
Abstract
Background
The interpretation of results from transcriptome profiling experiments via RNA sequencing (RNA-seq) can be a complex task, where the essential information is distributed among different tabular and list formats—normalized expression values, results from differential expression analysis, and results from functional enrichment analyses. A number of tools and databases are widely used for the purpose of identification of relevant functional patterns, yet often their contextualization within the data and results at hand is not straightforward, especially if these analytic components are not combined together efficiently.
Results
We developed the GeneTonic software package, which serves as a comprehensive toolkit for streamlining the interpretation of functional enrichment analyses, by fully leveraging the information of expression values in a differential expression context. GeneTonic is implemented in R and Shiny, leveraging packages that enable HTML-based interactive visualizations for executing drilldown tasks seamlessly, viewing the data at a level of increased detail. GeneTonic is integrated with the core classes of existing Bioconductor workflows, and can accept the output of many widely used tools for pathway analysis, making this approach applicable to a wide range of use cases. Users can effectively navigate interlinked components (otherwise available as flat text or spreadsheet tables), bookmark features of interest during the exploration sessions, and obtain at the end a tailored HTML report, thus combining the benefits of both interactivity and reproducibility.
Conclusion
GeneTonic is distributed as an R package in the Bioconductor project (https://bioconductor.org/packages/GeneTonic/) under the MIT license. Offering both bird’s-eye views of the components of transcriptome data analysis and the detailed inspection of single genes, individual signatures, and their relationships, GeneTonic aims at simplifying the process of interpretation of complex and compelling RNA-seq datasets for many researchers with different expertise profiles.
Similar content being viewed by others

Background
In modern life and clinical sciences, RNA-sequencing (RNA-seq) is an essential tool for studying gene expression and its regulation [1]. High-throughput sequencing technologies generate readouts for a large number of molecular entities simultaneously, posing challenges to proper hypothesis generation and data interpretation [2]. Among the typical bioinformatic workflows, differential expression (DE) analysis is often employed to identify the genes showing evidence for statistically significant changes, thus being candidate effectors for regulation across the sampled experimental conditions [3].
Most studies where these techniques are being adopted result in a list containing tens to thousands of gene candidates, with their associated effect size and significance level—often reported as log2 fold change (log2FC) and adjusted p-values, respectively. Putting these results into biological context by leveraging existing knowledge is essential for facilitating the interpretation of data at a systemic level, and enabling novel discoveries [4].
Commonly used knowledge bases for the purpose of functional enrichment analysis include Gene Ontology (GO) [5, 12].
A large variety of computational methods and software have been designed for functional enrichment analysis [18, 41].
Datasets and gene set collections increase constantly in their size and complexity, constituting a major barrier for the interpretability of transcriptomic data and their enrichment results, to the point that a potential bottleneck for omics data is the so-called tertiary analysis, opposed to map** and quantification (primary analysis) and statistical testing (secondary analysis) [42]. Efficient platforms that enable advanced workflows for a wide range of users can play a big role in providing the required level of interactivity, while guaranteeing the adherence to gold standard methods and to best practices for reproducible analyses [43,44,45].
The different atomic elements for a typical RNA-seq analysis (expression table, results from differential expression, functional enrichment results) can stem from different pipeline outputs, yet they need to be combined together, e.g. in a report created following the rules of literate programming [46]. By providing accessible summaries with proper data visualization and interpretation methods, in formats that facilitate dynamic shareable outputs, such frameworks can greatly reduce the time to generate novel hypotheses and insight. Often, this task is not straightforward to carry out, as different software solutions or environments might be chosen, resulting in different file formats, thus increasing the difficulty for practitioners to explore all relevant aspects of the data at hand, even if common sets of gene and pathway identifiers are adopted.
A number of solutions have been developed in diverse languages (mostly R, Python, Java) to address the challenges listed above, but no software package provides a comprehensive framework for assisting the proper interpretation of RNA-seq data; interested readers can find a comparative overview of the features of the above mentioned tools in Additional file 1: Table S1.
Here we present GeneTonic, an R/Bioconductor package aiming to streamline the identification of relevant functional patterns, as well as their contextualization in the data and results at hand, by combining in a seamless way all the pieces of information relevant for a transcriptomic analysis. The GeneTonic package is composed by a Shiny web application, with a variety of standalone functions to perform the analysis both interactively as well as in a programmatic way. GeneTonic requires as input the results generated by each analytic step (quantification, DE testing, functional enrichment), which are usually shared as separate tables or spreadsheets by bioinformaticians and core facility service providers, in formats that are suitable to standardization.
GeneTonic makes it easy to generate visualizations, starting from bird’s eye perspective summaries (gene-geneset graphs, enrichment maps, also linked to interactive tables in the web application), as well as getting in-depth dedicated summaries for each geneset of interest. User actions enable further insight and deliver additional information (e.g. gene info boxes, geneset summaries, and signature heatmaps), with drilldown tasks activated by simple mouse clicks. While simple operations within the call to the GeneTonic() main function makes the result set more interpretable, our package also supports built-in RMarkdown reporting as a foundation for computational reproducibility, to conclude an interactive exploration session [47, 48]. We carefully designed the user interface, enabling the required tasks in a straightforward way, as a result of an open and continuous dialogue with researchers adopting this tool in its early development. Users can learn-by-doing the functionality of GeneTonic via guided tours, creating a common ground for experimentalists and analysts to explore transcriptomic data at the desired depth and efficiently generate novel insights [49].
GeneTonic connects together a number of R/Bioconductor packages, implementing the current best practices in RNA-seq data analysis, and facilitates the communication between experts of different disciplines. Harmonizing the output of the many analysis steps, possibly performed also with a variety of approaches, GeneTonic is a powerful tool for digesting and enjoying any RNA-seq dataset: the interactivity is a compelling means to empower end users for the exploration of many features of interest, and by providing a report with full code snippets, we support analyses that are reproducible and easily extendable. The GeneTonic package is available at https://bioconductor.org/packages/GeneTonic/, and a public instance is available for demonstration purposes at http://shiny.imbei.uni-mainz.de:3838/GeneTonic.
Implementation
General design of GeneTonic
The GeneTonic package is written in the R programming language, leveraging many existing packages currently available in the Bioconductor project, which constitute the foundation for a broad spectrum of analytic workflows in computational biology and bioinformatics [50, 51], and the Shiny framework for interactivity [2D, E) to facilitate drilldown tasks and better understand the whole data components of the project. A number of automatically generated action buttons link directly to external databases, such as AmiGO [backbone package [81] to highlight the salient edges of the bipartite projections for each type of features included, as a way to summarize information contained in large networks (Fig. 2F, G).
Additional insight can be extracted by drilling down the interactive Enrichment Map [62, 78], either by focusing on the selected nodes (checking out signature heatmaps or bookmarking the genesets for inserting them into the report), or also by running a variety of community detection algorithms on the graph object returned by the enrichment_map() function (Additional file 4: Fig. S1C). Together with the community membership information, it is then possible to obtain a more compact summary for the functional enrichment results, where the most representative genesets for each subpartition of the graph are selected and returned in tabular format. This network-based approach can be exploited to detect the handful of overarching themes, which might give a more immediate snapshot than the many, often redundant, categories, commonly returned by pathway enrichment algorithms (Additional file 4: Fig. S1E-F-G).
Summarizing the enrichment results
GeneTonic provides numerous ways to summarize the enrichment results, often leveraging the effectiveness of visual representations to extract insights. The Overview and GSViz panels serve this purpose, showcasing different views on the dataset at hand, with the main controls provided in the right sidebar.
The geneset volcano plot (Fig. 3A) displays all genesets from the res_enrich object and labels the most relevant (or any subset of interest). We use one of the aggregated scores (geneset Z-score, or average log2 fold change) to determine the horizontal position in the plot. To avoid clutter, it is also possible to reduce the terms based on an overlap threshold, retaining only the most representative ones, and provide this more compact summary to the following visualization routines.
The enhanced table (Fig. 3B) summarizes the top genesets by displaying the log2FC of each set’s components along a line (one for each set). On top of the static version, this is provided also as an interactive widget, where tooltips activated with the mouse deliver extra information on each dot, representing a single gene.
The complex relationships among genesets and their behavior across samples are just two aspects one can inspect in depth with the implemented methods. Among these, users can generate a genesets-by-sample heatmap, showing the standardized expression values of the members (via the gs_scoresheat() function, Fig. 3C), or alternatively a summary heatmap (with gs_summary_heat(), Additional file 3), which aims to display the redundancy between different sets, while encoding the values of the expression changes. A multi-dimensional scaling (MDS) plot (Fig. 3D) delivers a 2d visualization of the distance among genesets, based on a similarity measure, e.g. their overlap or other criteria, such as their semantic similarity. In a similar fashion, a dendrogram for genesets enables the possibility to use node color, node size, and branch color to encode relevant features, with the tree structure mirroring the distance matrix based on a similarity measure. GeneTonic simplifies the creation of simple summaries for the enrichment, where the essential columns are encoded as graphical parameters of the points, extendable to the case of comparing the same genesets in more than one scenario (e.g. if it is possible to extract more than one contrast from the expression matrix). Switching to polar coordinates, this can be captured in spider plots for one or more res_enrich objects (see Additional file 3 for more examples of usage).
These visual summaries constitute appealing alternatives to the commonly reported tabular formats, which often fail to provide an overall view for the affected functional landscape.
Wrap** up the session
The Bookmarks panel offers the possibility to review and inspect the shortlisted features of interest, where both genes (on the left side of the interface) and genesets (right side) can be exported to text files.
A more comprehensive report, with dynamically generated content based on the user selections, is compiled when starting the happy_hour() function. This is made possible by a template RMarkdown document, included in the GeneTonic package, which accesses the input elements and the reactive values for the Shiny components. Notably, this functionality can also be used outside an interactive usage session, specifying as parameters the values for the genes and genesets to focus on. In either case, a full HTML document is rendered, whose content mirrors the structure of the info boxes, and can be later shared or stored as a reproducible artifact for the performed analyses.
A set of buttons below each generated visualization displays in a modal window the code necessary to reproduce a specific output—these snippets can be copied from the in-app editor into more comprehensive analysis scripts or notebooks.
Another action button creates the serialized version of a SummarizedExperiment object, ready to be provided as the main input to iSEE [74], for further tailored visualizations, either with standard or custom panels of the web application.
Discussion
Interpreting the results of transcriptomic studies can be a complex task, where differential expression analysis is combined with a higher-level pathway enrichment analysis, in order to robustly define the molecular actors that display expression changes, and also to identify the underlying functional patterns. Geneset functional enrichment has been successfully applied to thousands of works, and for this step many methods and approaches have been developed. These tasks are also often shared with alternative workflows other than DE analysis, whereas the aim is to extract meaningful information from large lists of genes, yet it is still a prohibitive task to combine in a straightforward way all the single results from each step. This can be for example due to disjoint sets of identifiers, different output and file formats, and to the difficulties in extracting knowledge while handling large numbers of redundant genesets. Providing concise and biologically meaningful views of the underlying cellular processes, defined via differential expression, is essential in many applications, and a proper visualization framework plays a fundamental role in transforming the otherwise tedious and error/bias-prone task of navigating large textual tables into a more compelling activity [18, 78].
In this work, we introduced GeneTonic as a solution to explore all the components of a transcriptome dataset in a more integrative way, instead of having to process them as separated outputs. As such, GeneTonic is not structured as an end-to-end workflow including quantification, preprocessing, exploratory data analysis, and DE modeling—all operations that are also time consuming, but in many scenarios need to be carried out only once. GeneTonic is focused on the analytic step devoted to the interpretation of data, rather than on the implementation of additional methods for detection of functionally enriched biological processes or pathways. Consequently, GeneTonic implements a variety of summary and visual representations, while accommodating the output of many commonly adopted enrichment tools, making efficient use of the Shiny framework to deliver interactivity and enable drilldown operations. These would otherwise need to be laboriously addressed in multiple iterations of scripted analyses, either done by the user itself or in collaboration with an external unit, such as a bioinformatics core facility. This approach liberates and empowers both experienced analysts and bench scientists, providing a common ground to efficiently transform data and results into interpretable discoveries.
Several software packages and web-based portals exist for providing similar functionality, and a comprehensive overview of their salient features is presented in Additional file 1: Table S1. Naturally, these tools differ in terms of implementation, range of applicability, ease of use, with many proposals offering embedded versions of enrichment tests. Since we developed GeneTonic in the R programming language, where many such testing procedures are natively available, we instead focused on the support and integration of their output formats into a common workflow. This can be easily combined with existing analysis pipelines, making our tool well suit for potential wide adoption. The comparison with other tools is also available online (https://federicomarini.github.io/GeneTonic_supplement), linked to a Google Sheet where the individual characteristics of each tool can be updated, in order to provide guidance for users who might be seeking advice on which solution best fits their needs (accessible at https://docs.google.com/spreadsheets/d/167XV0w18P0FSld1dt6owN4C2Esxl5FU2QTo4D-wclz0/edit?usp=sharing).
While currently focused on the output of single ORA and FCS enrichment methods, future developments of GeneTonic will implement functionality for combined and ensemble approaches, such as EnrichmentBrowser [82] or EGSEA [83]. Moreover, extending such visualizations and interactive summaries to scenarios where multiple omics layers are collected will be a promising avenue for GeneTonic, given the growing number of such datasets becoming available. Finally, we intend to address more refined similarity measurements among genesets, e.g. accounting for information contained in protein-protein interaction networks databases [84], in order to better capture the functional relatedness of the affected pathways.
As bioinformatics evolves constantly into a highly interdisciplinary field, it will become increasingly important to develop common platforms usable by many profiles with substantial differences in their level of programming skills, and GeneTonic’s design guidelines adhere to this principle. Serving as a bridge between experts from different disciplines, applications such as GeneTonic will have a significant didactic effect by making comprehensive analyses more open, transparent, and easy to share—especially in the steps of interpreting transcriptome datasets, unlocking their full potential for medical and biological research. Moreover, GeneTonic could make it easier for bioinformatics skilled users to better understand the systems under investigation, prompting e.g. the development of further tailored methods, which could be a key in obtaining a deeper knowledge of the experimental scenarios.
Conclusion
The identification of relevant functional patterns for the features identified in the differential expression analysis, accounting for the available expression data, remains one of the common bottlenecks for transcriptome-based workflows. GeneTonic provides a web application and many underlying functions to assemble the pieces together, supporting the exploration both interactively as well as in a programmatic way. Combining together the results for quantification, DE testing, and functional enrichment (either generated autonomously, or obtained from collaborators), GeneTonic assists in the unmet yet increasing need of extracting novel knowledge and insights, which can become daunting especially on larger datasets.
GeneTonic has the potential to become an ideal interface between experimental and computational scientists, with the HTML report built via RMarkdown as a milestone for reproducibility, upon conclusion of an interactive session. GeneTonic can be integrated in a wide spectrum of existing bioinformatic pipelines, as it provides functions to convert and input the results of many pathway enrichment tools. This aligns with the principle of interoperability at the heart of the Bioconductor project, which enables a large number of such workflows.
The experience of enjoying transcriptomic data analysis and exploration can be easily shared with reduced communication burden, with both experimental and computational sides empowered in the tasks of realizing complex summaries and visualizations. This will significantly facilitate and democratize the discovery process, bridging the gaps existing between technical and domain expertise.
Availability and requirements
-
Project name: GeneTonic.
-
Project home page: https://bioconductor.org/packages/GeneTonic/ (release), https://github.com/federicomarini/GeneTonic/ (development version).
-
Archived version: https://doi.org/10.5281/zenodo.5519859, package source as gzipped tar archive of the version reported in this article
-
Project documentation: rendered at https://federicomarini.github.io/GeneTonic/.
-
Operating systems: Linux, Mac OS, Windows.
-
Programming language: R.
-
Other requirements: R-4.0.0 or higher, Bioconductor 3.11 or higher.
-
License: MIT.
-
Any restrictions to use by non-academics: none.
Availability of data and materials
The datasets used in this manuscript and its supplements are available from the following articles: The data set on the macrophage immune stimulation is included in PubMed ID: 29379200 (https://doi.org/10.1038/s41588-018-0046-7). Dataset deposited at the ENA (ERP020977, project id: PRJEB18997) and accessed from the Bioconductor experiment package macrophage package (https://bioconductor.org/packages/macrophage/, version 1.7.2). The data set on murine A20-deficient microglia is included in PubMed ID: 32023471 (https://doi.org/10.1016/j.celrep.2019.12.097). Dataset deposited at the GEO (GSE123033, project id: PRJNA507355) and accessed from the https://github.com/federicomarini/GeneTonic_supplement/ repository. The GeneTonic package can be downloaded from its Bioconductor page https://bioconductor.org/packages/GeneTonic/ or the GitHub development page https://github.com/federicomarini/GeneTonic/. GeneTonic is also provided as a recipe in Bioconda (https://anaconda.org/bioconda/bioconductor-genetonic). The repository available at https://github.com/federicomarini/GeneTonic_supplement/ contains the code used to generate the supplemental material, and the required input data to replicate the analyses presented in the use cases.
Abbreviations
- DE:
-
Differential expression
- FCS:
-
Functional class scoring
- FDR:
-
False discovery rate
- GO:
-
Gene ontology
- GSEA:
-
Gene set enrichment analysis
- HGNC:
-
HUGO (Human Genome Organisation) Gene Nomenclature Committee
- log2FC:
-
Base-2 logarithm of the fold change
- MA plot:
-
M (log ratio) versus A (mean average) plot
- MDS:
-
Multi-dimensional scaling
- MSigDB:
-
Molecular Signatures Database
- NCBI:
-
National Center for Biotechnology Information
- ORA:
-
Over-representation analysis
- PT:
-
Pathway topology
- RNA-seq:
-
RNA sequencing
References
Van den Berge K, Hembach KM, Soneson C, Tiberi S, Clement L, Love MI, Patro R, Robinson MD. RNA sequencing data: Hitchhikers guide to expression analysis. Annu Rev Biomed Data Sci. 2019;2(1):139–73. https://doi.org/10.1146/annurev-biodatasci-072018-021255.
Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17(1):13. https://doi.org/10.1186/s13059-016-0881-8.
Love MI, Anders S, Kim V, Huber W. RNA-Seq workflow: gene-level exploratory analysis and differential expression. F1000Research. 2015;4:1070. https://doi.org/10.12688/f1000research.7035.1.
Chen Y, Lun ATL, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research. 2016;5:1438. https://doi.org/10.12688/f1000research.8987.2.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9. https://doi.org/10.1038/75556.
Carbon S, Douglass E, Dunn N, Good B, Harris NL, Lewis SE, Mungall CJ, Basu S, Chisholm RL, Dodson RJ, Hartline E, Fey P, Thomas PD, Albou LP, Ebert D, Kesling MJ, Mi H, Muruganujan A, Huang X, Poudel S, Mushayahama T, Hu JC, LaBonte SA, Siegele DA, Antonazzo G, Attrill H, Brown NH, Fexova S, Garapati P, Jones TEM, Marygold SJ, Millburn GH, Rey AJ, Trovisco V, Dos Santos G, Emmert DB, Falls K, Zhou P, Goodman JL, Strelets VB, Thurmond J, Courtot M, Osumi DS, Parkinson H, Roncaglia P, Acencio ML, Kuiper M, Lreid A, Logie C, Lovering RC, Huntley RP, Denny P, Campbell NH, Kramarz B, Acquaah V, Ahmad SH, Chen H, Rawson JH, Chibucos MC, Giglio M, Nadendla S, Tauber R, Duesbury MJ, Del NT, Meldal BHM, Perfetto L, Porras P, Orchard S, Shrivastava A, **e Z, Chang HY, Finn RD, Mitchell AL, Rawlings ND, Richardson L, Sangrador-Vegas A, Blake JA, Christie KR, Dolan ME, Drabkin HJ, Hill DP, Ni L, Sitnikov D, Harris MA, Oliver SG, Rutherford K, Wood V, Hayles J, Bahler J, Lock A, Bolton ER, De Pons J, Dwinell M, Hayman GT, Laulederkind SJF, Shimoyama M, Tutaj M, Wang SJ, D’Eustachio P, Matthews L, Balhoff JP, Aleksander SA, Binkley G, Dunn BL, Cherry JM, Engel SR, Gondwe F, Karra K, MacPherson KA, Miyasato SR, Nash RS, Ng PC, Sheppard TK, Shrivatsav Vp A, Simison M, Skrzypek MS, Weng S, Wong ED, Feuermann M, Gaudet P, Bakker E, Berardini TZ, Reiser L, Subramaniam S, Huala E, Arighi C, Auchincloss A, Axelsen K, Argoud GP, Bateman A, Bely B, Blatter MC, Boutet E, Breuza L, Bridge A, Britto R, Bye-A-Jee H, Casals-Casas C, Coudert E, Estreicher A, Famiglietti L, Garmiri P, Georghiou G, Gos A, Gruaz-Gumowski N, Hatton-Ellis E, Hinz U, Hulo C, Ignatchenko A, Jungo F, Keller G, Laiho K, Lemercier P, Lieberherr D, Lussi Y, Mac-Dougall A, Magrane M, Martin MJ, Masson P, Natale DA, Hyka NN, Pedruzzi I, Pichler K, Poux S, Rivoire C, Rodriguez-Lopez M, Sawford T, Speretta E, Shypitsyna A, Stutz A, Sundaram S, Tognolli M, Tyagi N, Warner K, Zaru R, Wu C, Chan J, Cho J, Gao S, Grove C, Harrison MC, Howe K, Lee R, Mendel J, Muller HM, Raciti D, Van Auken K, Berriman M, Stein L, Sternberg PW, Howe D, Toro S, Westerfield M. The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019;47(D1):330–8. https://doi.org/10.1093/nar/gky1055.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):353–61. https://doi.org/10.1093/nar/gkw1092.
Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47(D1):590–5. https://doi.org/10.1093/nar/gky962.
Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46(D1):649–55. https://doi.org/10.1093/nar/gkx1132.
Liberzon A., Subramanian A., Pinchback R., Thorvaldsdottir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40. https://doi.org/10.1093/bioinformatics/btr260.
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database Hallmark gene set collection. Cell Syst. 2015;1(6):417–25. https://doi.org/10.1016/j.cels.2015.12.004.
Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012;8(2):1002375. https://doi.org/10.1371/journal.pcbi.1002375.
**e C, Jauhari S, Mora A. Popularity and performance of bioinformatics software: the case of gene set analysis. BMC Bioinform. 2021;22(1):191. https://doi.org/10.1186/s12859-021-04124-5.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545–50. https://doi.org/10.1073/pnas.0506580102.
Nguyen T, Mitrea C, Draghici S. Network-based approaches for pathway level analysis. Curr Protoc Bioinform. 2018;61(1):8–25182524. https://doi.org/10.1002/cpbi.42.
Geistlinger L, Csaba G, Santarelli M, Ramos M, Schiffer L, Turaga N, Law C, Davis S, Carey V, Morgan M, Zimmer R, Waldron L. Toward a gold standard for benchmarking gene set enrichment analysis. Brief Bioinform. 2020. https://doi.org/10.1093/bib/bbz158.
Villaveces JM, Koti P, Habermann BH. Tools for visualization and analysis of molecular networks, pathways, and -omics data. Adv Appl Bioinform Chem. 2015;8(1):11–22. https://doi.org/10.2147/AABC.S63534.
Supek F, Škunca N, Visualizing GO annotations. In: The gene ontology handbook, vol. 1446. Humana Press; 2017. p. 207–20. https://doi.org/10.1007/978-1-4939-3743-1.
Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess over representation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–9. https://doi.org/10.1093/bioinformatics/bti551.
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–3. https://doi.org/10.1093/bioinformatics/btp101.
Mlecnik B, Galon J, Bindea G. Comprehensive functional analysis of large lists of genes and proteins. J Proteomics. 2018;171:2–10. https://doi.org/10.1016/j.jprot.2017.03.016.
Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009;10(1):48. https://doi.org/10.1186/1471-2105-10-48.
Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6(7):21800. https://doi.org/10.1371/journal.pone.0021800.
Walter W, Sánchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31(17):2912–4. https://doi.org/10.1093/bioinformatics/btv300.
Tian T, Liu Y., Yan H, You Q., Yi X., Du Z., Xu W., Su Z. AgriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017;45(W1):122–9. https://doi.org/10.1093/nar/gkx382.
Wei Q, Khan IK, Ding Z, Yerneni S, Kihara D. NaviGO: interactive tool for visualization and functional similarity and coherence analysis with gene ontology. BMC Bioinform. 2017;18(1):177. https://doi.org/10.1186/s12859-017-1600-5.
Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47(W1):199–205. https://doi.org/10.1093/nar/gkz401.
Kuznetsova I, Lugmayr A, Siira SJ, Rackham O, Filipovska A. CirGO: an alternative circular way of visualising gene ontology terms. BMC Bioinform. 2019;20(1):84. https://doi.org/10.1186/s12859-019-2671-2.
Zhu J, Zhao Q, Katsevich E, Sabatti C. Exploratory gene ontology analysis with interactive visualization. Sci Rep. 2019;9(1):1–9. https://doi.org/10.1038/s41598-019-42178-x.
Hale ML, Thapa I, Ghersi D. FunSet: an open-source software and web server for performing and displaying gene ontology enrichment analysis. BMC Bioinform. 2019;20(1):359. https://doi.org/10.1186/s12859-019-2960-9.
Federico A, Monti S. hypeR: an R package for geneset enrichment workflows. Bioinformatics. 2020;36(4):1307–8. https://doi.org/10.1093/bioinformatics/btz700.
Liu X, Han M, Zhao C, Chang C, Zhu Y, Ge C, Yin R, Zhan Y, Li C, Yu M, He F, Yang X. KeggExp: a web server for visual integration of KEGG pathways and expression profile data. Bioinformatics. 2019;35(8):1430–2. https://doi.org/10.1093/bioinformatics/bty798.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. https://doi.org/10.1038/s41467-019-09234-6.
Ulgen E, Ozisik O, Sezerman O.U. pathfindR: an R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Front Genet. 2019;10(SEP):1–33. https://doi.org/10.3389/fgene.2019.00858.
Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020;36(8):2628–9. https://doi.org/10.1093/bioinformatics/btz931.
Brionne A, Juanchich A, Hennequet-Antier C. ViSEAGO: a bioconductor package for clustering biological functions using gene ontology and semantic similarity. BioData Min. 2019;12(1):1–13. https://doi.org/10.1186/s13040-019-0204-1.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, von Mering C. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):607–13. https://doi.org/10.1093/nar/gky1131.
Tokar T, Pastrello C, Jurisica I. GSOAP: a tool for visualisation of gene set over-representation analysis. Bioinformatics. 2020. https://doi.org/10.1093/bioinformatics/btaa001.
Wang G, Oh D-H, Dassanayake M. GOMCL: a toolkit to cluster, evaluate, and extract non-redundant associations of gene ontology-based functions. BMC Bioinform. 2020;21(1):139. https://doi.org/10.1186/s12859-020-3447-4.
Kim J, Yoon S, Nam D. netGO: R-Shiny package for network-integrated pathway enrichment analysis. Bioinformatics. 2020. https://doi.org/10.1093/bioinformatics/btaa077.
Calura E, Martini P. Summarizing RNA-Seq data or differentially expressed genes using gene set, network, or pathway analysis. In: Picardi E, editor. RNA bioinformatics, chap 9, vol. 2284. Humana; 2021. p. 147–79. https://doi.org/10.1007/978-1-0716-1307-8.
Akhmedov M, Martinelli A, Geiger R, Kwee I. Omics Playground: a comprehensive self-service platform for visualization, analytics and exploration of big omics data. NAR Genom Bioinform. 2020;2(1):1–10. https://doi.org/10.1093/nargab/lqz019.
Sandve GK, Nekrutenko A, Taylor J, Hovig E. Ten simple rules for reproducible computational research. PLoS Comput Biol. 2013;9(10):1003285. https://doi.org/10.1371/journal.pcbi.1003285.
Marini F, Binder H. Development of applications for interactive and reproducible research: a case study. Genom Computl Biol. 2016;3(1):39. https://doi.org/10.18547/gcb.2017.vol3.iss1.e39.
Brito JJ, Li J, Moore JH, Greene CS, Nogoy NA, Garmire LX, Mangul S. Recommendations to enhance rigor and reproducibility in biomedical research. GigaScience. 2020;9(6):1–6. https://doi.org/10.1093/gigascience/giaa056.
Knuth DE. Literate programming. Comput J. 1984;27(2):97–111. https://doi.org/10.1093/comjnl/27.2.97.
Marini F, Binder H. pcaExplorer: an R/Bioconductor package for interacting with RNA-seq principal components. BMC Bioinform. 2019;20(1):331. https://doi.org/10.1186/s12859-019-2879-1.
Marini F, Linke J, Binder H. ideal: an R/Bioconductor package for interactive differential expression analysis. BMC Bioinform. 2020;21(1):565. https://doi.org/10.1186/s12859-020-03819-5.
Poplawski A, Marini F, Hess M, Zeller T, Mazur J, Binder H. Systematically evaluating interfaces for RNA-seq analysis from a life scientist perspective. Brief Bioinform. 2016;17(2):213–23. https://doi.org/10.1093/bib/bbv036.
Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, Gottardo R, Hahne F, Hansen KD, Irizarry R, Lawrence M, Love MI, MacDonald J, Obenchain V, Oleś AK, Pagès H, Reyes A, Shannon P, Smyth GK, Tenenbaum D, Waldron L, Morgan M. Orchestrating high-throughput genomic analysis with bioconductor. Nat Methods. 2015;12(2):115–21. https://doi.org/10.1038/nmeth.3252.
Amezquita R, Carey V, Carpp L, Geistlinger L, Lun A, Marini F, Rue-Albrecht K, Risso D, Soneson C, Waldron L, Pagès H, Smith M, Huber W, Morgan M, Gottardo R, Hicks S. Orchestrating single-cell analysis with bioconductor. BioRxiv. 2019. https://doi.org/10.1101/590562.
Chang W, Cheng J, Allaire J, **e Y, McPherson J. Shiny: web application framework for R. (2020). R package version 1.4.0.2. https://CRAN.R-project.org/package=shiny.
Alasoo K, Rodrigues J, Mukhopadhyay S, Knights AJ, Mann AL, Kundu K, Hale C, Dougan G, Gaffney DJ. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet. 2018;50(3):424–31. https://doi.org/10.1038/s41588-018-0046-7.
Mohebiany AN, Ramphal NS, Karram K, Di Liberto G, Novkovic T, Klein M, Marini F, Kreutzfeldt M, Härtner F, Lacher SM, Bopp T, Mittmann T, Merkler D, Waisman A. Microglial A20 protects the brain from CD8 T-cell-mediated immunopathology. Cell Rep. 2020;30(5):1585–15976. https://doi.org/10.1016/j.celrep.2019.12.097.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, Bhai J, Billis K, Boddu S, Marugán JC, Cummins C, Davidson C, Dodiya K, Fatima R, Gall A, Giron CG, Gil L, Grego T, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, Kay M, Lavidas I, Le T, Lemos D, Martinez JG, Maurel T, McDowall M, McMahon A, Mohanan S, Moore B, Nuhn M, Oheh DN, Parker A, Parton A, Patricio M, Sakthivel MP, Abdul Salam AI, Schmitt BM, Schuilenburg H, Sheppard D, Sycheva M, Szuba M, Taylor K, Thormann A, Threadgold G, Vullo A, Walts B, Winterbottom A, Zadissa A, Chakiachvili M, Flint B, Frankish A, Hunt SE, IIsley G, Kostadima M, Langridge N, Loveland JE, Martin FJ, Morales J, Mudge JM, Muffato M, Perry E, Ruffier M, Trevanion SJ, Cunningham F, Howe KL, Zerbino DR, Flicek P. Ensembl 2020. Nucleic Acids Res. 2019;48(D1):682–8. https://doi.org/10.1093/nar/gkz966.
Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, Barnes I, Berry A, Bignell A, Carbonell Sala S, Chrast J, Cunningham F, Di Domenico T, Donaldson S, Fiddes IT, García Girón C, Gonzalez JM, Grego T, Hardy M, Hourlier T, Hunt T, Izuogu OG, Lagarde J, Martin FJ, Martínez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Ruffier M, Schmitt BM, Stapleton E, Suner MM, Sycheva I, Uszczynska-Ratajczak B, Xu J, Yates A, Zerbino D, Zhang Y, Aken B, Choudhary JS, Gerstein M, Guigó R, Hubbard TJP, Kellis M, Paten B, Reymond A, Tress ML, Flicek P. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47(D1):766–73. https://doi.org/10.1093/nar/gky955.
Granjon D. bs4Dash: a ‘Bootstrap 4’ Version of ‘shinydashboard’. 2019. https://rinterface.github.io/bs4Dash/index.html, https://github.com/RinteRface/bs4Dash.
Chang W, Borges Ribeiro B. Shinydashboard: create dashboards with ‘Shiny’. (2018). R package version 0.7.1. https://CRAN.R-project.org/package=shinydashboard.
Ganz C. rintrojs: a wrapper for the intro. js library. J Open Source Softw. 2016;1(6):2016. https://doi.org/10.21105/joss.00063.
Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22(13):1600–7. https://doi.org/10.1093/bioinformatics/btl140.
Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J Integr Biol. 2012;16(5):284–7. https://doi.org/10.1089/omi.2011.0118.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. https://doi.org/10.1038/nprot.2008.211.
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):90–7. https://doi.org/10.1093/nar/gkw377.
Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, Wadi L, Meyer M, Wong J, Xu C, Merico D, Bader GD. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc. 2019;14(2):482–517. https://doi.org/10.1038/s41596-018-0103-9.
Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):191–8. https://doi.org/10.1093/nar/gkz369.
Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021. https://doi.org/10.1101/060012.
Agarwala R, Barrett T, Beck J, Benson DA, Bollin C, Bolton E, Bourexis D, Brister JR, Bryant SH, Canese K, Charowhas C, Clark K, DiCuccio M, Dondoshansky I, Feolo M, Funk K, Geer LY, Gorelenkov V, Hlavina W, Hoeppner M, Holmes B, Johnson M, Khotomlianski V, Kimchi A, Kimelman M, Kitts P, Klimke W, Krasnov S, Kuznetsov A, Landrum MJ, Landsman D, Lee JM, Lipman DJ, Lu Z, Madden TL, Madej T, Marchler-Bauer A, Karsch-Mizrachi I, Murphy T, Orris R, Ostell J, O’Sullivan C, Palanigobu V, Panchenko AR, Phan L, Pruitt KD, Rodarmer K, Rubinstein W, Sayers EW, Schneider V, Schoch CL, Schuler GD, Sherry ST, Sirotkin K, Siyan K, Slotta D, Soboleva A, Soussov V, Starchenko G, Tatusova TA, Todorov K, Trawick BW, Vakatov D, Wang Y, Ward M, Wilbur WJ, Yaschenko E, Zbicz K. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2017;45(D1):12–7. https://doi.org/10.1093/nar/gkw1071.
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, Kaplan S, Dahary D, Warshawsky D, Guan-Golan Y, Kohn A, Rappaport N, Safran M, Lancet D. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinform. 2016;54(1):1–30113033. https://doi.org/10.1002/cpbi.5.
Gamazon ER, Segrè AV, van de Bunt M, Wen X, ** HS, Hormozdiari F, Ongen H, Konkashbaev A, Derks EM, Aguet F, Quan J, Nicolae DL, Eskin E, Kellis M, Getz G, McCarthy MI, Dermitzakis ET, Cox NJ, Ardlie KG. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet. 2018;50(7):956–67. https://doi.org/10.1038/s41588-018-0154-4.
**e Y. Dynamic Documents with R and Knitr, p. 188. Chapman & Hall/CRC; 2013. https://doi.org/10.18637/jss.v056.b02. ar**v:ar**v:1501.0228. http://www.crcpress.com/product/isbn/9781482203530.
Rule A, Birmingham A, Zuniga C, Altintas I, Huang SC, Knight R, Moshiri N, Nguyen MH, Rosenthal SB, Pérez F, Rose PW. Ten simple rules for writing and sharing computational analyses in Jupyter Notebooks. PLoS Comput Biol. 2019;15(7):1–8. https://doi.org/10.1371/journal.pcbi.1007007.
Stodden V, Miguez S. Best practices for computational science: software infrastructure and environments for reproducible and extensible research. J Open Res Softw. 2014;2(1):21. https://doi.org/10.5334/jors.ay.
Rue-Albrecht K, Marini F, Soneson C, Lun ATL. iSEE: interactive summarized experiment explorer. F1000Research. 2018;7:741. https://doi.org/10.12688/f1000research.14966.1.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017. https://doi.org/10.1038/nmeth.4197. ar**v:1505.02710.
Lun ATL, Chen Y, Smyth GK. It’s DE-licious: a recipe for differential expression analyses of RNA-seq experiments using quasi-likelihood methods in edgeR. In: Mathé E, Davis S, editors. Statistical genomics, chap. 19. Humana Press; 2016. p. 391–416.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013;14:12. https://doi.org/10.1186/1471-2105-14-7.
Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE. 2010;5(11):13984. https://doi.org/10.1371/journal.pone.0013984.
Pomaznoy M, Ha B, Peters B. GOnet: a tool for interactive gene ontology analysis. BMC Bioinform. 2018;19(1):1–8. https://doi.org/10.1186/s12859-018-2533-3.
Almende BV, Thieurmel B, Robert T. visNetwork: network visualization using ‘vis.js’ library. (2019). R package version 2.0.9. https://CRAN.R-project.org/package=visNetwork.
Domagalski R, Neal ZP, Sagan B. Backbone: an R package for extracting the backbone of bipartite projections. PLoS ONE. 2021;16(1):0244363. https://doi.org/10.1371/journal.pone.0244363.
Geistlinger L, Csaba G, Zimmer R. Bioconductor’s EnrichmentBrowser: seamless navigation through combined results of set- & network-based enrichment analysis. BMC Bioinform. 2016;17(1):45. https://doi.org/10.1186/s12859-016-0884-1.
Alhamdoosh M, Ng M, Wilson NJ, Sheridan JM, Huynh H, Wilson MJ, Ritchie ME. Combining multiple tools outperforms individual methods in gene set enrichment analyses. Bioinformatics. 2016;33:623. https://doi.org/10.1093/bioinformatics/btw623.
Yoon S, Kim J, Kim S-K, Baik B, Chi S-M, Kim S-Y, Nam D. GScluster: network-weighted gene-set clustering analysis. BMC Genom. 2019;20(1):352. https://doi.org/10.1186/s12864-019-5738-6.
Acknowledgements
This work has been supported by the computing infrastructure provided by the Core Facility Bioinformatics at the University Medical Center Mainz, used also for deploying the demo instance. The authors thank the members of the Core Facility Bioinformatics at the Institute of Molecular Biology Mainz, Miguel Andrade (IOME, Johannes Gutenberg University of Mainz), Gerrit Toenges and Arsenij Ustjanzew (IMBEI Mainz), and Francesca Finotello (ICBI, Medical University of Innsbruck) for valuable feedback and suggestions.
Funding
Open Access funding enabled and organized by Projekt DEAL. The work of FM is supported by the German Federal Ministry of Education and Research (BMBF 01EO1003). The funding body had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
FM—conceptualization, data curation, formal analysis, funding acquisition, methodology, project administration, resources, software, supervision, visualization, writing—original draft, writing—review and editing. AL—data curation, formal analysis, methodology, software, visualization, writing—review and editing. JL—data curation, software, writing—review and editing. KS—conceptualization, funding acquisition, resources, supervision, writing—original draft, writing—review and editing. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Comparison of tools for assisting the interpretation of transcriptome data via functional enrichment analyses, including link to the related publications and to the source code repositories (if available). Evaluation criteria are included in the dedicated sheet. The information contained in this table are also available online at https://federicomarini.github.io/GeneTonic_supplement, displaying the content of the Google Sheet, accessible at https://docs.google.com/spreadsheets/d/1EQD8gNk6NID_8CJ-xetQm31XB4eRCXIsvVd4X0oaoPo/edit?usp=sharing).
Additional file 2.
Complete use case for the GeneTonic package, based on the RNA-seq dataset for macrophage immune stimulation (Interferon Gamma treatment vs naive cells, ERP020977).
Additional file 3.
Complete use case for the GeneTonic package on the RNA-seq A20-deficient microglia dataset (A20 deficiency vs control, GSE123033).
Additional file 4: Fig. S1.
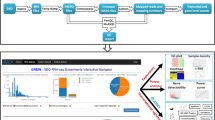
Screenshot of the Enrichment Map panel in the GeneTonic application. The sidebar menu (A) controls the main navigation in the app, and a common set of options is toggled with the cogs icon (B). The main area of the Enrichment Map panel (C) contains an interactive graph for the enrichment map of the genesets, connected according to their similarity, and color coded according to the specified geneset property (here, the Z-score). Upon clicking on any geneset, a Geneset Box (D) is displayed for further exploration (e.g. to show a volcano plot with the geneset members labelled). The geneset distillery (E) enables the exploration of meta-genesets, derived by computing clusters on the graph object underlying the enrichment map. From the tabular representation, it is possible to visualize meta-genesets as heatmaps (F), or display a modal popup containing the enrichment map where the cluster assignments of the genesets are shown (G).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Marini, F., Ludt, A., Linke, J. et al. GeneTonic: an R/Bioconductor package for streamlining the interpretation of RNA-seq data. BMC Bioinformatics 22, 610 (2021). https://doi.org/10.1186/s12859-021-04461-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-021-04461-5