
Abstract
Dermatophagoides farina (D. farinae) and Dermatophagoides pteronyssinus (D. pteronyssinus) are the prevalent kinds of house dust mites (HDMs). HDMs are common inhalant allergens that cause a range of allergic diseases, such as rhinitis, atopic dermatitis, and asthma. The epidemiology of these diseases is associated with exposure to mites. Therefore, in the present study, a method named multiplex loop-mediated isothermal amplification (LAMP) was developed to detect environmental dust mites. The multiplex LAMP assay allows amplification within a single tube and has an ITS plasmid detection limit as low as 40 fg/µL for both single dust mites and mixed dust mites (D. pteronyssinus and D. farinae), which is up to ten times more sensitive than classical PCR techniques. Furthermore, the multiplex LAMP method was applied to samples of single dust mites and clinical dust to confirm its validity. The multiplex LAMP assay exhibited higher sensitivity, simpler instrumentation, and visualization of test results, indicating that this method could be used as an alternative to traditional techniques for the detection of HDMs.
Similar content being viewed by others


Introduction
House dust mites (HDMs) have been named for their inhabitation of domestic dust. HDMs readily obtain food from the skin debris that people in daily. Dermatophagoides farinae (D. farinae) and Dermatophagoides pteronyssinus (D. pteronyssinus) are prevalent and widespread among all HDMs1. Dust mite allergens, including eggs, feces, bodies, and corpse residues, are common allergens and might cause allergic rhinitis, atopic dermatitis, and allergic asthma2,3,4. According to previous reports, approximately 10% of the average public and over 80% of allergic asthmatics undergo anaphylactic reactions when exposed to HDMs5. In the Asia–Pacific region of allergic diseases, HDMs have been reported to be one of the major allergens involved in allergic rhinitis6. In Hong Kong, sensitivity to HDMs is prevalent in more than 80% of children diagnosed with asthma7. Amin et al.8 investigated the prevalence of sensitivity to aeroallergens in patients with atopic dermatitis and reported that patients exhibited greater sensitivity to D. pteronyssinus (50%) and D. farinae (34%) than to birch pollen, cat epithelium, and cockroaches. Therefore, it is crucial to detect these mites using multiple diagnostic tests with high specificity, sensitivity, and simultaneous detection.
HDMs can be detected by morphological, immunological, and molecular biology approaches9,10,11,12. Morphological detection is a low-cost method that mostly involves the detection of HDMs under a microscope in the laboratory. However, HDMs tend to hide in dust and are tiny. Therefore, morphological tests for the detection of HDMs are time-consuming and prone to missed detection1,2. Immunological studies have shown that the amino acid sequences of the D. pteronyssinus allergen Der p 1 and D. farinae allergen Der f 1 are extremely similar, up to 83%; thus, there is a large degree of homology, and cross-reactivity between these allergens is present13,14. Molecular biological methods with high specificity and sensitivity have been widely used for the diagnosis of allergic diseases. In molecular biology, PCR is one of the most classical and representative amplification techniques. Yang et al.15 previously used PCR to taxonomically identify six distinct mite types. Oliveira et al.12 established a multiplex PCR for HDMs. The above methods, however, are time-consuming and cumbersome. They also require costly laboratory equipment, dedicated laboratory facilities, and trained staff16.
Loop-mediated isothermal amplification (LAMP) has been successfully carried out in diverse fields of study, such as for ABO genoty**, Salmonella, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), Toxoplasma gondii, and shrimp allergens, in a rapid, highly efficient and low-detection-limit manner compared to conventional PCR17,18,19,20,21. LAMP technology was reported by Notomi et al. in 200022. This amplification methodology involves the addition of four to six specific primers that are capable of recognizing up to six to eight fragment sequences on the target gene within the same reaction (Fig. 1), which is different from classical PCR amplification techniques. LAMP technology also uses a thermostable polymerase, Bst-DNA polymerase, with a high displacement capacity, in which the second strand is displaced without thermal denaturation during the reaction. The reaction can be completed within 30–60 min under constant temperature conditions (60–68 °C) and can be performed without a DNA amplicon. The specificity of the method is a result of the use of four or six primers in the reaction process22,23. The amplification reaction products generated at the reaction endpoint may be detected using various methods, including gel electrophoresis, measurement of the turbidity derived from magnesium pyrophosphate precipitation, or the addition of fluorescent chemicals to the reaction compounds for visual observation24,25. To increase detection efficiency and decrease experimental costs, several researchers have used LAMP technology to detect multiple target genes of the same pathogen or screen multiple pathogens26,27. When using multiplex LAMP for detection, cross-reaction between primers must be prevented, which necessitates designing multiple complex primers in advance. Kumar et al.28 combined the LAMP technique with magnetic particle visualization for the detection of chikungunya and dengue viruses. Joshi et al.29 established a multiplex LAMP method for the differential diagnosis of Leishmania donovani and Mycobacterium leprae by means of a real-time fluorescence analyzer. Wang et al.26 used real-time turbidimetry to establish multiplex LAMP for the primary screening of Perkinsus and Bonamia infections in shellfish parasites present in aquatic products.

Schematic representation of the LAMP reaction. DNA synthesis relied on DNA polymerase with high strand substitution activity, two outer primers (F3 and B3), and a set of two specially designed inner primers (FIP and BIP). When FIP (5’-F1c-F2) combines with F2c, complementary strand synthesis is initiated. When F3 combines with F3c, strand-substituted DNA synthesis is initiated, which releases the FIP-linked complementary strand that is used in single-stranded DNA. BIP (5’-B1c-B2) initiates strand displacement DNA synthesis, and subsequently, B3 initiates strand-substituted DNA synthesis. This leads to the production of dumbbell-shaped DNA, which serves as the foundation for subsequent reactions. This single-stranded DNA is used for BIP-initiated DNA synthesis and subsequent B3-primed strand-replacement DNA synthesis, resulting in the production of dumbbell-shaped DNA. This dumbbell-shaped DNA then serves as the foundation for the LAMP cycle, which proceeds continuously through chain substitution reactions with the inner primer cycle, repetitively and spontaneously, yielding amplification products of different lengths. This schematic illustrates only the single-target response mechanism.
We aimed to establish a multiplex LAMP method to increase the detection efficiency and decrease the experimental cost of D. pteronyssinus and D. farinae. The experimental results were illustrated using real-time turbidity curves and fluorescence detection reagents (FDRs). The developed method is expected to become popular in the field of point-of-care detection and on-site diagnosis of dust mites present in the environment as allergens owing to its rapid, efficient, and highly specific qualities and the simplicity of the equipment required for detection.
Materials and methods
Sample collection and DNA extraction
Dust mites were obtained from floor dust, air-conditioned filter dust, and bed dust from a student dormitory of Wannan Medical College (Anhui, China). The samples were examined under a light microscope for preliminary morphological identification of individual mites. D. farinae was cultured in the laboratory of Wannan Medical College (Anhui, China) in an incubator at 25 ± 2 °C and 70% ± 5% RH.
Specificity testing was conducted using mites belonging to other mite species, Cheyletus malaccensis (C. malaccensis), Scheloribates laevigatus (S. laevigatus), Aleuroglyphus ovatus (A. ovatus) and Lepidoglyphus destructor (L. desctructor), and other organisms commonly present in living environment dust, Musca domestica (M. domestica), Anopheles sinensis (An. sinensis), and Periplaneta americana (P. americana). The above samples were deposited in the Department of Medical Parasitology, Wannan Medical College, Anhui Province, China.
For all experimental samples, genomic DNA was extracted following the procedure provided by the manufacturer of the DNeasy Blood and Tissue Kit (Tiangen Biotech Bei**g Co. Ltd., China). Moreover, for the dust samples collected, genomic DNA was extracted following the procedure provided by the manufacturer of the TIANamp Soil DNA Kit (Tiangen Biotech Bei**g Co. Ltd., China). The above extracted genomic DNA samples were stored at − 20 °C until use.
Primer design
The internal transcribed spacer (ITS) region of conserved rDNA sequences has a certain degree of interspecies specificity and intraspecies conservation and is therefore used as the target point for interspecies identification. Therefore, the ITS gene was selected as the target gene in this study, and the ITS sequence of D. pteronyssinus (KC215340) was downloaded from the NCBI GenBank database. Five sets of LAMP candidate primer sets against the different regions of the target gene were designed online and automatically using Primer Explorer V5 (http://primerexplorer.jp/e/) (Eiken Chemical Co., Tokyo, Japan). The primers used for D. farinae were previously reported by Xue et al.30. All primers in our study were synthesized at Sangon Biotechnology (Sangon Biotech Shanghai Co. Ltd., China) and later diluted following the instructions provided by the manufacturer. Finally, the diluted primers were stored at − 20 °C until use.
Establishment of a single LAMP assay
Each LAMP reaction system was 25 µL, which contained 12.5 µL of 2 × LAMP reaction buffer (including 0.1% Tween 20, 20 mM Tris–HCl (pH 8.8), 10 mM (NH4)2SO4, 10 mM KCl, 0.8 M betaine, 8 mM MgSO4, and 1.4 mM dNTPs), 1 µL of Bst-DNA polymerase (8 U), 1 µL of FDRs (LAMP DNA Amplification Kit; Eiken Chemical Co. Ltd., Tochigi, Japan), and 2 µL of DNA template. All of the above reagent compositions were consistent during the configuration of the LAMP reaction system for D. pteronyssinus and D. farinae. While the single LAMP primer volume of D. pteronyssinus was 4 µL (40 µM FIP and BIP, 5 µM F3 and R3), the single LAMP primer volume of D. farinae was 5 µL (5 µM F3 and R3, 20 µM LB, 40 µM FIP and BIP). A negative control was selected for the DNAzyme-free water.
Primer screening was conducted using five sets of primers designed for D. pteronyssinus DNA, which were initially amplified using a 65 °C heating block for 60 min and then enzymatically extinguished at 80 °C for 10 min. The results were visualized through real-time turbidity formation curves and FDRs. A suitable temperature favors stimulation by Bst-DNA polymerase activity.
Temperature screening was conducted by incubating the reaction at a temperature of 62 °C to 69 °C (1 °C as a gradient) for 60 min, after which the reaction was terminated by incubation at 80 °C for 10 min. The single LAMP reaction conditions for D. farinae used in the present study were designed previously in our laboratory, as reported by Xue et al.30.
The specificity of the single LAMP method was then assessed using the ITS plasmids of D. pteronyssinus and D. farinae as positive controls. The genomic DNA templates of A. ovatus, C. malaccensis, L. destructor, S. laevigatus, P. americana, An. sinensis, and M. domestica were used as specific control samples. A negative control was selected for the DNAzyme-free water. The sensitivity of the single LAMP method was assessed by preparing ten-fold dilutions of the ITS plasmids of both species in the concentration range of 40 ng/µL to 400 ag/µL and then using these plasmids as templates for the assay.
Establishment of the multiplex LAMP assay
The best primers selected from the single LAMP reaction system, as described above, were assembled into the multiplex LAMP reaction system to establish the multiplex LAMP assay. Each LAMP reaction mixture had a volume of 25 µL, which contained 12.5 µL of two-fold LAMP reaction buffer, 1 µL of Bst-DNA polymerase (8 U), 1 µL of FDRs, 3.5 µL of the primer for D. farinae (10 µM F3 and R3, 40 µM FIP and BIP, 40 µM LB), 3 µL of the primer for D. pteronyssinus (10 µM F3 and R3, 40 µM FIP and BIP), 2 µL of the positive plasmid template (1 µL for D. pteronyssinus and 1 µL for D. farinae), and the remaining volume of the DNAzyme-free water. A negative control was selected for the DNAzyme-free water.
Detection of the LAMP products
The LAMP amplification results were calibrated for 6 s using a loopamp real-time turbidimeter (LA-500; Eken Chemical Co. Ltd., Tochigi, Japan). The reaction results obtained with the Loopamp real-time turbidimeter were standardized using a positive curve. At the same time, to allow immediate visualization, the results were visually inspected by adding 1 µL of FDRs (Eiken Chemical Co. Ltd., Tochigi, Japan) to the reaction system, followed by observing the color change in the reaction samples (Fig. 2). Positive and negative LAMP products were defined as fluorescent green and fluorescent orange signals, respectively, as observed by the naked eye under natural light.

The image is an outline of this study. (a) Outline illustrations of sample preparation and DNA extraction. (b) Outline illustrations of LAMP detection. Amplification unit: a positive reaction is depicted in fluorescent green, and a negative reaction is depicted in fluorescent orange with no discolouration. Control unit: A positive sample amplification produces significant amounts of insoluble magnesium pyrophosphate precipitate, and the turbidity profile (x-axis is time, y-axis is turbidity) is displayed on the operating panel of the control unit.
Evaluation of multiplex LAMP specificity
The specificity of the multiplex LAMP detection method was verified using the ITS plasmids of D. pteronyssinus, D. farinae, D. pteronyssinus, and D. farinae as positive controls. The genomic DNA samples of A. ovatus, C. malaccensis, L. destructor, S. laevigatus, P. americana, An. sinensis, and M. domestica were used as specific control samples. A negative control was selected for the DNAzyme-free water. The results were validated by repeating the assessment three times for each sample concentration.
Evaluation of multiplex LAMP sensitivity
The sensitivity and LODs of the multiplex LAMP assay were obtained by mixing equal volumes of the same concentrations of the two plasmids. The ITS plasmids of D. pteronyssinus, D. farinae and D. pteronyssinus and D. farinae were serially diluted to prepare different concentrations (40 ng/µL, 4 ng/µL, 400 pg/µL, 40 pg/µL, 4 pg/µL, 400 fg/µL, 40 fg/µL, 4 fg/µL, and 400 ag/µL). The results were validated by repeating the assessment three times for each sample concentration. A negative control was selected for the DNAzyme-free water.
Detection of clinical samples
The effectiveness of the multiplex LAMP assay was determined by examining the detection of a single D. pteronyssinus sample, a single D. farinae sample, and a mixed single mite sample (D. pteronyssinus and D. farinae). In each group, ten samples were prepared for testing. Mite DNA was extracted according to the instructions of the DNeasy Blood & Tissue Kit (Tiangen Biotech Bei**g Co. Ltd., China). The effectiveness of the multiplex LAMP method and the PCR method was detected for all samples.
A total of 55 floor dust, air-conditioned filters and bed dust samples collected from the student dormitories were collected in November 2023 and December 2023. The floor dust and air-conditioned filter dust were collected by a small brush, and a bed brush was used to pat down the bed surfaces and bed mattresses to collect dust. All collected samples were discarded for visible large particles of debris or hair and then placed under a stereomicroscope for observation and classification. DNA was extracted from the samples using the TIANamp Soil DNA Kit (Tiangen Biotech Bei**g Co. Ltd., China) according to the manufacturer’s instructions. All samples were detected by the multiplex LAMP method, after which the percentage of positive samples was determined. IBM SPSS statistical software version 27 (SPSS Inc., Chicago, IL) was used for the statistical analysis of the results, and χ2 value and P-value were determined to reveal significant differences.
PCR assay
Comparison of clinical sample specificity, sensitivity and detection efficacy using PCR assays. The PCR system had a volume of 25 µL, which contained 12.5 µL of the PrimeSTAR Max DNA Polymerase (8 mM Mg2+ and 0.4 mM each for dNTPs) (Takara Biomedical Technology Co. Ltd., Bei**g, China), 2 µL of the primer for D. farinae (10 µM F3 and R3), 2 µL of the primer for D. pteronyssinus (10 µM F3 and R3) 1 µL each of F3 and B3 (10 µM), and 2 µL of the DNA template, with the remaining volume made up of the DNAzyme-free water. The primers used were the F3 primer and B3 primer. The cycling conditions were as follows: initial 5 min at 94 °C; 35 cycles of 15 s at 94 °C, 30 s at 55 °C, and 30 s at 72 °C; and final 5 min at 72 °C. The PCR products were then subjected to electrophoresis on a 2% agarose gel, followed by staining with 4S Red Plus nucleic acid stain (Sangon Biotech Shanghai Co. Ltd., China). Images were analyzed semiquantitatively using a Bio-Rad Gel Doc EQ imaging system (Bio-Rad, Hercules, CA, USA).
Results
Optimization of the single LAMP assay
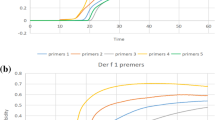
The optimal primers for D. pteronyssinus were determined by conducting LAMP reactions using different sets of primers. Amplification was performed at 65 °C for 60 min, after which the turbidity of the LAMP products was measured. Although each primer set produced amplification product curves with significant turbidity, the amplification efficiency of each primer differed. In the turbidity analysis, primer 3 appeared to be the fastest, but it also amplified D. farinae significantly (Supplementary Fig. S1 online). Therefore, the primer with the second highest amplification efficiency, primer 4, was selected as the optimal primer for D. pteronyssinus. Next, the reaction temperature was optimized. The reaction amplification curves revealed that the best amplification efficiency was achieved at 64 °C (Fig. 3). Therefore, 64 °C was selected as the optimal reaction temperature for subsequent experiments. In the case of D. farinae, the report by Xue et al.30 was referred to for selecting the optimum primer (primer 4) and temperature (64 °C). The sequences and positions of the optimal primers determined through optimization experiments are presented in Table 1 and Fig. 4.

Optimization of the reaction conditions for the single LAMP assay for D. pteronyssinus. A negative control was selected for the DNAzyme-free water. (a) Screening of the designed 5 primer pairs. The primer screening results revealed that the 4th primer pair had the highest amplification efficiency. (b) Reaction temperature optimization: Eight different ladders were tested at different temperatures—62 °C, 63 °C, 64 °C, 65 °C, 66 °C, 67 °C, 68 °C, and 69 °C—and 64 °C was identified as the optimal temperature. The results were observed within 60 min of the reaction.

Schematic representation of primer positions. (a) D. pteronyssinus ITS sequence (GenBank accession number KC215340). (b) D. farinae ITS sequence (GenBank accession number MH793957).
Specificity and sensitivity of the single LAMP assay
Optimized reaction systems were utilized to determine the specificity and sensitivity of the single LAMP assay for D. pteronyssinus and D. farinae. The specificity analysis of the single LAMP assay revealed that only D. pteronyssinus produced an amplification curve, while no amplification was noted for the other samples (Fig. 5a). Purified genomic DNA plasmids of D. pteronyssinus were assayed by serial dilution of ten-fold at a known concentration. The single LAMP primer sets exhibited a limit of detection (LOD) of 40 fg/µL (Fig. 5b). The specificity analysis of the single LAMP assay for D. farinae revealed that only D. farinae presented an amplification curve, while none of the other samples exhibited amplification (Fig. 5c). The sensitivity of the single LAMP assay for D. farinae was determined to be 40 fg/µL (Fig. 5d).

Real-time turbidity formation curves obtained for the single LAMP assays. (a) Specificity assay conducted for D. pteronyssinus alone. (b) Sensitivity assay conducted for D. pteronyssinus alone. (c) Specificity assay conducted for D. farinae alone. (d) Sensitivity assay conducted for D. farinae alone.
Specificity of the multiplex LAMP assay
Standard plasmids were utilized as templates to ensure the specificity of the multiplex LAMP assay. The results revealed that the amplification signal could be detected for the positive plasmids (D. pteronyssinus, D. farinae, D. pteronyssinus, and D. farinae) containing ITS, while all the remaining other templates were negative. In addition, the results of the FDR method were consistent with those of the turbidity assay (Figs. 6a and 7a). Subsequent comparisons with conventional PCR (Fig. 8a) also revealed bands only for the positive plasmids.

Real-time turbidity curves obtained after the multiplex LAMP assay. (a) Specificity of mixed ITS plasmids (D. pteronyssinus and D. farinae), D. farinae ITS plasmids, and D. pteronyssinus ITS plasmids. (b) Sensitivity to mixed ITS plasmids (D. pteronyssinus and D. farinae). (c) Sensitivity of D. farinae to ITS plasmids. (d) Sensitivity of D. pteronyssinus to ITS plasmids. (b–d) Dilutions within the range of 40 ng/µL to 400 ag/µL.

Colorimetric indication of the multiplex LAMP assay results. (a) Specificity of mixed ITS plasmids (D. pteronyssinus and D. farinae), D. farinae ITS plasmids, and D. pteronyssinus ITS plasmids. Samples 1–11 correspond to mixed ITS plasmids (D. pteronyssinus and D. farinae), D. farinae, D. pteronyssinus, A. ovatus, An. sinensis, S. laevigatus, M. domestica, C. malaccensis, L. destructor, P. americana and a negative control. (b) Sensitivity to mixed ITS plasmids (D. pteronyssinus and D. farinae). (c) Sensitivity of D. farinae to ITS plasmids. (d) Sensitivity of D. pteronyssinus to ITS plasmids. (b), (c), and (d) Samples 1–10 represent concentrations of 40 ng/µL, 4 ng/µL, 400 pg/µL, 40 pg/µL, 4 pg/µL, 400 fg/µL, 40 fg/µL, 4 fg/µL, and 400 ag/µL of the ITS plasmid, respectively, and the negative control.

Gel electrophoresis by PCR. (a) Specificity of mixed ITS plasmids (D. pteronyssinus and D. farinae), D. farinae ITS plasmids, and D. pteronyssinus ITS plasmids. Lanes 1–11 represent mixed ITS plasmids (D. pteronyssinus and D. farinae), D. farinae, D. pteronyssinus, A. ovatus, An. sinensis, S. laevigatus, M. domestica, C. malaccensis, L. destructor, P. americana and a negative control. (b) Sensitivity to mixed ITS plasmids (D. pteronyssinus and D. farinae). (c) Sensitivity of D. farinae to ITS plasmids. (d) Sensitivity of D. pteronyssinus to ITS plasmids. (b), (c), and (d) Lanes 1–10 represent the concentrations 40 ng/µL, 4 ng/µL, 400 pg/µL, 40 pg/µL, 4 pg/µL, 400 fg/µL, 40 fg/µL, 4 fg/µL, and 400 ag/µL of the ITS plasmid, respectively, and the negative control.
Sensitivity of the multiplex LAMP assay
The sensitivity of the LAMP assay was evaluated, and it was revealed that the lowest detection level was 40 fg/µL for the mixed plasmids (D. pteronyssinus and D. farinae), D. pteronyssinus and D. farinae (Fig. 6b–d). The obtained turbidity curves were consistent with the findings of the FDR dye method (Fig. 7b–d). In the case of the conventional PCR method, the lowest detection level achieved was 400 fg/µL for mixed plasmids (D. pteronyssinus and D. farinae), D. pteronyssinus and D. farinae (Fig. 8b–d).
Detection of clinical samples
Clinical sensitivity is an important criterion for assessing the usefulness of an assay. In the present study, 10 single mite clinical samples were assessed. The multiplex LAMP assay detected 40% and 70% of the single samples of the two mite species evaluated, respectively. In addition, the assay detected 80% of the mixed single mite samples. The detection validity of PCR was only 30% for D. pteronyssinus, 50% for D. farinae, and 50% for the mixed single mite samples (Table 2).
Furthermore, 55 dust samples collected from student dormitories were evaluated. A total of 46 positive samples were detected, with a positive detection rate of 83.64% (46/55). In contrast, PCR detected only 35 positive samples and exhibited a positive detection rate of only 63.64% (35/55). The chi-squared test conducted using SPSS statistical software revealed χ2 = 15.687, with P < 0.001, indicating that the difference between the LAMP assay and PCR assay results was statistically significant (Table 3).
Discussion
HDMs are tiny organisms that dwell on bedding and carpets and feed on skin scales shed by humans and pets. When temperature and humidity are high, dust mites survive throughout the year. D. pteronyssinus and D. farinae are two common species of dust mites1,31. Since certain people can develop allergic diseases when contact dust mites bodies and excretions, dust mites are considered to be one of the allergenic sources in humans2,32. Since epidemics of HDM-associated clinical diseases are associated with exposure to dust mites, it is important to be aware of the presence of HDMs inside the human household12,30,33,34. The accurate and efficient detection of common allergenic mites in clinical samples is essential for timely and effective control of parasitic dust mites infestation, which can mitigate the incidence of allergic diseases and improve people's quality of life standards.
The experimental results of the present study were recorded as amplification curves based on LoopAmp real-time turbidimetry and visual observation of the color change in the FDR. Next, the best primers for D. pteronyssinus and D. farinae were utilized to establish the multiplex LAMP assay. Furthermore, the optimal primers for both species were combined to optimize the development of a multiplex LAMP reaction system for the simultaneous detection of both D. pteronyssinus and D. farinae, followed by assessments of the specificity and sensitivity of this multiplex system. The findings indicated that the multiplex LAMP method had excellent specificity and presented no cross-reactivity between the multiplex LAMP primers. The LOD of the multiplex LAMP assay for both single and mixed mite samples was 40 fg/µL, which was 10 times higher than the value achieved using the PCR assay. In a previous study, Joshi et al.29 established a multiplex LAMP assay that achieved sensitivities of 100 fg/µL and 1 fg/µL for Mycobacterium leprae and Leishmania donovani, respectively. In China, Wang et al.26 successively established single LAMP and multiple LAMP assays for aquatic products of the shellfish parasites Perkinsus and Bonamia. Both Perkinsus and Bonamia presented consistent LODs (10 copies/µL for Perkinsus and 100 copies/µL for Bonamia) in both single and multiple LAMP assays. The multiplex LAMP assay developed in their study also presented good accuracy in the detection of actual samples26.
Clinical sensitivity is an important criterion for determining the practicality of a detection method. Thus, the multiplex LAMP assay established in this study was applied to clinical samples to determine its clinical sensitivity. Among the 10 single mite and mixed mite (D. pteronyssinus and D. farinae) samples evaluated separately, PCR detected only 3 positive samples of D. pteronyssinus and 5 positive samples of D. farinae. In addition, 5 positive samples were detected among the mixed single D. pteronyssinus samples and single D. farinae samples. On the other hand, the multiplex LAMP assay detected 4 positive samples for D. pteronyssinus and 7 positive samples for D. farinae, in addition to 8 positive samples for mixed single D. pteronyssinus samples and single D. farinae samples. In the validity test conducted with 55 clinical samples, the positive detection rate of the developed multiplex LAMP assay was 83.64%, which was significantly higher than that of PCR (63.64%). This further indicated that the developed multiplex LAMP assay had higher sensitivity and effectiveness than conventional PCR. Moreover, Fraser's group reported the presence of Sarcoptes scabiei DNA in several samples that were negative according to conventional PCR results. This finding corroborates that LAMP might prove to be more sensitive than the conventional PCR method for detecting Sarcoptes scabiei35. Sheng et al.21 demonstrated that the LAMP method was superior, more sensitive, and more effective than PCR for the detection of Toxoplasma gondii oocyst.
In summary, compared to conventional PCR, the multiplex LAMP assay developed in the present study offers the following advantages: (1) higher amplification efficiency; higher sensitivity, and higher specificity; (2) substantially shorter test duration; (3) simpler operation and relatively portable instrumentation, without the need for specific instruments such as agarose electrophoresis equipment and gel presenters; and (4) easy detection, visualization with the naked eye or the use of a turbidimeter that detects the precipitate to directly evaluate whether amplification occurred. In comparison, the single LAMP assay involves testing each dust mites individually in a clinical trial, which increases the gross cost and duration of diagnosis. LAMP is limited by the detection of a single target, which limits its scope and utility in real-world scenarios. Multiplex LAMP technology allows for the simultaneous detection of multiple target mites (D. pteronyssinus and D. farinae), resulting in high multiplex applicability and considerable reductions in time, cost, and effort.
The present study was the first to use the dust mites ITS region as a target gene for multiplex LAMP. Within the same set of samples, multiplex LAMP detected positive samples relatively more rapidly, accurately, and efficiently than PCR. The validity of multiplex LAMP was further confirmed with clinical samples. The present study provided strong evidence in favor of the potential of the multiplex LAMP assay in the detection of dust mites present in the indoor environment of individuals with allergy concerns. The design of effective primers is critical to the success of the LAMP assay. It is essential to use specific primers to prevent cross-reactivity with other organisms or genes.
A multiplex LAMP assay with high sensitivity and specificity for the simultaneous detection of dust mites of D. pteronyssinus and D. farinae was developed. Furthermore, D. pteronyssinus and D. farinae are two common species of dust mites that breed in domestic rooms. Therefore, the method established in this study is suitable for detecting the presence of dust mites dwelling in indoor living environments and is convenient for monitoring allergy-prone populations.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request. The ITS sequences of D. pteronyssinus (KC215340) and D. farinae (MH793957) were retrieved from the NCBI GenBank database.
References
Sarwar, M. In Parasitology and Microbiology Research 26 (IntechOpen, 2020). https://doi.org/10.5772/intechopen.91891.
Xue, Q. et al. Detection and assessment of dust mite allergens in an indoor environment in Anhui, China. Environ. Sci. Pollut. Res. 30, 3045–3055. https://doi.org/10.1007/s11356-022-22395-0 (2023).
Siwak, E. et al. Allergens of mites. Adv. Hyg. Exp. Med. 68, 369–374. https://doi.org/10.5604/17322693.1098023 (2014).
Cao, H. & Liu, Z. Clinical significance of dust mite allergens. Mol. Biol. Rep. 47, 6239–6246. https://doi.org/10.1007/s11033-020-05613-1 (2020).
Li, L., Qian, J., Zhou, Y. & Cui, Y. Domestic mite-induced allergy: Causes, diagnosis, and future prospects. Int. J. Immunopathol. Pharmacol. 32, 2058738418804095. https://doi.org/10.1177/2058738418804095 (2018).
Abdullah, B. et al. Pharmacological management of allergic rhinitis: A consensus statement from the malaysian society of allergy and immunology. J. Asthma Allergy 15, 983. https://doi.org/10.2147/JAA.S374346 (2022).
Leung, C. W. M., Chu, H. C., Leung, J. C. H. & Leung, T. F. Indications for house dust mite allergen-specific immunotherapy. Hong Kong Med. J. 29, 469–471. https://doi.org/10.12809/hkmj2310696 (2023).
Amin, M. M. & Elkady, H. M. Prevalence of common sensitizing aeroallergens in patients with atopic dermatitis. Cytokine 162, 156087. https://doi.org/10.1016/j.cyto.2022.156087 (2023).
Cui, Y., Wang, Q. & Jia, H. Consideration of methods for identifying mite allergens. Clin. Transl. Allergy 8, 1–11. https://doi.org/10.1186/s13601-018-0200-4 (2018).
Ibrahim, R. et al. Species identification and seasonal prevalence of house dust mites in Assiut City, Egypt: A descriptive study in an urban area. Persian J. Acarol. 11, 83–99 (2022).
Liu, Y. et al. Serological analysis of allergic components of house dust mite provides more insight in epidemiological characteristics and clinical symptom development in North China. Front. Immunol. 14, 1083755. https://doi.org/10.3389/fimmu.2023.1083755 (2023).
Oliveira, A. S. et al. Development of a new multiplex PCR to detect prevalent species of house dust mites in house dust. Int. J. Environ. Health Res. 32, 1500–1512. https://doi.org/10.1080/09603123.2021.1893282 (2022).
Chruszcz, M. et al. Molecular determinants for antibody binding on group 1 house dust mite allergens. J. Biol. Chem. 287, 7388–7398. https://doi.org/10.1074/jbc.M111.311159 (2012).
Shafique, R. H., Klimov, P. B., Inam, M., Chaudhary, F. R. & Oconnor, B. M. Group 1 allergen genes in two species of house dust mites, Dermatophagoides farinae and D. pteronyssinus (Acari: Pyroglyphidae): Direct sequencing, characterization and polymorphism. PloS One 9, e114636. https://doi.org/10.1371/journal.pone.0114636 (2014).
Yang, B., Cai, J. & Cheng, X. Identification of astigmatid mites using ITS2 and COI regions. Parasitol. Res. 108, 497–503. https://doi.org/10.1007/s00436-010-2153-y (2011).
Rako, L. et al. LAMP (Loop-mediated isothermal amplification) assay for rapid identification of Varroa mites. Sci. Rep. 13, 11931. https://doi.org/10.1038/s41598-023-38860-w (2023).
Choi, H. et al. Rapid ABO genoty** method using loop-mediated isothermal amplification (LAMP) and real-time PCR. Int. J. Legal Med. 138, 751–756. https://doi.org/10.1007/s00414-023-03144-5 (2024).
Sheu, S.-C., Yu, M.-T., Lien, Y.-Y. & Lee, M.-S. Development of a specific isothermal nucleic acid amplification for the rapid and sensitive detection of shrimp allergens in processed food. Food Chem. 332, 127389. https://doi.org/10.1016/j.foodchem.2020.127389 (2020).
Yang, Z. et al. Rapid and convenient detection of SARS-CoV-2 using a colorimetric triple-target reverse transcription loop-mediated isothermal amplification method. PeerJ 10, e14121. https://doi.org/10.7717/peerj.14121 (2022).
da Silva, E. C. et al. Salmonella detection with LAMP and qPCR and identification of serovars of interest by multiplex qPCR in poultry carcasses. Braz. J. Microbiol. 54, 2173–2182. https://doi.org/10.1007/s42770-023-01095-y (2023).
Sheng, L. et al. Rapid and visual detection of Toxoplasma gondii oocyst in cat feces using loop-mediated isothermal amplification (LAMP) assay. Sci. Rep. 13, 17269. https://doi.org/10.1038/s41598-023-44658-7 (2023).
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63–e63. https://doi.org/10.1093/nar/28.12.e63 (2000).
Razuvaeva, A., Ulyanova, E., Skolotneva, E. & Andreeva, I. Species identification of spider mites (Tetranychidae: Tetranychinae): A review of methods. Vavilov J. Genet. Breed. 27, 240. https://doi.org/10.18699/vjgb-23-30 (2023).
Mori, Y., Nagamine, K., Tomita, N. & Notomi, T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 289, 150–154. https://doi.org/10.1006/bbrc.2001.5921 (2001).
Tian-yu, R., **, S., Zhi-wei, Z., Wei, X. & **-hong, Z. Morphological and molecular characteristics of Ascaridia galli isolates from Huangshan area. J. Trop. Dis. Parasitol. 18, 87 (2020).
Wang, C. et al. Establishment of duplex loop-mediated isothermal amplification method for simultaneous detection of shellfish infected with perkinsus and bonamia. China Anim. Husbandry Vet. Med. 42, 1935–1942 (2015).
Song, M., Hong, S. & Lee, L. P. Multiplexed ultrasensitive sample-to-answer RT-LAMP chip for the identification of SARS-CoV-2 and influenza viruses. Adv. Mater. 35, 2207138. https://doi.org/10.1002/adma.202207138 (2023).
Kumar, S. et al. Magnetic Multiplex Loop Mediated Isothermal Amplification (MM-LAMP) technique for simultaneous detection of dengue and chikungunya virus. J. Virol. Methods 300, 114407. https://doi.org/10.1016/j.jviromet.2021.114407 (2022).
Joshi, S. et al. Rapid multiplex loop-mediated isothermal amplification (m-LAMP) assay for differential diagnosis of leprosy and post-kala-azar dermal leishmaniasis. Am. J. Trop. Med. Hyg. 104, 2085–2090. https://doi.org/10.4269/ajtmh.19-0313 (2021).
Xue, Q. et al. Rapid visual detection of the allergen Dermatophagoides farinae in house dust by loop-mediated isothermal amplification. Environ. Sci. Pollut. Res. Int. 30, 84674–84685. https://doi.org/10.1007/s11356-023-28398-9 (2023).
Sarwar, M. Mites (Arachnida: Acarina) affecting humans and steps taking for the solution of problematics. Int. J. Res. Mech. Eng. 1, 1–14 (2016).
Chermprapai, S. & Thengchaisri, N. A descriptive study of allergen-specific IgE serological tests for canine atopic dermatitis in Thailand. BMC Vet. Res. 16, 475. https://doi.org/10.1186/s12917-020-02684-x (2020).
Thomas, W. R., Hales, B. J. & Smith, W.-A. House dust mite allergens in asthma and allergy. Trends Mol. Med. 16, 321–328. https://doi.org/10.1016/j.molmed.2010.04.008 (2010).
Migueres, M. et al. Types of sensitization to aeroallergens: Definitions, prevalences and impact on the diagnosis and treatment of allergic respiratory disease. Clin. Transl. Allergy 4, 16. https://doi.org/10.1186/2045-7022-4-16 (2014).
Fraser, T. A. et al. A Sarcoptes scabiei specific isothermal amplification assay for detection of this important ectoparasite of wombats and other animals. PeerJ 6, e5291. https://doi.org/10.7717/peerj.5291 (2018).
Acknowledgements
We are grateful to the technologist Zouliang Xu, the staff of EIKEN China Co., LTD. for their valuable support during the study. This work was supported by grants from the Academic Aid Program for Top-notch Talents in Provincial Universities (gxbjZD2020071), Wuhu Key Research and Development Program of China (2021yf391) and Young and Middle-aged Research Fund of Wannan Medical College (WK202216).
Author information
Authors and Affiliations
Contributions
H.Z. and J.Z.: Study concept and design, investigation, resource, visualization, supervision, reviewing of the manuscript, study administration. Y.S., Q.X., H.T., J.G. and Q.T: Drafting of the manuscript, experiment performance, visualization, collected and screened clinical samples. Y.X., J.X. and Y.L.: Statistical analysis, data collection and critically reviewing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shuai, Y., Xue, Q., Tu, H. et al. Multiplex LAMP assay for detecting the prevalent species of dust mites Dermatophagoides farinae and Dermatophagoides pteronyssinus in the domestic environment. Sci Rep 14, 16156 (2024). https://doi.org/10.1038/s41598-024-66043-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-66043-8
- Springer Nature Limited