
Abstract
Pediatric neoplasms in the central nervous system (CNS) show extensive clinical and molecular heterogeneity and are fundamentally different from those occurring in adults. Molecular genetic testing contributes to accurate diagnosis and enables an optimal clinical management of affected children. Here, we investigated a rare, molecularly distinct type of pediatric high-grade neuroepithelial tumor (n = 18), that was identified through unsupervised visualization of genome-wide DNA methylation array data, together with copy number profiling, targeted next-generation DNA sequencing, and RNA transcriptome sequencing. DNA and/or RNA sequencing revealed recurrent fusions involving the capicua transcriptional repressor (CIC) gene in 10/10 tumor samples analyzed, with the most common fusion being CIC::LEUTX (n = 9). In addition, a CIC::NUTM1 fusion was detected in one of the tumors. Apart from the detected fusion events, no additional oncogenic alteration was identified in these tumors. The histopathological review demonstrated a morphologically heterogeneous group of high-grade neuroepithelial tumors with positive immunostaining for markers of glial differentiation in combination with weak and focal expression of synaptophysin, CD56 and CD99. All tumors were located in the supratentorial compartment, occurred during childhood (median age 8.5 years) and typically showed early relapses. In summary, we expand the spectrum of pediatric-type tumors of the CNS by reporting a previously uncharacterized group of rare high-grade neuroepithelial tumors that share a common DNA methylation signature and recurrent gene fusions involving the transcriptional repressor CIC. Downstream functional consequences of the fusion protein CIC::LEUTX and potential therapeutic implications need to be further investigated.
Similar content being viewed by others


Introduction
Pediatric neoplasms in the central nervous system (CNS) are extremely heterogeneous and diagnostically challenging tumors. According to data from the Central Brain Tumor Registry of the United States (CBTRUS), CNS tumors have become the leading cause of cancer-related death in childhood1. Accurate diagnosis is crucial for an optimal management of children with these diseases. During the last several years, remarkable advances in our understanding of the molecular underpinnings of these tumors have occurred as a result of comprehensive (epi-)genetic profiling and led to substantial progress in the classification and therapy of pediatric CNS tumors2. In addition, several novel and extremely rare tumor types have been identified using state-of-the-art molecular methods such as DNA methylation arrays and genetic profiling3. More recently, a wide range of different oncogenic gene fusions outside the mitogen-activated protein kinase (MAPK) pathway have shown to play an important role in driving tumorigenesis of pediatric CNS tumors4,5,6, some of them with the potential to provide novel therapeutic options.
DNA methylation profiling of CNS tumors has been demonstrated to be a powerful tool for molecular tumor classification with the additional evaluation of copy number profiles being extremely useful for the identification of oncogenic gene fusions4,7,8,9. Such an approach is particularly valuable for the discovery and characterization of rare and novel tumor types that show a wide variety of clinicopathological appearances5.
Here, we describe a novel molecular CNS tumor type, primarily occurring in children, identified through unsupervised visualization of a large cohort of genome-wide DNA methylation profiling data, together with targeted next-generation DNA sequencing, and RNA transcriptome sequencing.
Results
DNA methylation profiling reveals an epigenetically distinct group of pediatric-type neuroepithelial tumors
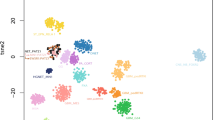
Through unsupervised visualization of genome-wide DNA methylation data from a large cohort of approximately 90,000 pediatric and adult CNS tumor samples, we identified an epigenetically distinct group of tumors (n = 16), that did not match any known DNA methylation class. This group was comprised of tumors with a wide spectrum of original histological diagnoses, including predominantly high-grade glioma (such as glioblastoma, anaplastic astrocytoma, or ganglioglioma), with many tumors considered as not classifiable or with a descriptive diagnosis (Supplementary Table 1). In addition, two further CNS tumor samples harboring a CIC::LEUTX fusion (see below) that have already been published were included into subsequent molecular profiling10. A more selected visualization (t-SNE) of DNA methylation patterns of tumors in this novel cluster compared with well-characterized reference samples (tumor samples included in the current version of the Heidelberg DNA methylation brain tumor classifier with a calibrated score >0.9; Supplementary Table 2) confirmed a clearly distinct grou** (Fig. 1a). Importantly, no similarity was seen with the recently described tumor type “CIC-rearranged sarcoma” (previously CNS Ewing sarcoma family tumor with CIC alteration; Fig. 1b)11. Further analysis of differentially methylated regions between tumors within the novel group and CIC-rearranged sarcoma showed aberrant methylation patterns, including promoter region hypomethylation amongst others of CD44, EMP3, and VIM in these tumors (Fig. 1c). This was supported by an inverse expression profile of the respective markers by immunohistochemistry (n = 4; Fig. 1d). Analysis of copy number profiles derived from the raw intensities of the DNA methylation array probes revealed recurrent structural aberrations on chromosome 19q around the genetic loci of the capicua transcriptional repressor (CIC) and leucine twenty homeobox (LEUTX) in all samples (Fig. 2a and Supplementary Fig. 1). Further recurrent copy number alterations included: loss of chromosome 1p, 13q, 14q, and 22q (Fig. 2b). Gain of chromosome 8, typically present in CIC-rearranged sarcoma (Fig. 2c), was seen in a high proportion of cases as well (Fig. 2b). A summary of detected structural aberrations is given in Supplementary Table 3.

a Unsupervised, nonlinear t-distributed stochastic neighbor embedding (t-SNE) projection of DNA methylation array profiles from 1059 tumors. DNA methylation profiling reveals a molecular distinct group of high-grade neuroepithelial tumors (HGNET, CIC fusion-positive; n = 18). b t-SNE analysis of DNA methylation array profiles of HGNET, CIC fusion-positive, and CIC-rearranged sarcoma (CNS SARC, CIC). For DNA methylation class abbreviations, see Supplementary Table 2. c Volcano plot comparing differentially methylated probes between HGNET, CIC fusion-positive and CNS SARC, CIC. d Immunohistochemical expression of vimentin (VIM), CD44, and EMP3 in HGNET, CIC fusion-positive and CNS SARC, CIC. Scale bars 300 μm.

a Copy number profile derived from DNA methylation array data of a HGNET, CIC fusion-positive showing structural alterations affecting chromosome 19q around the CIC and LEUTX locus. b, c Summary plot of copy number alterations in HGNET, CIC fusion-positive and CIC-rearranged sarcoma (CNS SARC, CIC). d Visualization of the CIC::LEUTX gene fusion detected by RNA sequencing, in which exons 1–20 of CIC, as the 5′ partner, are fused to exon 3 of LEUTX.
CIC gene rearrangements are a characteristic feature of tumors within the novel group
By targeted next-generation DNA sequencing and/or RNA sequencing, 9 out of 10 tumors analyzed (including the two previously published samples) demonstrated gene fusions between CIC and leucine twenty homeobox (LEUTX) as the 3′ partner, both located on chromosome 19q13.2 (Fig. 2d). In all of the tumors, exons 1–20 of CIC (NM_015125.5) were fused in frame to exon 3 of LEUTX (NM_001143832.2), retaining the DNA-binding high-mobility group (HMG) box of CIC and the suggested 9aaTAD domain of LEUTX12. These findings are in line with the breakpoints detected in a previously reported pediatric embryonal tumor of the CNS13 (Supplementary Table 4). In addition, a fusion between exons 1–20 of CIC and NUT midline carcinoma family member 1 (NUTM1, located on chromosome 15q14) exons 2–7 (NM_001284293.1) was observed, very similar to the CIC::LEUTX rearrangement (Supplementary Fig. 2 and Supplementary Table 4). Apart from the detected fusion events, no additional oncogenic alteration was identified in these tumors based on sequencing.
Clinical characteristics and morphological features indicate pediatric‐type high-grade neuroepithelial tumors
Analysis of available clinical data demonstrated that all tumors were located in the supratentorial compartment, mainly in the parietal and occipital lobe. The median age at presentation was 8.5 years (range 1–19) and the sex distribution was not significantly biased when considering the small number of patients. Clinical outcome data were available for only six patients. Median PFS was 13.5 months (range 6–16 months) with all of the patients experiencing a relapse during the follow-up period. Only one of the patients died of the disease during the follow-up period at 15 months after diagnosis. Together, these initial data suggest an intermediate malignancy grade (Supplementary Fig. 3). Initial histopathologic diagnoses comprised various tumor types of mainly high-grade glioma. More detailed descriptions of the cases are given in Supplementary Table 1. A histopathological review was performed on a subset of the tumors with available material (n = 9) that revealed a morphologically heterogeneous group. Histologically, all reviewed tumors shared a high cellular density with most neoplasms showing slightly pleomorphic neoplastic cells often with remarkably condensed chromatin (Fig. 3a). A more pronounced cellular pleomorphism with multinucleated cells were seen in single cases (Fig. 3b–d). An oligodendrocyte-like phenotype with perinuclear clearing was focally found in four of the cases (Fig. 3e, f). Microcystic changes were present in half of the tumors. Tumors were highly vascularized with hypertrophic or proliferated vessels in most of the cases (Fig. 3g). In three of the tumors, perivascular anucleate zones (pseudorosettes) were observed (Fig. 3h). Necrosis was present in five tumor samples (Fig. 3d). Mitotic activity was generally high, with the exception of one case. Immunostaining for markers of glial differentiation (GFAP and OLIG2) was positive in all tumors (Fig. 3i, j). However, GFAP expression was only weakly positive or restricted to a minor proportion of neoplastic cells in some of the cases. In 4/4 tumors, a focal immunoreactivity for MAP2 was detected (Fig. 3k). All tumors showed a weak and focal positivity for synaptophysin (n = 9; Fig. 3l). CD56 was expressed in all samples analyzed (n = 4). All evaluated tumors had absent immunostaining for NeuN (n = 5). CD34 (n = 6) expression was restricted to the vessels (Fig. 3m). A focal positivity for CD99 was observed in all evaluated samples (n = 4; Fig. 3n). Ki-67 labeling indices ranged from 10 to 70% (Fig. 3o, p).

a Histologically, tumors show a high increase in cellular density of slightly pleomorphic neoplastic cells. b, c A more pronounced cellular pleomorphism with multinucleated cells is present in a subset of cases. d Tumor necrosis. e, f An oligodendroglial morphology with perinuclear halos is focally present in a minor proportion of tumors. g, h Tumors are highly vascularized with a subset of cases demonstrating perivascular anucleate zones (pseudorosettes). i, j Positive immunostaining for markers of glial differentiation (GFAP and OLIG2). k, l Tumor cells show focal immunoreactivity for MAP2 and synaptophysin. m CD34 expression is restricted to the vessels. n CD99 expression is focally present in all evaluated samples. o, p Ki-67 labeling indices range from about 10 to 70% of the neoplastic cells. Scale bars 200 μm.
Discussion
Here, we describe a previously uncharacterized group of rare, pediatric CNS tumors that was discovered through unsupervised visualization of genome-wide DNA methylation profiles. This novel group of tumors, epigenetically distinct from all known CNS neoplasms, shows recurrent gene fusions involving the transcriptional repressor CIC (most commonly with LEUTX) as an additional unifying feature.
CIC, a human homolog of capicua in Drosophila, acts as a transcriptional repressor with a DNA-binding high-mobility group (HMG) box domain that normally inhibits ETV1/4/5 expression and counteracts activation of genes downstream of receptor tyrosine kinase (RTK) signaling14. Aberrations in CIC have been identified in various types of cancer, with loss-of-function mutations frequently observed in oligodendroglioma15,16, leading to activation of downstream RTK signaling. Intriguingly, rearrangements involving CIC and the double homeobox 4 (DUX4) commonly found in high-grade round cell undifferentiated sarcoma17,18, have been shown to enhance the transcriptional activity of CIC downstream targets, including the member of the ETS family of transcription factors, such as ETV1/4/519,20. Consistent with that, an upregulation of members of the ETS transcription factor family have been reported in CIC-rearranged sarcoma (“Ewing sarcoma family of tumors with CIC alterations”) harboring oncogenic fusions between CIC and NUTM111.
In contrast, LEUTX is a member of the paired (PRD)-like homeobox gene family of transcription factors and is expressed almost exclusively in early embryos where it is thought to play a role during preimplantation development21,22. Although rearrangements between CIC and LEUTX have been reported recently10,9 were used to calculate the 1-variance weighted Pearson correlation between samples. The resulting distance matrix was used as input for t-SNE analysis (Rtsne package version 0.13). The following non-default parameters were applied: is_distance = T, theta = 0, pca = F, max_iter = 10,000, perplexity = 30. Estimation of differential methylated positions (DMP) was done in R by using the function “dmpFinder” from the minfi package (v1.43). The Illumina EPIC platform was used to annotate CpGs by their position in the genome and associated genes. The tests were carried out on the M-values of all promoter-associated genes as well as the top 100k CpGs according to mean average deviance. CpGs with an FDR q-value smaller than 0.05 were considered as significant differential methylated. Volcano plots of the DMPs were generated by the R-package ggplot2 (v3.3.6). CpGs are distributed according to their −log10 Q values and fold change (intersect). Further significant different methylated CpGs are depicted in red while the associated gene top100 DMP is shown.
Targeted next-generation DNA sequencing
For a subset of samples with DNA available (n = 9), DNA sequencing using a customized enrichment/hybrid-capture-based next-generation sequencing (NGS) gene panel were performed on a NextSeq 500 or NovaSeq 6000 instrument (Illumina) at the Department of Neuropathology of the University Hospital Heidelberg (Heidelberg, Germany)27. The NGS panel comprised the entire coding (all exons + /– 25 bp) and selected intronic and promoter regions of 170 genes of particular relevance in CNS tumors, and was designed to detect single nucleotide variants (SNV), small insertions/deletions (InDel), exonic rearrangements, and recurrent fusion events. Paired-end sequencing was applied to increase the detection sensitivity of duplicates and possible gene fusions. Sequence reads were mapped to the reference human genome build GRCh37 (hg19) using the Burrows–Wheeler aligner (BWA).
RNA sequencing and analysis
RNA sequencing for the purpose of gene fusion detection of samples for which RNA of sufficient quality and quantity was available (n = 8) was performed as previously described28. In brief, RNA sequencing libraries were prepared using the TruSeq RNA Library Prep for Enrichment kit (Illumina) and paired-end reads were sequenced on a NextSeq 500 or NovaSeq 6000 instrument (Illumina). After adapter trimming, reads were aligned to the human genome (GRCh37) with the STAR aligner29 and counted using RSEM30. Fastq files from transcriptome sequencing were used for de novo annotation of fusion transcripts using the Arriba (v1.2.0) algorithm31 with standard parameters, which removes recurrent alignment artifacts, transcript variants also observed in normal tissue, reads with low sequence complexity, and events with short anchors or breakpoints in close proximity or a low number of supporting reads relative to the overall number of predicted events in a gene.
Survival analysis
Survival analysis was performed using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA). Data on survival could be retrospectively retrieved for six patients. Overall survival (OS) and progression-free survival (PFS) probabilities were displayed using the Kaplan–Meier method.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
DNA methylation data generated during this study has been deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) under accession number GSE223546. DNA methylation data used as a reference has been deposited under accession number GSE90496. Consent for public data sharing of sequencing data was not obtained from the patients, so datasets are available upon IRB-approved collaboration from the corresponding author (PS) and will only be shared for research-related, non-commercial purposes. The remaining data are available within the article and supplementary material.
Code availability
R packages and scripts used to analyze the data, along with input data, are explained in the methods and supplementary material. The code underlying random forest-based algorithm for DNA methylation-based CNS tumor classification was previously described9.
References
Ostrom, Q. T., Cioffi, G., Waite, K., Kruchko, C. & Barnholtz-Sloan, J. S. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol. 23, iii1–iii105 (2021).
Pfister, S. M. et al. A summary of the inaugural WHO classification of pediatric tumors: transitioning from the optical into the molecular era. Cancer Discov. 12, 331–355 (2022).
Pratt, D., Sahm, F. & Aldape, K. DNA methylation profiling as a model for discovery and precision diagnostics in neuro-oncology. Neuro Oncol. 23, S16–S29 (2021).
Sievers, P. et al. Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial neuroepithelial tumors. Acta Neuropathol. 142, 827–839 (2021).
Alhalabi, K. T. et al. PATZ1 fusions define a novel molecularly distinct neuroepithelial tumor entity with a broad histological spectrum. Acta Neuropathol. 142, 841–857 (2021).
Bandopadhayay, P. et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat. Genet. 48, 273–282 (2016).
Sievers, P. et al. GOPC:ROS1 and other ROS1 fusions represent a rare but recurrent drug target in a variety of glioma types. Acta Neuropathol. 142, 1065–1069 (2021).
Clarke, M. et al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 10, 942–963 (2020).
Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018).
Lake, J. A. et al. Targeted fusion analysis can aid in the classification and treatment of pediatric glioma, ependymoma, and glioneuronal tumors. Pediatr. Blood Cancer 67, e28028 (2020).
Sturm, D. et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164, 1060–1072 (2016).
Katayama, S. et al. Phylogenetic and mutational analyses of human LEUTX, a homeobox gene implicated in embryogenesis. Sci. Rep. 8, 17421 (2018).
Hu, W. et al. Case Report: a unique case of pediatric central nervous system embryonal tumor harboring the CIC-LEUTX fusion, germline NBN variant and somatic TSC2 mutation: expanding the spectrum of CIC-rearranged neoplasia. Front. Oncol. 10, 598970 (2020).
Jimenez, G., Shvartsman, S. Y. & Paroush, Z. The Capicua repressor—a general sensor of RTK signaling in development and disease. J. Cell Sci. 125, 1383–1391 (2012).
Bettegowda, C. et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 333, 1453–1455 (2011).
Sahm, F. et al. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. 123, 853–860 (2012).
Antonescu, C. R. et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am. J. Surg. Pathol. 41, 941–949 (2017).
Italiano, A. et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer 51, 207–218 (2012).
Kawamura-Saito, M. et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 15, 2125–2137 (2006).
Specht, K. et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 53, 622–633 (2014).
Jouhilahti, E. M. et al. The human PRD-like homeobox gene LEUTX has a central role in embryo genome activation. Development 143, 3459–3469 (2016).
Tohonen, V. et al. Novel PRD-like homeodomain transcription factors and retrotransposon elements in early human development. Nat. Commun. 6, 8207 (2015).
Song, K., Huang, Y., **a, C. D., Zhu, H. Q. & Wang, J. A case of CIC-rearranged sarcoma with CIC-LEUTX gene fusion in spinal cord. Neuropathology https://doi.org/10.1111/neup.12850 (2022).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Christians, A., Poisel, E., Hartmann, C., von Deimling, A. & Pusch, S. Characterization of the epithelial membrane protein 3 interaction network reveals a potential functional link to mitogenic signal transduction regulation. Int. J. Cancer 145, 461–473 (2019).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Sahm, F. et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 131, 903–910 (2016).
Stichel, D. et al. Routine RNA sequencing of formalin-fixed paraffin-embedded specimens in neuropathology diagnostics identifies diagnostically and therapeutically relevant gene fusions. Acta Neuropathol. 138, 827–835 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Uhrig, S. et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 31, 448–460 (2021).
Acknowledgements
The authors thank L. Dörner and L. Hofmann for excellent technical assistance, and the DKFZ Genomics and Proteomics Core Facility for support in DNA methylation analysis. This study was supported by the Hertie Network of Excellence in Clinical Neuroscience. P. Sievers is supported by the Else Kröner Fresenius Stiftung and a fellow of the Hertie Academy of Excellence in Clinical Neuroscience.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Experimental design: P.S., F.S., and D.T.W.J. Implementation: P.S., F.S., and D.T.W.J. Data analysis/interpretation: P.S., M. Sill, D. Schrimpf, D.F., S.M.P., A.V.D., F.S., and D.T.W.J. Sample collection: P.S., Z.A., A.M.D., J.A.L., D. Scheie, O.T., T.R., K.L.V., D. Samuel, R.C., R.G.G., K.W.P., M. Snuderl, T.S.J., K.A., W.W., and A.K. Approved final manuscript: all authors.
Corresponding author
Ethics declarations
Competing interests
M. Sill, D. Schrimpf, M. Snuderl, S.M.P., A.V.D., F.S., and D.T.W. J. are co-founders and shareholders in Heidelberg Epignostix, GmbH. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sievers, P., Sill, M., Schrimpf, D. et al. Pediatric-type high-grade neuroepithelial tumors with CIC gene fusion share a common DNA methylation signature. npj Precis. Onc. 7, 30 (2023). https://doi.org/10.1038/s41698-023-00372-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00372-1
- Springer Nature Limited
This article is cited by
-
Pediatric central nervous system tumor with CIC::LEUTX fusion: a diagnostic challenge
Acta Neuropathologica Communications (2024)
-
Molecular and clinicopathologic characteristics of CNS embryonal tumors with BRD4::LEUTX fusion
Acta Neuropathologica Communications (2024)
-
Small blue round cell tumor with focal epithelial differentiation harboring a novel YAP1::LEUTX fusion with poor prognosis
Virchows Archiv (2024)