in RNA detection compared to Illumina RiboZero. Hierarchical clustering genes with fold changes > 2 and Benjamini corrected p-values < 0.001 are shown (union of all comparisons). d) Count of genes with fold changes > 2 and Benjamini corrected p-values < 0.001 are shown for each kit as compared to Illumina RiboZero. Increased detection shown in red, decreased detection shown in green. e) Distribution of read lengths for transcripts detected at higher (red) or lower (green) rate relative to RiboZero. f) Distribution of GC% for transcripts detected at higher (red) or lower (green) rate relative to RiboZero
While the total number of protein coding genes detected was quite similar, many genes appear to be detected at significantly different rates. To better understand this observation, we performed differential gene expression analysis on the intact RNA samples that passed our QC metrics, comparing each preparation back to Illumina RiboZero Gold (Fig. 3c). The Qiagen rRNA depletion kit was excluded as only two replicates passed these criteria. Hierarchical clustering of the differentially detected genes, clusters the kits first by their RNAseq library prep methodology, with all three kits prepared using the NEB Ultra II Directional RNA Library kit clustering together, followed by the Kapa RiboErase kit and finally the low input Takara/Clontech SMARTer pico. Generally, several hundred genes could be easily observed as differentially detected between each of the kits and Illumina’s RiboZero (fold changes > 2, Benjamini corrected p-values < 0.001, Fig. 3d). Testing the physical properties of these differentially detected genes found that gene length appears to be a large contributor to the direction of the bias with shorter transcripts better detected by RiboZero and longer transcripts better detected by the other kits (Fig. 3e). Many of the most variably detected genes across the data set are quite small, such as mitochondrial proteins and ribosomal proteins (Additional file 3: Figure S2). The libraries themselves did not display any particular size bias with the RiboZero samples having an average length distribution similar to the other kits (Additional file 4: Figure S3). Bias in GC percentage was also observed in a few samples, with the Kapa RiboErase kit having the strongest bias against high GC transcripts (though the underlying gene list is quite small, Fig. 3f).
Ribosomal depletion is a key methodology used in studying noncoding RNAs as many of these transcripts lack polyadenylation sites [5]. We focused on lincRNAs (long intervening noncoding RNAs), one type of noncoding RNAs, since they do not overlap with any protein coding or other long non-coding RNA genes. Using the ENSEMBL annotation, approximately 4% of the non rRNA reads map to lincRNAs using RiboZero (Fig. 4a). Similar numbers are observed with the SMARTer Pico kit and Kapa RiboErase. The other kits, all of which were prepared with NEB Ultra II directional RNA library kit, show less than 3% of reads map** to lincRNAs. This global decrease in number of lincRNA reads appears to reflect a general decrease in the number of map** reads rather than a specific bias against a subset of lincRNAs. Two lines of evidence support this conclusion. First, the number of lincRNAs detected at > = 0.01 RPM remains ~ 3500 for all of the different kits tested and no bias is seen against the NEB prepped kits (Fig. 4b). Second, while the majority of lincRNAs detected can be assigned to 4 specific lincRNAs (MALAT1, SNORD3A, RNRP and NEAT1), the remaining fraction remains constant among the different kits, suggesting a global decrease in map** (Fig. 4c). The precise set of lincRNAs detected varies somewhat but the core of 3200 lincRNAs are detected by all of the kits (Fig. 4d).

LincRNA Detection in rRNA Depleted Libraries: a) Mean fraction of non-rRNA reads assigned to lincRNAs based on the ENSEMBL annotation. b) Number of lincRNAs detected over specified RPM levels. c) Fraction of lincRNA mapped reads assigned to the top 4 lincRNAs detected for each sample. Data sets ordered by site then intact/degraded status within each kit left to right. Average RPM counts for each lincRNA was calculated and the top four lincRNAs were shown kee** remaining lincRNAs in ‘Others’ category. d) Overlap of lincRNAs detected by the three core library prep methodologies: ribosomal pulldown (RLQ), RNAse H (NCK), and ZapR (CZ). Average RPM counts for each lincRNA for all samples in each of the three core library methods (RLQ = RZ, LX, Q; NCK = NE, CR, K; CZ = CZ) was calculated and lincRNAs with average RPM > 0 were compared among the methods
While comparison of different detection rates of mRNAs in UHR can point to possible differences between the kits’ chemistries, the spike-in controls provide an absolute metric to evaluate their effectiveness. Both SIRV (co-degraded with the RNA) and ERCC (not degraded) control spike-ins were added to the sample before library preparation and should give an unbiased look at the behavior of the different kits. As an initial test, we examined the ratio between the two spike-in types to model the impact of degradation on efficiency of library formation. Importantly, the kits do treat degraded RNA differently in their protocols and the not degraded ERCCs in the degraded samples were processed along the path proscribed for the bulk RNA, suggesting they are under-fragmented relative to the bulk population. Examining the ratios between the ERCC and SIRVs, we find that all the intact samples show ~ 60% SIRV reads (Fig. 5a). By comparison, the degraded samples show significant bias between the ERCCs and SIRV, generally favoring the intact ERCC. The RiboZero Gold and Kapa RiboErase kits show the least bias based on degradation, while the kits using the NEB stranded RNAseq kits showed a bias against shorter RNA fragments, which is similar to what was observed for protein coding genes (Fig. 3e). The SMARTer Pico kit is biased against the intact ERCC spike-ins in the degraded sample, which is likely due to not pre-degrading the ERCCs in the context of the degraded total RNA, emphasizing the importance of this step. Inserting not degraded spike-in controls into variably degraded experimental samples may confound the ability of these spike-ins to serve as a normalization tool, as previously observed [6, 7].

Effect of rRNA Depletion Chemistry on Spike-In Controls: a) Effect of degradation of SIRVs on the ratio of SIRV reads to ERCC reads in each replicate. Percent of reads map** to SIRVs out of total reads map** to Spike-ins in shown. Data sets ordered by intact/degraded status followed by site within each kit left to right. b) Relative ratio of reads map** to SIRV1. Fraction of reads map** to each isoform of SIRV1 are shown for each replicate. Expected fractions shown as dark horizontal lines. Light horizontal lines show 2-fold changes in fraction observed (log scale). Each SIRV1 isoform is shown in a different color. Replicates ordered as in a. c) Boxplot of normalized transcripts per million (TPM) for subsets of SIRVs. SIRVs present at 4×, 1× and 1/4× in the pool were randomized and normalized TPM scores for all sites for two sets of 10 SIRVs were plotted across different chemistries using intact samples. 5th, 25th,Median, 75th and 95th percentile are shown. d) Boxplot as in c except red boxplot highlights shortest quintile (len < 480 nt) and green includes longest quintile (len > 2200). e) Boxplot as in c except red boxplot highlights lowest GC quintile (< 36% GC) and green includes highest quintile (> 44.5% GC). f) Boxplot as in c except red highlights random set of 10 transcripts included at 4× while green includes set of 10 transcripts at 1/4×
Distinct transcripts are present at defined ratios in the spike-in control allowing direct visualization of over and under-representation of transcripts. Within the SIRV spike-ins, the distinct transcripts were largely at equal ratios between kits and sites, though deviation from the expected values is observed (Fig. 5b). The SMARTer Pico kit was particularly susceptible to variation, possibly due to the low total input (1:100th of the other kits), and some transcripts (e.g. purple at 1/4×) show loss of signal in the degraded sample. By comparison, the ERCC spike-ins showed significantly more variability across sites, even within the intact RNA samples (Additional file 5: Figure S4). Overall, the Lexogen and Takara/Clontech RiboGone kits generally had the most consistent and even performance on both the ERCC and SIRV spike-in controls across their test sites for intact samples.
Using the transcript diversity within the SIRV spike-in control, we further examined the general trends observed with respect to transcript length, GC content, and abundance. The 50 highest expressed SIRV transcripts from the intact samples were individually quantified using STAR/RSEM and the TPMs normalized to the expected frequency of the transcripts. We then compared the normalized frequencies for all of the transcripts highest and lowest quintile for different parameters to look for differences. Randomly selected transcripts show minimal differences (Fig. 5c), nor do we observe biases when the transcripts are sorted by GC percentage or amount of expected transcript (Fig. 5e, f). Transcript length does appear to show a bias, with the longest 10 transcripts (len > 2200 nt) showing lower levels of detection than the shortest transcripts (len < 480 nt, Fig. 5d) and three of the poorest detected transcripts in the SIRVs are in the set of longest transcripts. However, the overall distribution remains very broad and size alone is not predictive of the behavior of the transcript.
