
Abstract
Humicola insolens is an excellent producer of pH-neutral active, thermostable cellulases that find many industrial applications. In the present study, we developed an efficient Agrobacterium tumefaciens-mediated transformation system for H. insolens. We transformed plasmids carrying the promoter of the glyceraldehyde-3-phosphate dehydrogenase gene of H. insolens driving the transcription of genes encoding neomycin phosphotransferase, hygromycin B phosphotransferase and enhanced green fluorescent protein. We optimized transformation efficiency to obtain over 300 transformants/106 conidia. T-DNA insertional mutagenesis was employed to generate an H. insolens mutant library and we isolated a transformant termed T4 with enhanced cellulase and hemicellulase activities. The FPase, endoglucanase, cellobiohydrolase, β-glucosidase and xylanase activities of T4, measured at the end of fermentation, were 60%, 440%, 320%, 41% and 81% higher than those of the wild-type strain, respectively. We isolated the sequences flanking the T-DNA insertions and thus identified new genes potentially involved in cellulase and hemicellulase production. Our results show that it is feasible to use T-DNA insertional mutagenesis to identify novel candidate genes involved in cellulase production. This will be valuable when genetic improvement programs seeking to enhance cellulase production are planned and will also allow us to gain a better understanding of the genetics of the thermophilic fungus H. insolens.
Similar content being viewed by others

Introduction
Lignocellulose is the most abundant renewable biological resource on Earth and can be converted to mixed sugars prior to fermentation to yield biofuels and many other useful biomaterials. The use of enzymes active on carbohydrates to catalyze lignocellulose degradation has long been considered to be the most promising strategy. Enzyme-catalyzed processes, in contrast to acid hydrolysis, afford high yields of fermentable sugars and minimize environmental pollution. Presently, most commercially available cellulases used for biomass degradation are derived from Trichoderma reesei1. Although many studies have used classical mutagenesis and genetic modification in efforts to obtain hypercellulolytic T. reesei mutants2,3, the relatively low β-glucosidase activity of the strain, poor hemicellulose production, the need for acidophilic culture conditions and poor thermal stability remain major obstacles when seeking to employ T. reesei to efficiently hydrolyze lignocellulose and in many other applications.
Filamentous fungi of the genus Humicola are excellent producers of cellulases for industrial applications4. Humicola insolens is an innocuous non-toxic fungus producing a comprehensive profile of cellulases and hemicellulases, including at least two cellobiohydrolases, seven endoglucanases, two β-glucosidases, five xylanases and three xylosidases5,6,7,8,9,10,11,12,13. Recently, two further novel β-glucosidases of the glycosyl hydrolase family 3 were identified in H. insolens strain Y1 (unpublished data). Unlike most enzymes from acidophilic and mesophilic fungi, the cellulases and hemicellulases of H. insolens are active under neutral conditions, are alkali-tolerant and exhibit good thermostability5,7,11. Such properties render enzymes secreted by H. insolens outstanding choices for applications in many industries. EGV (Carezyme; Novozymes) has dominated the laundry market for several years and a multi-active β-glucanase preparation from H. insolens (Ultraflo L, Novozymes) is used by breweries in the mashing process. In addition, hemicellulases and cellulases from H. insolens degrade lignocellulose-rich materials, such as rice straw or wood chips, much more efficiently than do enzymes from T. reesei14. H. insolens also has served as an excellent host for overproduction of heterologous enzymes, especially neutral cellulases15. Thus, H. insolens should be seriously considered as an alternative to T. reesei in terms of biomass degradation.
However, despite the dramatic developments in biotechnology over the past few decades, few reports to date have focused on the genetics or molecular biology of H. insolens. In the present study, we establish an efficient Agrobacterium tumefaciens-mediated transformation (ATMT) system for H. insolens and create a mutant library using this system. Further, we isolate a mutant (termed T4) with enhanced cellulolytic capacity and identify the sequences flanking the T-DNA insertion sites. These results will help us gain a better understanding of the genetics of the organism and will greatly facilitate future genetic engineering of the fungus to obtain strains producing high levels of cellulase.
Materials and Methods
Strains, media and growth conditions
H. insolens Y116 was used as the recipient for transformation. Strain AGL-1 of A. tumefaciens was used to transform H. insolens.
To trigger sporulation, H. insolens Y1 was grown in potato-dextrose agar (PDA) medium for 7–12 days at 42 °C. The minimal medium (MM), induction medium (IM) and co-cultivation medium (CM) used for A. tumefaciens-mediated transformation (ATMT) were prepared as described previously17. MNN medium (per liter: 1 g tryptone, 20 g yeast extract, 0.6 g MgSO4·7H2O, 0.3 g CaCl2·2H2O and 20 g Avicel) was used for fermentation. A. tumefaciens was routinely grown at 28 °C.
Sensitivity testing for geneticin and hygromycin
The sensitivity of H. insolens to geneticin and hygromycin B was monitored by growing the strain on PDA medium. Spore suspensions (about 100 μL of a suspension of 1.0 × 105 spores/mL) were spread on PDA plates supplemented with various concentrations of geneticin (0, 25, 50, 100 and 150 μg/mL) or hygromycin B (0, 5, 10, 25 and 50 μg/mL). Growth was monitored during incubation at 42 °C for 3–5 days.
Plasmid construction
The promoter and terminator regions of the glyceraldehyde 3-phosphate dehydrogenase (gpd) gene were amplified with primers Pgpd-F/Pgpd-R and Tgpd-F/Tgpd-R (shown in Table 1), respectively. The PCR fragments were ligated into pEASY-Blunt (Transgen, China) to generate pB-Pgpd and pB-Tgpd, respectively. Next, pB-Tgpd was digested with EcoRV and XhoI and ligated into the corresponding sites of pBluescript I KS (+) to yield pTgpd. Next, pB-Pgpd was digested with NotI and BamHI and subcloned into the corresponding sites of pTgpd to generate pPgpd-T. A geneticin-resistance gene (neo) was amplified from pEGFP-N1 (Clontech) with the aid of primers npt-F/npt-R. After cloning into pEASY-Blunt, the amplified DNA fragments were digested with BamHI-EcoRV and subcloned into the corresponding sites of the vector pPgpd-T, to generate pP-neo-T. Then, the entire Pgpd-neo-Tgpd fragment was amplified from pP-neo-T using Pgpd-F-2/Tgpd-R. The fragment was subcloned into pEASY-Blunt, digested with SpeI and XhoI and ligated into the corresponding sites of pAg1-H318 to generate pAg1-neo, in which the hph gene driven by the A. nidulans trpC promoter was thus replaced by the neo gene driven by the H. insolens gpd promoter. We also constructed a plasmid that could be used to express heterogeneous genes. The strategy was as follows: First, pAg1-H3 was digested with BglII and self-ligated to remove the PtrpC-hph-TtrpC cassette, yielding pAg1. Next, the hph fragment was amplified from pAg1-H3 using the primers hph-F/hph-R. After ligation into pEASY-Blunt, the hph fragment was digested with BamHI and EcoRV and subcloned into the corresponding sites of the pPgpd-T-2 vector. This vector was constructed in the same manner as pPgpd-T except that Pgpd was amplified with Pgpd-F-3/Pgpd-R to remove the SmaI site located upstream of Pgpd-F-3, to yield pP-hph-T. The entire Pgpd-hph-Tgpd cassette was obtained by PvuII digestion and ligated into the corresponding site of pAg1 to generate pAg1-hyg. Next, Pgpd and Tgpd fragments were obtained from pPgpd-T-2 via PvuII digestion and ligated into the SwaI site of pAg1-hyg, to yield pAg1-hyg-P-T. As only one SmaI site lies between Pgpd and Tgpd [the site is derived from pBluescript I KS(+)] in pAg1-hyg-P-T, any gene can be blunt-end ligated into that site. The egfp gene was amplified from pEGFP-N1 (Clontech) using primers gfp-F/gfp-R and ligated into pEASY-Blunt. After digestion with EcoRV, the fragment was cloned into the SmaI site of pAg1-hyg-P-T, in the appropriate direction, to yield pAg-egfp. Plasmids pAg1-neo and pAg-egfp were introduced into wild-type H. insolens by ATMT.
A. tumefaciens-mediated transformation
The transformation procedure was based on a previously described protocol19 with some modifications. Spores were obtained on PDA medium after culture at 42 °C for 10 days. A. tumefaciens was grown at 28 °C for 2 days in liquid MM medium supplemented with 50 μg/mL kanamycin. Bacterial cell suspensions were subsequently diluted to an optical density at 600 nm (OD600) of 0.2; induction medium (IM) with 200–500 μM acetosyringone (AS) was used for dilution. The cells were grown for an additional 6 h to an OD600 of 0.4–0.8. Next, 100 μL of bacterial culture previously induced with 400 μM AS were added to CM plates containing fungal mycelia that had been allowed to germinate for various periods (6–30 h) on cellophane paper. Co-cultivation proceeded at various temperatures (22 °C, 25 °C, or 28 °C) for 24–96 h. Next, the mixed cultures were transferred to PDA medium supplemented with 200 μg/mL cefotaxime and 100 μg/mL geneticin to select fungal transformants. Each factor was varied with all other factors held constant.
Fluorescence microscopy
The hyphae of randomly selected H. insolens transformant harboring pAg-egfp were prepared on PDA plates without hygromycin B. After 2 d growth, the edge of the colony was observed. The green fluorescence emission from EGFP was detected using a Nikon Eclipse Ni microscope (Nikon, Japan). Images were taken under 40 × objective and processed with NIS-Elements BR 3.0 imaging software (Nikon, Japan).
Molecular analysis of transformants
Fungal genomic DNA was extracted from mycelia grown on PDA plates. The mycelia were harvested, dried with filter paper and ground in liquid nitrogen using a sterilized mortar and pestle. Genomic DNA was isolated with the aid of a DNA Quick Plant System (TianGen, China). The neo gene was detected via PCR using primers neo-F/neo-R, which amplified a 555-bp sequence spanning the gene. Routine PCR amplification consisted of initial denaturation at 95 °C; followed by 30 cycles of amplification (95 °C for 30 s, 58 °C for 30 s and 72 °C for 40 s); followed by an additional 10 min at 72 °C.
Mitotic stability of transformants
To evaluate stability, 20 randomly selected transformants were cultured on PDA plates without geneticin for 7 d. Mycelia from the edges of the cultures were transferred to fresh PDA plates and grown for another 7 d. After repeating this procedure 5 times, germinating mycelia from each transformant were transferred to PDA plates containing 100 μg/mL geneticin.
Screening for mutants with improved cellulase activity
T-DNA inserted mutants and the wild-type H. insolens Y1 strain were inoculated onto screening plates (15 g wheat bran, 0.8 g bean pulp and 0.8 g grass meal pellets were boiled in 1 L ddH2O for 10 min, the supernatant was harvested and the volume adjusted to 1 L with ddH2O, followed by addition of 2% Avicel and 1.5% agar). After growth at 42 °C for 3–5 days, the target mutants were selected on the basis of colony size and hyphal morphology using the wild-type strain as the control.
TAIL-PCR analysis of integration junctions
TAIL-PCR was performed as previously described20. Genomic DNA from selected transformants was extracted and purified as described above. The degenerate primers (LAD1-1, LAD1-3, LAD1-5 and LAD1-11) and the nested LB-specific primers (LB1, LB2 and LB3) and RB-specific primers (RB-A, RB-B and RB-C) used are listed in Table 1. Reaction products were recovered and sequenced.
Cellulase assay
The wild-type and T4 strains were fermented in 40 mL of MNN medium inoculated with 1.0 × 106 12-day-old spores at 42 °C with shaking at 200 rpm. Cellulase activities in supernatants were assayed at various intervals. FPase, CMCase and xylanase were assayed using the 3,5-dinitrosalicylic acid (DNS) method21. Whatman No.1 filter papers (1 × 6 cm in area) were immersed in 2 mL of appropriately diluted supernatants rendered to 100 mM in Na2HPO4-citric acid (pH 6.0) at 60 °C for 30 min. Next, 3 mL of DNS were added to terminate the reaction. The mixtures were boiled for 5 min and the absorbances at 540 nm determined. The standard assays for CMCase and xylanase featured the addition of 100 μL of appropriately diluted supernatants to 900 μL of 100 mM Na2HPO4-citric acid (pH 6.0) containing 1.0% (w/v) CMC-Na or birchwood xylan, followed by incubation at 60 °C for 10 min. Each reaction was terminated by the addition of 1.5 mL of DNS reagent and boiling for 5 min. The absorbance at 540 nm was determined when the reaction mixture had cooled to room temperature. Glucosidase activity was measured using p-nitrophenyl-β-D-glucopyranoside (pNPG) as a substrate. The reaction featured the addition of 250 μL of 4 mM pNPG in 100 mM Na2HPO4-citric acid (pH 6.0) to 250 μL of appropriately diluted solutions, followed by incubation at 60 °C for 10 min. Finally, 1.5 mL of 1.0 M Na2CO3 were added to terminate the reaction and the liberated p-nitrophenyl was detected by measuring the absorbance at 420 nm. Cellobiohydrolase activity was measured in the same way, except that p-nitrophenyl-β-D-cellobioside (pNPC) served as a substrate. One unit of enzyme activity was defined as the amount of enzyme required to release 1 μmoL of reduced sugar (for FPase, CMCase and xylanase) or 1 μmoL of p-nitrophenyl (for glucosidase and cellobiohydrolase) from the substrate, per min, under the conditions described above. Glucose and p-nitrophenyl served as standards.
SDS-PAGE and protein assay
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) was performed as described by Laemmli22 with 12% polyacrylamide gel. Proteins were stained with Coomassie Brilliant Blue G-250. Protein concentration was measured by the Bradford method23 using a protein assay kit (Bio-Rad).
Nucleotide sequence accession numbers
The nucleotide sequences of gpd, proA and tdiD from H. insolens Y1 have been deposited in GenBank under accession nos. KU847960, KU836630 and KU836631, respectively.
Results
Construction of binary vectors for A. tumefaciens-mediated transformation of H. insolens Y1
To aid in the development of a rapid and efficient transformation method for H. insolens Y1, we tested geneticin and hygromycin B currently used as selectable markers in fungal transformations. We found that geneticin at ≥50 μg/mL or hygromycin B at ≥10 μg/mL completely inhibited conidial germination by the H. insolens Y1 wild-type strain (Figure S1). Thus, both antibiotics were used to select H. insolens transformants.
To transform H. insolens Y1, we constructed the binary vector pAg1-neo using the backbone of plasmid pAg1-H3 (Fig. 1A). To efficiently drive the expression of the antibiotic-resistance gene, we cloned the promoter and terminator of the glyceraldehyde 3-phosphate dehydrogenase gene (gpd) from H. insolens and used this promoter to drive the neo gene. We also constructed the binary vector pAg1-hyg-P-T for expression of heterologous genes (Fig. 1B). To determine the utility of this plasmid, the gene encoding enhanced green fluorescent protein (egfp) was cloned into the vector, yielding pAg-egfp.

Schematic representation of pAg1-neo (A) and pAg-egfp (B). The positions of Pgpd and Tgpd (the promoter and terminator of the H. insolens Y1 gpd gene, respectively), neo (geneticin-resistance gene), hph (hygromycin B-resistance gene); egfp (enhanced green fluorescent protein gene) and RB and LB (the right and left borders of T-DNA, respectively) are shown.
Transformation competency of H. insolens with the constructed vectors
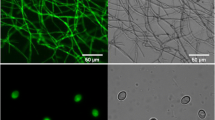
A. tumefaciens strain AGL-1, harboring the binary vector pAg1-neo, was used to transform H. insolens Y1. Plasmid pAg1-neo was confirmed to be transformation-competent; the transformants grew on PDA plates containing 100 μg/mL geneticin (data not shown). After transformation of pAg-egfp into the wild-type strain, strong green fluorescence was observed by fluorescence microscopy (Fig. 2), suggesting that the egfp gene was successfully expressed.

Fluorescence of mycelia of H. insolens transformed with the egfp gene.
One transformant (gfp) was randomly selected. Hyphae were observed by both bright-field (BF) microscopy and fluorescence (GFP) microscopy after growth on PDA plates for 2 days. Bar: 10 μm.
Optimization of an ATMT system for H. insolens
The ATMT procedure for H. insolens was optimized by determining the effects of various parameters on transformation efficiency. Pre-germination is crucial for successful transformation. No transformants were obtained using non-germinated spores. Commencing at 6 h of pre-germination, transformation efficiency increased to 24 h and fell thereafter (Fig. 3A). Co-cultivation time also affected transformation efficiency. Maximum efficiency was observed after 48 h of co-cultivation and was maintained to 72 h (Fig. 3B). The optimal level of AS was found to be 300–400 μM (Fig. 3C). A. tumefaciens cell concentration notably affected the transformation efficiency of H. insolens; the maximum transformation efficiency was observed at an OD600 of 0.6 (Fig. 3D). As the cell concentration increased, the transformation efficiency fell significantly. The optimal temperature for co-cultivation was 25 °C (Fig. 3E).

Factors affecting the transformation efficiency of H. insolens.
(A) Effect of pre-germination on transformation efficiency. 400 μM AS, OD600 of 0.6 (A. tumefaciens cells), 25 °C (co-cultivation temperature) and 2 days (co-cultivation time) were used in the experiments. Colonies that grew were considered to be transformants. (B) Effect of co-cultivation time on transformation efficiency. 400 μM AS, 24 h (pre-germination time), OD600 of 0.6 (A. tumefaciens cells) and 25 °C (co-cultivation temperature) were used in the experiments. (C) Effect of AS concentration on transformation efficiency. 24 h (pre-germination time), OD600 of 0.6 (A. tumefaciens cells), 25 °C (co-cultivation temperature) and 2 days (co-cultivation time) were used in the experiments. (D) Effect of A. tumefaciens cell concentration on transformation efficiency. 400 μM AS, 24 h (pre-germination time), 25 °C (co-cultivation temperature) and 2 days (co-cultivation time) were used in the experiments. (E) Effect of co-cultivation temperature on transformation efficiency. 400 μM AS, 24 h (pre-germination time), OD600 of 0.6 (A. tumefaciens cells) and 2 days (co-cultivation time) were used in the experiments. Error bars represent standard deviations from three independent experiments.
Molecular analysis of transformants
To confirm that T-DNA had become integrated into the H. insolens genome, seven putative geneticin-resistant transformants were randomly selected for PCR analysis using the neo-specific primers neo-F and neo-R. In all mutants, a 555-bp neo-specific fragment was amplified, indicating that the T-DNA was in fact integrated (Fig. 4).

PCR products of neo from the genomic DNA of H. insolens transformants.
All strains were grown on PDA plates for 2 days and genomic DNA extracted. Primers neo-F/neo-R were used. M, 100 bp DNA ladder; WT, the wild-type strain; 1–7, randomly selected transformants; N, negative control.
Mitotic stability of transformants
In other fungi, DNA integrated via ATMT was relatively stable during growth in the absence of selective antibiotics24,25. This is an important feature of an effective mutagenesis system. After five subcultures in the absence of geneticin, all randomly selected H. insolens transformants tested grew on PDA with 100 μg/mL geneticin, confirming the genetic stability of integrated DNA (Figure S2).
T-DNA insertional mutagenesis and screening for mutants with improved cellulase activity
A T-DNA tagged mutant library of H. insolens was obtained by one-time ATMT transformation of about 1 × 107 fungal spores. 1000 randomly selected transformants were screened by screening plates. Transformants exhibiting phenotypic changes on screening plates were fermented in MNN medium for 5 d and the cellulase activity of the supernatants was determined. One promising strain (designated T4) with obvious phenotypic changes compared to the WT (Figure S3), exhibited improved cellulolytic activity (FPase activity; 50–60% increase) when grown in fermentation medium for 4–6 days, compared to the parental strain Y1 (Fig. 5B). The endoglucanase and cellobiohydrolase activities of T4 were 4.4- and 3.2-fold higher, respectively, than those of the wild-type strain after 6 days of fermentation (Fig. 5C,D). The T4 glucosidase and xylanase activities were also elevated (by 41% and 81% at 6 days of growth, respectively) (Fig. 5E,F). T4 was further characterized in terms of the protein expression profile. SDS–PAGE showed that the T4 fermentation supernatant contained more secreted proteins than did the wild-type supernatant (Figure S4A). As the biomasses attained by the T4 and wild-type strains did not differ significantly (Fig. 5A), the enhanced cellulolytic capacity of T4 may be attributable to the secretion of more cellulolytic enzymes by that strain (compared to the wild-type strain; Figure S4B).

Characterization of strain T4.
Over a 6-day fermentation period (A) dry weight, (B) total cellulase activity (FPase), (C) endoglucanase activity (CMCase), (D) cellobiohydrolase activity, (E) xylanase activity and (F) glucosidase activity were measured. Error bars represent standard deviations from three independent experiments.
Cloning of genomic DNA flanking T-DNA insertion sites
TAIL-PCR was used to isolate T4 genomic DNA segments adjacent to the T-DNA inserts. Sequencing of the flanking T-DNA junctions showed that the T4 genome had two T-DNA integration sites. One was in the promoter region (85 bp upstream of the starting ATG) of a gene encoding a putative Zn(2)-Cys(6) transcriptional regulatory protein, designated proA. The ProA polypeptide shares 67% sequence identity with Cryphonectria parasitica Pro1 and Sordaria macrospora Pro1 and 44% with Aspergillus nidulans NosA, which have previously been shown to play important roles in sexual reproduction26,27,28. The other integration site was in the middle of a putative aminotransferase-encoding gene, designated tdiD. The TdiD polypeptide shares 65% sequence identity with Aspergillus nidulans TdiD, which is involved in biosynthesis of the antitumor fungal metabolite terrequinone A29.
Discussion
H. insolens produces a variety of cellulases and hemicellulases that find ready applications in industry. Nevertheless, the fungus remains poorly known genetically. Development of efficient transformation and expression systems will facilitate molecular genetic analysis and gene manipulation, including heterologous gene expression, functional analysis of targeted genes and genetic engineering of the original strain.
ATMT is an essential tool when studying the functional genomics of filamentous fungi and has been reported to be applicable to many such fungi18,19. We describe successful A. tumefaciens-mediated transformation of H. insolens. The promoter of the endogenous glyceraldehyde 3-phosphate dehydrogenase gene efficiently drove the expression of exogenous genes, including geneticin- and hygromycin B-resistance genes and the gene encoding the enhanced green fluorescent protein. We thus constructed and tested a doubly functional binary Agrobacterium vector, pAg1-hyg-P-T, bearing a hygromycin B-resistance expression cassette and another copy of both Pgpd and Tgpd. pAg1-hyg-P-T can be used to express any gene of interest in H. insolens Y1. The plasmid can serve as an expression vector or as a backbone plasmid if gene knockout is planned. We optimized the transformation efficiency to obtain over 300 transformants/106 conidia. This efficiency is similar with those obtained for Fusarium oxysporum19 (300–500 transformants/106 conidia), Aspergillus terreus (350 transformants/106 conidia)25 and A. awamori (200–250 transformants/106 conidia)30 and much higher than many other fungi, such as A. fumigatus (100 transformants/107 conidia)31, Guignardia citricarpa (14–16 transformants/106 spores)32 and Lecanicillium lecanii (25 transformants/106 conidia)33. This will facilitate functional genetic analysis of and enhancement of cellulase synthesis by H. insolens.
In addition to the introduction of foreign genes, ATMT has also been an effective way for strain improvement, since non-homologous recombination of T-DNA into the host genome often triggers gene disruption, creating mutant phenotypes34. ATMT has been applied to eliminate mycotoxin production by an industrially important strain of Monascus purpureus35, to enhance the resistance of Lecanicillium lecanii to benzimidazole fungicides33 and to improve cellulase production by T. reesei36. Our T4 mutant exhibited a cellulolytic capacity that was dramatically greater than that of the wild-type. The FPase activity increased by 60%, attaining a level equal to that obtained when a multistep mutation strategy (alternating ultraviolet light and chemical treatments) was employed37. In particular, the endoglucanase and cellobiohydrolase activities of T4 were 4.4- and 3.2-fold higher than those of the wild-type strain, respectively. These figures were much higher than those obtained using a multistep mutation strategy37. Moreover, the glucosidase and xylanase activities also improved, by 41% and 81%, respectively, suggesting that T-DNA insertional mutagenesis globally enhanced cellulase and hemicellulase production. These results showed that ATMT effectively improved cellulase production by H. insolens.
The pivotal advantage of insertional mutagenesis compared to chemical or radiation mutagenesis is that the disrupted genes and their flanking sequences can be conveniently identified38. In the present study, we identified T-DNA insertions in two genes, proA and tdiD, from the cellulase-hyperproducing mutant T4. tdiD encodes a protein of the pyridoxal phosphate (PLP)-dependent aspartate aminotransferase superfamily, which participates in the secondary metabolism of Aspergillus nidulans29. proA encodes a putative transcription factor that contains a GAL4-like Zn(II)2Cys6-binuclear cluster DNA-binding domain and a fungal-specific transcription factor domain. Proteins homologous to ProA play important roles in many fungal biological processes. The first identified Pro1 (that of S. macrospora) plays a central role in sexual development26,39. In Alternaria brassicicola, disruption of pro1 resulted in significant reductions in virulence and the rate of vegetative growth40. ProA is an essential regulator of the mutual symbiotic interaction between Epichloë festucae and perennial ryegrass28. However, no study has yet found that ProA-like proteins function in cellulase production. Our results suggest that proA and/or tdiD are novel candidate genes that may be involved in cellulase and hemicellulase production by H. insolens, even though their contribution to these processes needs to be validated by further studies.
Additional Information
How to cite this article: Xu, X. et al. The use of T-DNA insertional mutagenesis to improve cellulase production by the thermophilic fungus Humicola insolens Y1. Sci. Rep. 6, 31108; doi: 10.1038/srep31108 (2016).
References
Lynd, L. R., Weimer, P. J., van Zyl, W. H. & Pretorius, I. S. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol R 66, 506–577 (2002).
Peterson, R. & Nevalainen, H. Trichoderma reesei RUT-C30–thirty years of strain improvement. Microbiology 158, 58–68, 10.1099/mic.0.054031-0 (2012).
Wang, S. et al. Enhancing cellulase production in Trichoderma reesei RUT C30 through combined manipulation of activating and repressing genes. J Ind Microbiol Biot 40, 633–641 (2013).
Maheshwari, R., Bharadwaj, G. & Bhat, M. K. Thermophilic fungi: their physiology and enzymes. Microbiol Mol Biol R 64, 461–488 (2000).
Du, Y. L. et al. Characterization of three novel thermophilic xylanases from Humicola insolens Y1 with application potentials in the brewing industry. Bioresour Technol 130, 161–167, 10.1016/j.biortech.2012.12.067 (2013).
Meleiro, L. P. et al. A novel β-glucosidase from Humicola insolens with high potential for untreated waste paper conversion to sugars. Appl Biochem Biotech 173, 391–408, 10.1007/s12010-014-0847-9 (2014).
Schulein, M. Enzymatic properties of cellulases from Humicola insolens. J Biotech 57, 71–81 (1997).
Shi, P. et al. Molecular characterization of a new alkaline-tolerant xylanase from Humicola insolens Y1. Biomed Res Int 2015, 149504, 10.1155/2015/149504 (2015).
Souza, F. H. M., Inocentes, R. F., Ward, R. J., Jorge, J. A. & Furriel, R. P. M. Glucose and xylose stimulation of a β-glucosidase from the thermophilic fungus Humicola insolens: A kinetic and biophysical study. J Mol Catal B-Enzym 94, 119–128, 10.1016/j.molcatb.2013.05.012 (2013).
**a, W. et al. High level expression of a novel family 3 neutral β-xylosidase from Humicola insolens Y1 with high tolerance to D-xylose. PloS one 10, e0117578, 10.1371/journal.pone.0117578 (2015).
Xu, X. et al. A Neutral Thermostable β-1,4-Glucanase from Humicola insolens Y1 with Potential for Applications in Various Industries. PloS one 10, e0124925, 10.1371/journal.pone.0124925 (2015).
Yang, X. et al. Two xylose-tolerant GH43 bifunctional β-xylosidase/α-arabinosidases and one GH11 xylanase from Humicola insolens and their synergy in the degradation of xylan. Food Chem 148, 381–387, 10.1016/j.foodchem.2013.10.062 (2014).
Yang, X. et al. A new GH43 α-arabinofuranosidase from Humicola insolens Y1: biochemical characterization and synergistic action with a xylanase on xylan degradation. Appl Biochem Biotech 175, 1960–1970, 10.1007/s12010-014-1416-y (2015).
Matsumoto, H. et al. Cell dispersion culture for the effective growth of Humicola insolens and efficient enzyme production. J Biosci Bioeng, 10.1016/j.jbiosc.2013.08.014 (2013).
Koga, J. et al. Purification and characterization of a new family 45 endoglucanase, STCE1, from Staphylotrichum coccosporum and its overproduction in Humicola insolens. App Environ Microb 74, 4210–4217, 10.1128/aem.02747-07 (2008).
Luo, H. et al. Gene cloning, expression and biochemical characterization of an alkali-tolerant β-mannanase from Humicola insolens Y1. J Ind Microbiol Biot 39, 547–555, 10.1007/s10295-011-1067-8 (2012).
Khang, C. H., Park, S.-Y., Rho, H.-S., Lee, Y.-H. & Kang, S. Filamentous fungi (Magnaporthe grisea and Fusarium oxysporum). Agrobacterium Protocols Volume 2, 403–420 (2007).
Zhang, A. et al. Efficient disruption of a polyketide synthase gene (pks1) required for melanin synthesis through Agrobacterium-mediated transformation of Glarea lozoyensis. Mol Genet Genomics 268, 645–655 (2003).
Mullins, E. D. et al. Agrobacterium-mediated transformation of Fusarium oxysporum: An efficient tool for insertional mutagenesis and gene transfer. Phytopathology 91, 173–180, 10.1094/phyto.2001.91.2.173 (2001).
Liu, Y. G. & Chen, Y. High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. BioTechniques 43, 649–650, 652, 654 passim (2007).
Miller, G. L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal chem 31, 426–428 (1959).
Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 (1970).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–254 (1976).
**g Zhang, J. et al. An efficient Agrobacterium-mediated transformation method for the edible mushroom Hypsizygus marmoreus. Microbiol. Res. 169, 741–748 (2014).
Wang, D. et al. An efficient tool for random insertional mutagenesis: Agrobacterium tumefaciens-mediated transformation of the filamentous fungus Aspergillus terreus. J Microbiol Meth 98, 114–118 (2014).
Masloff, S., Jacobsen, S., Pöggeler, S. & Kück, U. Functional analysis of the C6 zinc finger gene pro1 involved in fungal sexual development. Fungal Genet Biol 36, 107–116 (2002).
Sun, Q., Choi, G. H. & Nuss, D. L. Hypovirus-responsive transcription factor gene pro1 of the chestnut blight fungus Cryphonectria parasitica is required for female fertility, asexual spore development and stable maintenance of hypovirus infection. Eukaryot cell 8, 262–270 (2009).
Tanaka, A. et al. ProA, a transcriptional regulator of fungal fruiting body development, regulates leaf hyphal network development in the Epichloë festucae–Lolium perenne symbiosis. Mol Microbiol 90, 551–568 (2013).
Balibar, C. J., Howard-Jones, A. R. & Walsh, C. T. Terrequinone A biosynthesis through L-tryptophan oxidation, dimerization and bisprenylation. Nat Chem Biol 3, 584–592 (2007).
Michielse, C. B., Hooykaas, P. J., van den Hondel, C. A. & Ram, A. F. Agrobacterium-mediated transformation of the filamentous fungus Aspergillus awamori. Nat Protoc 3, 1671–1678 (2008).
Sugui, J. A., Chang, Y. C. & Kwon-Chung, K. Agrobacterium tumefaciens-mediated transformation of Aspergillus fumigatus: an efficient tool for insertional mutagenesis and targeted gene disruption. Appl Environ Microbiol 71, 1798–1802 (2005).
Figueiredo, J. et al. Agrobacterium tumefaciens-mediated transformation of Guignardia citricarpa. J Microbiol Meth 80, 143–147 (2010).
Zhang, Y.-J., Zhao, J.-J., **e, M. & Peng, D.-L. Agrobacterium tumefaciens-mediated transformation in the entomopathogenic fungus Lecanicillium lecanii and development of benzimidazole fungicide resistant strains. J Microbiol Meth 105, 168–173 (2014).
Zhang, P. et al. Agrobacterium tumefaciens-mediated transformation as a tool for insertional mutagenesis in the fungus Penicillium marneffei. Mycol Res 112, 943–949 (2008).
Jia, X. Q., Xu, Z. N., Zhou, L. P. & Sung, C. K. Elimination of the mycotoxin citrinin production in the industrial important strain Monascus purpureus SM001. Metab Eng 12, 1–7 (2010).
Zhong, Y., Wang, X., Yu, H., Liang, S. & Wang, T. Application of T-DNA insertional mutagenesis for improving cellulase production in the filamentous fungus Trichoderma reesei. Bioresour Technol 110, 572–577, 10.1016/j.biortech.2012.01.129 (2012).
Muhammad mohsin Javed, I.-U.-H. & Irfana, Mariyam Multistep mutagenesis for the over-expression of cellulase in Humicola insolens. Pak. J. Bot 43, 669–677 (2011).
Michielse, C. B., Hooykaas, P. J., van den Hondel, C. A. & Ram, A. F. Agrobacterium-mediated transformation as a tool for functional genomics in fungi. Curr Genet 48, 1–17 (2005).
Masloff, S., Pöggeler, S. & Kück, U. The pro1+ gene from Sordaria macrospora encodes a C6 zinc finger transcription factor required for fruiting body development. Genetics 152, 191–199 (1999).
Cho, Y. et al. Identification of novel virulence factors associated with signal transduction pathways in Alternaria brassicicola. Mol Microbiol 72, 1316–1333 (2009).
Acknowledgements
The authors sincerely thank Prof. Gang Liu and Prof. **ngzhong Liu (Institute of Microbiology, Chinese Academy of Sciences) for providing the plasmid pAg1-H3. This research was supported by the National High Technology Research and Development Program of China (2012AA022105, 2012AA022207).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: X.X., J.L., P.S., B.Y., Y.F. and W.Z. Performed the experiments: X.X., J.L. and W.J. Analyzed the data: X.X., J.L., P.S., B.L. and Y.Z. Contributed reagents/materials/analysis tools: W.Z. and B.Y. Wrote the paper: X.X. and J.L.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xu, X., Li, J., Shi, P. et al. The use of T-DNA insertional mutagenesis to improve cellulase production by the thermophilic fungus Humicola insolens Y1. Sci Rep 6, 31108 (2016). https://doi.org/10.1038/srep31108
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31108
- Springer Nature Limited
This article is cited by
-
A versatile system for fast screening and isolation of Trichoderma reesei cellulase hyperproducers based on DsRed and fluorescence-assisted cell sorting
Biotechnology for Biofuels (2018)
-
The use of Agrobacterium-mediated insertional mutagenesis sequencing to identify novel genes of Humicola insolens involved in cellulase production
3 Biotech (2018)