
Abstract
Microbial NAT enzymes, which employ acyl-CoA to acylate aromatic amines and hydrazines, have been well-studied for their role in xenobiotic metabolism. Some homologues have also been linked to secondary metabolism, but this function of NAT enzymes is not as well-known. For this comparative study, we surveyed sequenced microbial genomes to update the list of formally annotated NAT genes, adding over 4000 new sequences (mainly bacterial, but also archaeal, fungal and protist) and portraying a broad but not universal distribution of NATs in the microbiocosmos. Localization of NAT sequences within microbial gene clusters was not a rare finding, and this association was evident across all main types of biosynthetic gene clusters (BGCs) implicated in secondary metabolism. Interrogation of the MIBiG database for experimentally characterized clusters with NAT genes further supports that secondary metabolism must be a major function for microbial NAT enzymes and should not be overlooked by researchers in the field. We also show that NAT sequences can be associated with bacterial plasmids potentially involved in horizontal gene transfer. Combined, our computational predictions and MIBiG literature findings reveal the extraordinary functional diversification of microbial NAT genes, prompting further research into their role in predicted BGCs with as yet uncharacterized function.
Similar content being viewed by others


Introduction
In the course of evolutionary time, microorganisms have developed immense metabolic potential and adaptability, and their capabilities have attracted scientific interest for useful biotechnological applications. Through xenobiotic metabolism, bacteria and fungi can detoxify, degrade or biotransform exogenous compounds of natural or synthetic origin, surviving and even thriving in adverse chemical environments that would be toxic to more complex organisms1. Microbial xenobiotic metabolism involves a plethora of enzyme activities, and arylamine N-acetyltransferase (NAT, E.C. 2.3.1.5) is one of them2. Microbial NAT enzymes catalyze the N-acetylation of aromatic amines, leading to detoxification of many harmful by-products of industrial activity and farming (e.g. pharmaceuticals, dyes, pesticides, etc.)3,4,5,6,7,8. However, they can also bioactivate procarcinogenic N-hydroxyarylamines via O-acetylation (E.C. 2.3.1.118), an activity exploited by Ames and colleagues in the popular Salmonella mutagenicity test9. The study of Salmonella NAT was indeed groundbreaking, in that it additionally revealed the basic structure and catalytic mechanism of the enzyme family, which employs a cysteine-histidine-aspartate (Cys-His-Asp) protease-like catalytic triad to transfer an acetyl group from donor acetyl coenzyme A (CoA) to the amino group of the acceptor aromatic amine10,11.
An unexpected discovery was reported for the (AMYMS)NAT3 (alias symbol RifF, GenBank ID: AFO74156.1) homologue of the actinobacterium Amycolatopsis mediterranei str. S699, implicating NAT not only in xenobiotic, but also in secondary metabolism. That particular homologue, which acts as an amide synthase, is encoded by a gene located at the end of the core biosynthetic gene cluster (BGC) driving production of the antibiotic rifamycin B in the actinomycete12,13. The reaction is atypical for a NAT enzyme, in that it employs a large polyketide chain as substrate and does not utilize acetyl-CoA. Like xenobiotic metabolism, secondary metabolism is not generally associated with vital functions of cells, but rather enhances the biological fitness of microbes as a response to environmental stress (e.g., by generating chemical weapons against competitors)14,15. Due to their remarkable chemical properties and variety, the products of secondary metabolism have long been exploited as a natural source of pharmaceuticals (e.g., antibiotics, anticancer agents, immunomodulating substances, etc.) and other compounds of industrial utility16.
A common feature of specialized microbial pathways, such as those associated with xenobiotic or secondary metabolism, is that their enzymatic components are often encoded by co-regulated genes arranged in clusters17,18,19. Activation of those gene clusters is usually triggered by specific environmental stimuli, directing resources and products of primary metabolism towards xenobiotic biotransformation or the biosynthesis of secondary metabolites. Apart from the aforementioned (AMYMS)NAT3 (alias rifF) homologue of the rifamycin BGC in A. mediterranei, other actinobacterial NAT genes have also been localized in clusters associated with cholesterol degradation (specifically in slow-growing pathogenic mycobacteria) or vitamin biosynthesis (in fast-growing, free-living mycobacteria)20,21,22. Moreover, in the corn-pathogenic fungus Fusarium verticillioides (teleomorph Gibberella moniliformis), the (GIBMO)NAT1 (alias symbol FDB2, GenBank ID: EU552489.1) gene, encoding the N-malonyltransferase that is essential for detoxification of host phytoanticipin 2-benzoxazolinone, is also part of a well-characterized gene cluster18,23.
Other lines of evidence suggest that certain microbial NAT homologues could play a role in secondary metabolism. For example, acyl-CoA monomers (e.g., acetyl-CoA and malonyl-CoA) derived from acetate and propionate metabolism, are employed as starter and/or extender units during the biosynthesis of polyketides24,25, while they are also utilized by NAT enzymes. Specifically, in addition to acetyl-CoA, NAT enzymes can utilize propionyl-CoA, butyryl-CoA and acetoacetyl-CoA as donor substrates5,6,26,27,28,29, while certain microbial homologues have been shown to be selective for malonyl-CoA4,6,29 and others can non-selectively bind various short-chain acyl-CoA compounds6.
The enzymatic processes of xenobiotic and secondary metabolism are believed to share an overlap** evolutionary history, while some of their key components are also encountered in fatty acid metabolism24,30. Although it seems likely that different NAT homologues have diverged from their ancestral forms to serve such metabolic functions in microorganisms, evidence remains sporadic and the corresponding evolutionary relationships are elusive, particularly for those NAT proteins with roles other than xenobiotic metabolism. For this comparative computational genetic study, we surveyed microbial genomes to annotate NAT genes, then investigating their possible localization within clusters. We also looked for possible association of NAT genes with bacterial plasmids, as the enzymes of xenobiotic and secondary metabolism are often encoded by genes participating in horizontal gene transfer (HGT) events involving mobile genetic elements31.
Results and discussion
Identification and annotation of microbial NAT genes
Our previous genomic database surveys, published in 200832 and 201033, collectively retrieved and annotated 467 microbial NAT sequences (347 bacterial, 1 archaeal, 94 fungal and 25 protist), allowing the first overview of NAT gene distribution in the microbiocosmos. At the time of the second survey33, only 2,300 sequenced microbial genomes were accessible to screen, but this number has since multiplied very rapidly (Fig. 1). In view of this progress, a new survey was undertaken, to expand the earlier ones and support the analyses described later in this manuscript. The core dataset of annotated NAT sequences was retrieved through exhaustive database survey of approximately 34,500 prokaryotic genomes (98% bacterial, 2% archaeal; performed in 2015) and 1,400 eukaryotic genomes (68% fungal, 32% protist; performed in 2016). Additional searches were carried out later (2020–2021) to enrich the dataset, particularly with respect to previously underrepresented microbial taxa in the database. By the end of the survey, it was estimated that we had collectively covered about 324,000 prokaryotic (98% bacterial, 2% archaeal) and 8,700 eukaryotic (88% fungal, 12% protist) microbial genomes (Fig. 1). Searches were concluded for large taxonomic groups (e.g., mycobacteria, bacilli, staphylococci, burkholderias, enterobacteria, etc.) when the addition of new NAT genes effectively became redundant, expanding the existing set mainly with sequences from new strains of already described species. The final list (Fig. 2 and Supplementary Information S1) comprised about 4,600 annotated microbial NAT genes (92% bacterial, 1% archaeal, 6% fungal, 1% protist) representing 1,318 species (87% bacterial, 2.5% archaeal, 9% fungal and 1.5% protist), including the previously annotated prokaryotic and eukaryotic microbial NAT sequences32,33. The data is also available on the NAT website (http://nat.mbg.duth.gr/).

Increase in the numbers of sequenced microbial genomes deposited in the Genome database, monitored for bacteria, archaea, protists and fungi during the course of the study.

Overview of the NAT gene dataset compiled for the purposes of this study. The top panel depicts the main taxonomic groups represented in the dataset for prokaryotic (left) and eukaryotic (right) species. The bottom panel depicts the distribution of annotated NAT sequences in prokaryotes (left) and eukaryotes (right). Archaea (Halobacteria) and protists are indicated with blue font. The compiled NAT gene dataset also included 467 sequences annotated previously32,33.
In archaea, NAT genes were only found in the phylum of Euryarchaeota, specifically in the class of Halobacteria. In bacteria, NAT genes were found in the phyla of Acidobacteria (classes Blastocatellia, Holophagae, Vicinamibacteria), Actinobacteria, Armatimonadetes, Bacteroidetes (FCB group), Bdellovibrionota (class Oligoflexia), Calditrichaeota, Chlamydiae (PVC group), Chlorobi (FCB group), Chloroflexi, Cyanobacteria, Deferribacteres, Deinococcus-Thermus, Eremiobacteraeota, Firmicutes, Haloplasmatales/Tenericutes, Nitrospinae, Nitrospirae, Planctomycetes (PVC group), Proteobacteria (Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, Epsilonproteobacteria), Spirochaetes, Verrucomicrobia (PVC group), and various unclassified bacteria. NAT genes were not found in the sequenced genomes from the phyla of Aquificae, Chrysiogenetes, Coprothermobacterota, Dictyoglomi, Elusimicrobia, Fibrobacteres (FCB group), Fusobacteria, Krumholzibacteriota, Marinimicrobia, Synergistetes, Thermodesulfobacteria, Thermotogae and Caldiserica/Cryosericota group (Fig. 2).
In protists, NAT genes were found in the paraphyletic clades of Alveolata (Apicomplexa and Ciliophora), Amoebozoa (Mycetozoa/Dictyosteliida and Discosea/Centramoebida), Discoba (Euglenozoa and Heterolobosea), and Stramenopiles (Oomycetes, Pelagophyceae and Bacillariophyta). Finally, in fungi, NAT genes are present in the phyla of Ascomycota (only Pezizomycotina) and Basidiomycota, as well as in lower fungi (Fungi incertae sedis) and specifically in the phyla of Chytridiomycota and Zoopagomycota (Fig. 2).
Overall, the compiled list of annotated NAT genes complements the previous datasets4,29,32,33. In prokaryotes, several new bacterial taxa with NAT genes were identified, while all annotated NAT genes of archaea belonged to halophiles, consistent with previous observation33. The list of NAT genes in eukaryotic microorganisms also expanded considerably, with new taxons added for protists, but without major changes in taxon distribution for fungi, compared with previous surveys29,33. On the basis of the observed sequence redundancy, it is likely that the current dataset is now effectively saturated with information and is illustrative of a broad, but not universal, distribution of NAT genes in microbial genomes.
Localization of NAT genes in BGCs of prokaryotic microorganisms
The possible localization of annotated microbial NAT genes within genomic clusters was probed using the antibiotics and secondary metabolite analysis shell software (antiSMASH)34. Initially, the genomic region of 1,820 NAT genes was analyzed through the early antiSMASH version 3.0, and the investigation was later reiterated and expanded to include an additional 1,272 bacterial and archaeal annotated NAT genes, analyzed through the newer and more stringent version of antiSMASH 5.0. This screen identified 102 putative clusters bearing 103 NAT genes in 96 prokaryotic species, including one putative cluster with a NAT gene in the archaeon Halostella salina strain CBA1114 (Fig. 3 and Supplementary Information S2). Reanalysis of all the clusters identified with antiSMASH 5.0 was finally performed with the latest antiSMASH version 7.0, and all hits were verified, apart from four bacterial NAT genes which were predicted in BGCs by version 5.0, but not by version 7.0. Cluster type descriptions were also more complete with the latest version 7.0 (Fig. 3 and Supplementary Information S3). As the current version is the most accurate one, the predicted cluster coordinates and length are reported here only relative to the output of version 7.0. Refinement of BGC detection rules in the later versions provided a wider panel of predicted BGC classes, including furan, thiopeptide, linaridin, acyl-amino acid, β-lactone, arylpolyene, RiPP-like and several hybrid clusters (Fig. 3 and Supplementary Informations S2 and S3). It is, however, notable that the early (antiSMASH 3.0) version predicted several NAT1 mycobacterial clusters which were not found by the later versions. Those clusters have been described in the literature before for Mycobacterium bovis BCG, but they are known to play a role in cholesterol catabolism20. This lack of antiSMASH 3.0 cluster prediction stringency was useful, from the point of view of our study, as it allowed comparison of an already known type of cluster across a range of different mycobacteria (Supplementary Information S4).

Clusters with NAT genes per bacterial taxon, predicted during analyses with antiSMASH software versions 5.0 (top) and 7.0 (bottom). PKS: polyketide synthase; NRPS: non-ribosomal peptide synthase. The descriptions of clusters classified as “Other” are provided in Supplementary Informations S2 and S3, for the analyses performed with antiSMASH versions 5.0 and 7.0, respectively. Note that the streptomycetes are shown separately from other actinobacteria, as they are of major importance from the point of view of secondary metabolism and the biosynthesis of natural products35.
In view of the known association of (AMYMS)NAT3 (rifF) gene with the BGC of rifamycin in A. mediterranei12,13, it was expected that antiSMASH would detect NAT genes only in conserved actinobacterial polyketide synthase (PKS) clusters responsible for the biosynthesis of ansamycin antibiotics like rifamycin. Surprisingly, this was not the case, as the software predicted different NAT genes within a spectrum of BGC types (Fig. 3 and Supplementary Informations S2 and S3), implying that the enzymatic function of NAT proteins in secondary metabolism is unlikely to be restricted merely to the amide synthase activity reported for (AMYMS)NAT3 (RifF). The diversity in the gene content and organization of BGCs harbouring NAT homologues was indeed remarkable, with synteny between clusters observed just for different strains of the same species and only partially between closely related species of the same genus (see examples in Fig. 4 and Supplementary Information S5). It was also apparent that NAT genes are not associated with BGCs restricted to a specific taxonomic group of bacteria, as phylogenetic analyses demonstrated that the distribution of BGC-associated NAT homologues is intermixed, with low basal resolution across different taxa. More specifically, in the phylogenetic trees of Fig. 5 and Supplementary Information S6, the distribution of BGC-associated NAT sequences in different clades is neither according to taxonomy, nor according to BGC type. In contrast, BGC-associated NATs illustrate a mosaic distribution pattern that spans different bacterial groups, suggesting widespread HGT events, not just at the level of individual genes (as has been reported before4,33), but also at the level of whole BGCs. For example, the NATs of terpene BGCs appear to cluster together in the phylogenetic tree, although some of them belong to alphaproteobacteria and some to actinobacteria (Fig. 5b,c and Supplementary Information S6b–e). The same mosaic distribution of BGC-associated NATs is also observed in the sequence similarity networks (SSNs) of Fig. 6, showing a highly intermixed core group (whether it is viewed from the standpoint of taxonomy or of BGC type), connected with two more specialized groups of homologues. The first group contains certain Firmicutes NATs associated with non-ribosomal peptide synthase (NRPS) clusters, while the second group comprises the actinobacterial NATs associated with PKS or PKS-NRPS hybrid clusters that are responsible for the biosynthesis of ansamycins (Fig. 6). In those last BGCs, the NAT genes are likely to be orthologous to rifF.

Representative illustrations of synteny between putative bacterial clusters with NAT genes. An example of NRPS cluster synteny between different strains of Bacillus anthracis (phylum Firmicutes) is presented in the top panel, while the bottom panel shows synteny of a terpene cluster between different species of Bosea (phylum Proteobacteria). The NAT gene on each cluster is indicated with an asterisk. Additional examples are provided in Supplementary Information S5.

Distribution of NAT genes per prokaryotic taxon and type of biosynthetic gene cluster (BGC), determined during the antiSMASH 5.0 analyses (including MIBiG). The species tree (a) was constructed according to conventional taxonomy (NCBI Taxonomy Database common tree). The phylogenetic tree of BGC-associated NAT sequences (b) was constructed using the neighbour-joining method, and the leaves are coloured according to taxonomy. The same phylogenetic tree is also presented with leaves coloured according to cluster type (c). Note that, in a and b, the streptomycetes are shown with a different colour from other actinobacteria, as they are of major importance from the point of view of secondary metabolism and the biosynthesis of natural products35. The same trees are provided enlarged in Supplementary Information S6a–c, for additional clarity, alongside the corresponding trees generated with the maximum likelihood method (Supplementary Information S6d–e).

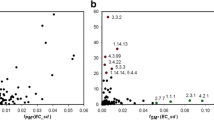
Sequence similarity network (SSN) demonstrating the relationships between different NAT genes found in biosynthetic gene clusters (BGCs) by antiSMASH 5.0 analyses (including MIBiG). Each node represents a BGC-associated NAT sequence and the edges connect close relatives, using an alignment score threshold of 29 (E-value = 10−29). The colouring of nodes is either according to taxonomic group (a) or according to cluster type (b). Note that, in a, the streptomycetes are shown with a different colour from other actinobacteria, as they are of major importance from the point of view of secondary metabolism and the biosynthesis of natural products35.
As the actinobacteria, and particularly the streptomycetes, represent the richest source of bacterial secondary metabolites36, it is perhaps unsurprising that the majority (66%) of BGCs with NAT genes were identified to belong to this particular taxonomic group (Fig. 3). Moreover, about 60% of those actinobacterial clusters were predicted to belong to the BGC types of NRPS, PKS, PKS-NRPS hybrid or NRPS hybrid. In those biosynthetic pathways, scaffold assembly is regarded to proceed through successive rounds of chain elongation, using acyl-CoA molecules (in PKS clusters) or amino acids (in NRPS clusters) as extension units24,25. The ability of NAT enzymes to accommodate aromatic amines and short-chain acyl-CoA molecules in their active site may partially explain the recruitment of microbial NAT genes by the NRPS/PKS system. Moreover, although those assembly lines are typically terminated by thioesterases, the example of the (AMYMS)NAT3 (RifF) amide synthase demonstrates that other homologous NATs could also serve the release of fully assembled scaffolds from the biosynthetic machinery37. It is also possible that NAT enzymes may be implicated in chemical modification of the peptide or polyketide core structure, contributing to chemical diversification of the end product.
About 17% of identified BGCs with NAT genes were found in Firmicutes, mainly bacilli. Most of those BGCs were of the NRPS type and were associated with NAT3 isoforms, such as those of Bacillus anthracis and Bacillus cereus which have been expressed in recombinant form and tested for catalytic activity against arylamines38,39,40. Although active, the (BACCE)NAT3 isoenzyme of B. cereus deviates from other functionally characterized NATs in that it has a catalytic triad with Glu instead of Asp39. In contrast, although endogenously expressed, the (BACAN)NAT3 of B. anthracis is substantially shorter and apparently non-functional as N-acetyltransferase, due to its gene being compromised by a frameshift mutation32. It is tempting to speculate whether those unusual features of NAT3 in bacilli could serve some specific function in the associated NRPS cluster, especially since studies have shown that truncation of the C-terminus may convert NATs into acetyl-CoA hydrolases41,42.
Unlike Actinobacteria and Firmicutes, in Proteobacteria only a few NAT genes were predicted within BGCs. In alphaproteobacteria, those are involved in the biosynthesis of terpenes which differs substantially from that of polyketides and non-ribosomal peptides. Therefore, the NAT enzymes participating in those pathways could differentiate functionally. For instance, as the core hydrocarbon skeleton of terpenes is modified, e.g. by addition of amino acids or fatty acids43, NAT could act as acyltransferase or as modulator of acyl-CoA availability, like it has been suggested before for mycobacteria44. It is also of note that two NAT genes of Bradyrhizobium oligotrophicum are localized within the same terpene BGC.
In betaproteobacteria, all three BGCs with NAT genes were predicted to direct the synthesis of acyl-amino acids. Those NAT enzymes could act as acyltransferases, and recent work has demonstrated human NAT2 to be capable of employing not just aromatic, but also aliphatic amines as substrates45. The remaining BGCs with NAT genes in gamma, delta and epsilonproteobacteria were of various types and only sporadic, most likely the outcome of HGT from other bacterial groups. The same is also probable for the β-lactone BGC found in the archaeon. In conclusion, it is likely that once associated with secondary metabolism, NAT genes had broad opportunity to diverge from their archetypal function to serve a range of biosynthetic processes.
Localization of NAT genes in BGCs of eukaryotic microorganisms
As BGCs are also known to drive secondary metabolism in fungi15,17, the 268 NAT genes annotated during the genomic survey described above (Supplementary Information S1) and previously33 were investigated as to their possible localization within clusters. The procedure was the same as for prokaryotes, and the results of the analyses with antiSMASH versions 3.0, 5.0 and 7.0 were compared. As in prokaryotes, the earliest less stringent version 3.0 localized certain functionally investigated NAT genes6 within clusters, namely, NAT1 (encoding for N-malonyltransferase) and NAT3 (encoding for N-acetyltransferase) found in Fusarium graminearum str. PH-1 and F. oxysporum f.sp. lycopersici str. 4287, as well as the (GIBMO)NAT3 of F. verticillioides str. 7600 and the (ASPFN)NAT3 of A. flavus str. NRRL 3357. The NAT4 homologue6 of various F. oxysporum types was also predicted to be associated with BGCs.
When the analysis was repeated with the later antiSMASH version 5.0, the number of recovered hits was considerably smaller, but much more accurately annotated (Supplementary Information S7). All 16 fungal BGCs, identified to harbour NAT genes, belonged to filamentous ascomycetes. Of those, 13 belonged to Eurotiomycetes and they were predicted to function as PKS or PKS hybrid clusters. Only 3 BGCs with NAT were predicted in Sordariomycetes, and these were mainly of the NRPS type (Supplementary Information S7). Reanalysis of those results with the latest version of antiSMASH 7.0 verified the hits, also updating matches with experimentally characterized BGCs like the PKS cluster for 8-methyldiaporthin of A. flavus str. RIB4046 (Table 1 and Supplementary Information S8). As expected, in the SSN of Fig. 6, the fungal and bacterial sequences were separate, consistent with the monophyletic origin of fungal NAT genes33.
Finally, no hits were provided by antiSMASH analyses of 51 annotated NAT sequences from protists, reported here (Supplementary Information S1) and in our previous study33. The only possible exception was (DICDI)NAT4 of Dictyostelium discoideum str. AX4 which could reside in a BGC. In addition to gene annotations provided by the GenBank, in the future it may be useful to also try different eukaryotic gene-calling algorithms, like Augustus47, to investigate the genomic context of NAT loci in fungi and protists.
Localization of NAT genes in bacterial plasmids
Although the Genome database reported almost 30,000 sequenced plasmids at the time of the study, those sequences were not accessible by BLAST via the NCBI and so instead we looked for them via the specialized PLSDB database48. A total of 92 bacterial plasmids were identified to carry 117 NAT genes in several actinobacteria, alphaproteobacteria, betaproteobacteria, gammaproteobacteria and bacilli (Table 2 and Supplementary Information S9). Those plasmids were either circular or linear, and their size varied from about 30.3 Kb (plasmid pYGD30 of Bacillus thuringiensis strain YGd22-03) to 2.8 Mb (plasmid of Cupriavidus campinensis strain MJ1). It is noteworthy that several of the identified plasmids carry more than one NAT gene, particularly in the bacilli which often display multiple NAT open reading frames (ORFs) in their plasmids, similarly to their genomic sequence. Those included ORFs with frameshift mutations, as has been reported previously for the genomic NAT3 homologues of certain bacilli32.
All plasmid-associated NAT genes were subsequently screened by antiSMASH 6.0 for possible localization within BGCs, and this was confirmed for five of them (Table 3). Finally, all identified plasmids were screened for the presence of genomic islands, which are indicative of exchanges between plasmid and chromosomal DNA in bacteria49. Such genomic islands were identified to harbour NAT genes in five different plasmids, but only the plasmids of the gammaproteobacterium Pantoea agglomerans were found to carry intact ORFs without frameshift mutations (Fig. 7).

Plasmid genomic islands harbouring NAT genes. The genomic islands of two plasmids carried by strains CFSAN047153 and CFSAN047154 of the gammaproteobacterium Pantoea agglomerans, were predicted by IslandViewer algorithms SIGI-HMM and IslandPath-DIMOB. The outer circle is the plasmid and the graphical illustration of the inner circle is the GC content of the corresponding sequence, as deviation from the expected GC content may be indicative of heterologous portions originated through horizontal gene transfer (HGT). The NAT gene is located between the black dots, within a low-GC genomic island.
Genes like NAT, implicated in xenobiotic and secondary metabolism, are often encountered in plasmids and are exchanged between bacterial cells enhancing adaptability to adverse environmental conditions. Moreover, BGCs introduced from plasmids can enhance the biosynthetic capabilities of hosts50,51. In that respect, plasmids with NAT genes may enhance the ability of bacterial cells to detoxify potentially harmful xenobiotics in their environment. Moreover, NAT genes carried by plasmids were also found to be associated with BGCs. For example, in plasmid II of Streptomyces reticuli str. TUE45, the NAT gene is located within a predicted BGC for the ansamycin antibiotic rubradirin52, where it is predicted to act as an amide synthase similar to (AMYMS)NAT3 (RifF). Furthermore, BLAST search of the NAT sequence found in the genomic island of the P. agglomerans plasmid, demonstrates a good match with chromosomal gene Pnp2A that is homologous to NAT and is part of a six-gene BGC responsible for antibiotic biosynthesis53.
Interrogation of the MIBiG database for NAT genes associated with experimentally characterized BGCs
A significant aim of the present work was to assess the amount of information available in the literature, regarding the genomic and functional links of microbial NAT genes with secondary metabolism. For decades, this information has been increasing in volume, but has effectively stayed under the radar of scientists dedicated to NAT research, because of a gap in gene nomenclature. Specifically, it is common practice for researchers characterizing new BGCs to name genes after the cluster they are located in and according to their genomic order. For example, (AMYMS)NAT3 of A. mediterranei was identified to be the sixth gene (F) on the core BGC for rifamycin (rif), so it was named rifF. Moreover, the protein product of this gene was described based on function (amide synthase), rather than homology to other NAT enzymes12,13,37,54. Consequently, using the keywords “NAT” or “arylamine N-acetyltransferase” to search PubMed cannot readily pick up relevant literature. Hence, with the exception of rifF55, studies directly connecting NATs with their BGC-associated homologues are effectively lacking and microbial NATs have been functionally investigated as xenobiotic metabolizing enzymes.
Modern databases provide access to the literature, enabling search with a gene/protein sequence instead of keywords. One such database is MIBiG (minimum information about a biosynthetic gene cluster)56, used in this study as part of the antiSMASH searches described above. In addition, the whole MIBiG sequence repository was downloaded and subjected to BLAST search with NAT sequences as query. This database is dedicated to depositing information about experimentally characterized BGCs and their chemical products, thus, any NAT sequences recovered would be expected to be part of an already characterized gene cluster.
Indeed, the interrogation of MIBiG database identified several characterized NAT homologues within bacterial BGCs, for which literature was already available (Table 4). Apart from A. mediterranei, the marine actinomycete Salinispora arenicola has been demonstrated to possess a rifamycin BGC carrying a NAT/rifF orthologue57,58. Other BGCs responsible for the production of ansamycin secondary metabolites have been experimentally characterized in actinomycetes and, based on sequence comparison and chemical analogy of the synthesized product, the corresponding NAT homologues are proposed to have an amide synthase function similar to RifF. Ansamycins are medicinally important compounds characterized by an aliphatic (ansa) chain linked to non-adjacent positions of a benzene- or naphthalene-based chromophore59,60. Benzenic ansamycins (e.g. geldanamycin, macbecin and ansamitocin in Table 4) are known for their cytotoxic action against eukaryotic cells, while naphthalene-based ansamycins (e.g. rifamycin and its congeners, rubradirin, streptovaricin and naphthomycin in Table 4) exhibit mainly antimicrobial activity. Despite the structural variation of the produced metabolites, biosynthetic pathways of ansamycins share crucial similarities, reflected in the organization of the corresponding BGCs. The main part of those clusters is typically occupied by genes encoding a PKS. Directly downstream there is usually a NAT gene, followed by genes responsible for 3-amino-5-hydroxybenzoic acid (3,5-AHBA) biosynthesis, which serves as the universal precursor for ansamycin polyketides synthesized by the PKS machinery59,60. The assembled linear product then serves as substrate for the NAT amide synthase, which links the carboxyl to the arylamine end of the polyketide chain, simulating the typical donor–acceptor substrate reaction of NAT enzymes. Consistent with the known NAT catalytic mechanism61, the first step for ansamycin macrolactamization is likely to involve covalent attachment of the polyketide aliphatic end to catalytic Cys62. Completion of the reaction requires that the two ends of the polyketide substrate come into close proximity, indicating that the catalytic pocket is large enough to accommodate such a bulky substrate. The modelled structure of RifF has a loop, instead of the typical helix, between domains II and III, potentially rendering entry to the active site less restricted relative to other NATs55.
Several of the NAT homologues of Table 4 are involved in biosynthetic pathways that link substrate molecules via an amide bond. For example, asuC2 of Streptomyces nodosus and colC2 of Streptomyces aureus encode NAT homologues that are proposed to participate in the biosynthesis of the pokyketides asukamycin and colabomycin E, respectively80,81. The metabolic phenotype of an asuC2 knockout strain indicates that NAT acts as the amide synthase performing the attachment of the upper polyketide chain to the amino group of 3-amino-4-hydroxybenzoic acid (3,4-AHBA)80. Similarly to its isomer 3,5-AHBA, this compound is a precursor in the biosynthesis of secondary metabolites, e.g. the terpene pigment grixazone produced by Streptomyces griseus. Although the NAT homologue of this actinomycete is not part of the grixazone BGC, the encoded protein can Ν-acetylate exogenous 3,4-AHBA, as well as other 2-aminophenol derivatives94. However, N-acetylated 3,4-AHBA was not detected under grixazone-producing conditions95.
Closer to the more familiar NAT-catalyzed acyl-CoA mediated acyltransfer reaction is the activity of seven NAT homologues in Table 4. Among them, the ptnC and ptmC genes of Streptomyces platensis encode NAT enzymes that can employ (thio)platensicyl- or (thio)platencinyl-CoA as donor substrates, catalyzing the last step in the biosynthesis of antibiotics platencin, platencimycin, and their thiocarboxylic congeners. More specifically, those enzymes form the amide bond which connects the ketolide with the 3-amino-2,4-dihydroxybenzoic acid moiety of the aforementioned products82,83,84. Another example is the nybK gene of Streptomyces albus, encoding a NAT homologue involved in biosynthesis of the antibiotic nybomycin, where it performs transfer of two acetoacetyl groups from CoA to 2,6-diaminophenol85. Acetoacetyl-CoA has been reported to serve as donor substrate for (MYCTU)NAT1 of Mycobacterium tuberculosis, but this particular homologue was shown to be part of a cholesterol catabolic gene cluster essential for microbial survival inside macrophages27. Furthermore, the NAT homologues daqS and daqT (Table 4) participate in the biosynthesis of diazaquinomycin antibiotics, transferring β-ketoacyl units from CoA to the amine groups of 2,6-diaminohydroquinone86. Deviating from the aforementioned acyl-transfer reactions, where the acceptor substrate is an aromatic amine, the product of cetD gene (Table 4) performs N-acetylation of an aminocyclitol during biosynthesis of the antitumor agent cetoniacytone A87,88.
Finally, some BGC-associated NAT homologues have been described to exert O-acyltransferase activity towards the hydroxyl group of acceptor substrates (Table 4). For instance, the tubG gene of the proteobacterium Archangium disciforme is located in the cluster responsible for biosynthesis of the cytotoxin tubulysin, where it encodes a NAT homologue that is proposed to O-acylate the pre-tubulysin molecule90. Similarly, in Streptomyces sp. RI18, the NAT product of bezG gene may O-acetylate p-hydroxyaminobenzoic acid, during the biosynthesis of benzastatins91.
Concluding remarks
Over the past twenty years, we have witnessed progress in genomics by researching the distribution of NAT homologues across the entire spectrum of (sequenced) prokaryotic and eukaryotic life2,3,32,33 and annotating new NAT genes on behalf of the NAT committee96. The present study is estimated to have surveyed over 300,000 sequenced microbial genomes and, although this number has almost doubled today, we believe that our portrayal of microbial NAT gene distribution, diversity and phylogeny is now comprehensive and unlikely to change substantially. Similarly exciting has been the progression of knowledge about the functional divergence of microbial NATs, captured by many research groups97 demonstrating multiple roles of NATs in xenobiotic, secondary and fatty acid metabolic pathways that arm bacteria and fungi to survive or modify their chemical environment and thrive within animal or plant hosts. Given the broad spectrum of functions attributed to microbial NAT enzymes, it is no wonder that scientists have been unable to connect all those homologues under the same consensus nomenclature. Modern databases are nowadays overcoming this difficulty, enabling literature searches using the sequence or other standardized identifiers of genes, proteins and families, while also providing accurate predictions of possible functions. Through the use of such tools, our knowledge of the different roles of NATs in microbes is expanding and the worlds of xenobiotic and secondary metabolism are converging, as recently demonstrated by a group of medicinal chemists characterizing the (STRPT)NAT1 (PtmC) homologue from Streptomyces platensis and comparing it with other NATs84.
Overall, the experimental evidence supports that the NAT activities associated with bacterial biosynthesis of secondary metabolites can be classified into two main types. The first is the amide synthase activity involved in the production of polyketide ansamycins, while the second is the acyltransferase activity encountered in the biosynthetic pathways of various polyketides, terpenes and other compounds. The association of NAT homologues with secondary metabolism is less evident for eukaryotic microorganisms, although NAT genes were predicted to participate in clusters relevant to other functions, in line with previous observations. It is also significant that, like other genes of xenobiotic and secondary metabolism, NAT sequences are associated with mobile genetic elements involved in HGT, consistent with the mosaic phylogenetic pattern observed for bacterial NATs.
Through our comparative application of different antiSMASH versions, we have been able to follow the advancement of this valuable computational tool. More importantly, the in silico predictions and the experimental findings of the literature retrieved via the MIBiG portal, revealed the extraordinary functional diversification of microbial NAT enzymes in the biosynthesis of secondary metabolites, prompting further research into the role of NAT genes in computationally predicted BGCs with as yet uncharacterized functions.
Methods
Genomic survey and annotation of microbial NAT homologues
NAT genes were mined from sequenced microbial genomes and annotated according to established criteria, as previously described32,33,96. Searches of the Genome database, accessed through the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/genome), were carried out using the tBLASTn algorithm with the appropriate reference sequence as query33. Specifically, genomes were interrogated with the following annotated amino acid sequences: (SALTY)NAT1 (GenBank ID: BAA14331.1) of Salmonella enterica subsp. enterica serovar Typhimurium str. LT2 for bacteria; (HALBP)NAT1 (GenBank ID: CBL43355.1) of Halogeometricum borinquense str. DSM 11551 for archaea; (GIBMO)NAT1 (GenBank ID: ACD88491.1) for fungi; (DICDI)NAT1 (GenBank ID: CBL43356.1) of Dictyostelium discoideum str. AX4 for protists. More focused searches were additionally performed, as necessary, using annotated NAT sequences found in microorganisms more closely related to each interrogated taxon. Reconstruction of NAT ORFs was performed computationally and/or manually, guided by individual GenBank entries, and annotation was based on inspection of the corresponding translated sequences for identification of the characteristic semi-conserved motifs “VPFENL”, “RGGYC”, “THRL” and “VDV”, where underlined residues indicate the Cys-His-Asp catalytic triad. Species-specific NAT gene symbols were assigned based on the percent identity of translated sequences with the corresponding reference sequence mentioned above, according to the guidelines of the NAT Gene Nomenclature Committee (http://nat.mbg.duth.gr/)96,98. Sequence handling was performed on BioEdit Sequence Alignment Editor 7.0.5.399 and Unipro UGENE100.
Microbial genome mining for BGCs with NAT genes
Computational investigation into the possible localization of microbial NAT homologues within BGCs was conducted using antiSMASH (https://antismash.secondarymetabolites.org/)34. The genomic coordinates of annotated microbial NAT genes were initially determined, in order to define the surrounding region. Prokaryotic NAT genes were then retrieved together with 500 kb of upstream and downstream flanking sequences (~ 1 Mb of total sequence length), whereas for eukaryotic NAT genes the flanking sequences were 1 Mb each (~ 2 Mb in total). Sequences were downloaded in full GenBank format with gene annotations incorporated as provided by the database. Those files were then uploaded to the antiSMASH platform version 3.0101, enabling the ClusterFinder algorithm option. The initial analyses were performed in 2016–2018 and were repeated with a larger dataset in 2020, using antiSMASH updated version 5.0102 with default parameters. The results were finally validated in 2023, using the new antiSMASH version 7.0103. When a NAT gene was found within the overlap** region of more than one protocluster, it was considered as part of all protoclusters sharing this region. It is also noted that, newer antiSMASH versions (5.0 and 7.0) fail to run the analysis, if the input sequence begins or terminates with a partial (truncated) ORF. Given the high gene density of microbial genomes, the input sequences thus required additional editing with Unipro UGENE, to remove any partial ORFs from the ends. The GenBank files of all putative clusters containing NAT genes were finally downloaded and saved as individual files compiling a comprehensive local dataset. The predictions and BGC definitions with the newer version 7.0 should be regarded as more accurate and complete compared with the previous versions.
Interrogation of the MIBiG database for BGCs bearing NAT genes
For NAT genes predicted by antiSMASH to localize within BGCs, the minimum information about a biosynthetic gene cluster (MIBiG, https://mibig.secondarymetabolites.org/)104 version 2.0 database was interrogated for previous publications associating NATs with experimentally characterized gene clusters. The content of the MIBiG database was initially downloaded in a FASTA file format. This file, containing all the amino acid sequences encoded by genes from MIBiG entries, was converted into a local database suitable for interrogation via the BLASTp algorithm, using the amino acid sequences of (SALTY)NAT1 or (GIBMO)NAT1 as query. When a NAT gene was found within the overlap** region of more than one protocluster, it was considered as part of all the protoclusters sharing this region. The accession numbers of BGC regions identified to harbour NAT genes were used to extract additional information regarding the experimental vs. computational characterization of the corresponding clusters through the MIBiG repository (https://mibig.secondarymetabolites.org/repository). MIBiG searches were also performed by selecting the MIBiG cluster comparison option in the newer antiSMASH versions (5.0–7.0) employed56.
Search for homology across genomic clusters with NAT genes
To assess homology between identified clusters with NAT genes, a custom database was first constructed using the cluster sequences in GenBank format. Searches were carried out with the MultiGeneBlast tool105, using the GenBank file of each gene cluster of interest as query. Based on the output of each individual search, a multi-sequence FASTA file was created, incorporating all the amino acid sequences encoded by genes found in homologous gene clusters. To visualize those results, this file was then used as query in SimpleSynteny version 1.4 software106 and the analysis was performed against a local database comprising the nucleotide sequence FASTA files of the corresponding gene clusters. To avoid redundancies, syntenic units demonstrating 100% conservation were grouped and represented by a single genomic sequence in graphical displays. All procedures were carried out with default program parameters.
Construction of phylogenetic trees and sequence similarity networks (SSNs)
For the construction of phylogenetic trees, a multiple protein sequence alignment was initially performed on ClustalW107. Phylogenetic trees were constructed with MEGAX108,109, using neighbor-joining110 or maximum likelihood111 methods with default parameters. The bootstrap replication number was set to 1000112. Common trees for microbial taxa were generated in PHYLIP format using the Common Taxonomy Tree tool of the NCBI (https://www.ncbi.nlm.nih.gov/Taxonomy/CommonTree/wwwcmt.cgi). Generated phylogenetic trees were visualized using the Interactive Tree of Life (iTOL) online resource (https://itol.embl.de/)113.
For the construction of SSNs, a FASTA file was created with all protein sequences of interest and an all-by-all BLAST analysis was executed using the EFI-enzyme similarity tool (EFI-EST; https://efi.igb.illinois.edu/efi-est/)114, setting the alignment score threshold (E-value) appropriately. The SSN was created by EFI-EST and visualized in Cytoscape115. In each SSN, the nodes represent individual proteins and the edges connect nodes when similarity is above the alignment score threshold set for the analysis.
Search for localization of NAT genes in bacterial plasmids
Sequenced bacterial plasmids were accessed via the PLSDB database in 2021 (https://ccb-microbe.cs.uni-saarland.de/plsdb/)48, using (SALTY)NAT1 amino acid sequence (GenBank ID: BAA14331.1) as query. Decreasing the High Scoring Pair (HSP) threshold value to as low as 40% retrieved the maximum number of non-redundant tBLASTn hits, which were then analysed and annotated as described above for other NAT homologues. Additional information was available through the PLSDB database, e.g., regarding surrounding genes on the same plasmid, the microbiological sample of origin, etc. The identified plasmid sequences were subsequently subjected to antiSMASH (version 6.0) search for BGCs, activating the MIBiG cluster comparison option. The specific features of plasmid BGCs with NAT genes were then recorded. The plasmids were further screened using IslandViewer version 4 (https://www.pathogenomics.sfu.ca/islandviewer/)116,117 for putative genomic islands, and those were inspected for the presence of NAT genes within them.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
Abbreviations
- 3,4-AHBA:
-
3-Amino-4-hydroxybenzoic acid
- 3,5-AHBA:
-
3-Amino-5-hydroxybenzoic acid
- antiSMASH:
-
Secondary metabolite analysis shell software
- BGC:
-
Biosynthetic gene cluster
- CoA:
-
Coenzyme A
- EFI-EST:
-
EFI-enzyme similarity tool
- HGT:
-
Horizontal gene transfer
- MIBiG:
-
Minimum information about a biosynthetic gene cluster
- NRPS:
-
Non-ribosomal peptide synthase
- ORF:
-
Open reading frame
- PKS:
-
Polyketide synthase
- SSN:
-
Sequence similarity network
References
van der Meer, J. R., de Vos, W. M., Harayama, S. & Zehnder, A. J. Molecular mechanisms of genetic adaptation to xenobiotic compounds. Microbiol. Rev. 56, 677–694 (1992).
Boukouvala, S. & Fakis, G. Arylamine N-acetyltransferases: What we learn from genes and genomes. Drug Metab. Rev. 37, 511–564 (2005).
Boukouvala, S. & Glenn, A. E. Arylamine N-acetyltransferases in eukaryotic microorganisms. In Arylamine N-acetyltransferases in Health and Disease (eds Laurieri, N. & Sim, E.) 255–281 (World Scientific, 2018). https://doi.org/10.1142/9789813232013_0010.
Garefalaki, V. et al. The actinobacterium Tsukamurella paurometabola has a functionally divergent arylamine N-acetyltransferase (NAT) homolog. World J. Microbiol. Biotechnol. 35, 174 (2019).
Garefalaki, V. et al. Comparative investigation of 15 xenobiotic-metabolizing N-acetyltransferase (NAT) homologs from bacteria. Appl. Environ. Microbiol. 87, e0081921 (2021).
Karagianni, E. P. et al. Homologues of xenobiotic metabolizing N-acetyltransferases in plant-associated fungi: Novel functions for an old enzyme family. Sci. Rep. 5, 12900 (2015).
Martins, M. et al. An acetyltransferase conferring tolerance to toxic aromatic amine chemicals: Molecular and functional studies. J. Biol. Chem. 284, 18726–18733 (2009).
Rodrigues-Lima, F. et al. Cloning, functional expression and characterization of Mesorhizobium loti arylamine N-acetyltransferases: Rhizobial symbiosis supplies leguminous plants with the xenobiotic N-acetylation pathway. Mol. Microbiol. 60, 505–512 (2006).
Ames, B. N., Gurney, E. G., Miller, J. A. & Bartsch, H. Carcinogens as frameshift mutagens: Metabolites and derivatives of 2-acetylaminofluorene and other aromatic amine carcinogens. Proc. Natl. Acad. Sci. U. S. A. 69, 3128–3132 (1972).
Watanabe, M., Sofuni, T. & Nohmi, T. Involvement of Cys69 residue in the catalytic mechanism of N-hydroxyarylamine O-acetyltransferase of Salmonella typhimurium. Sequence similarity at the amino acid level suggests a common catalytic mechanism of acetyltransferase for S. typhimurium and higher organisms. J. Biol. Chem. 267, 8429–8436 (1992).
Sinclair, J. C., Sandy, J., Delgoda, R., Sim, E. & Noble, M. E. Structure of arylamine N-acetyltransferase reveals a catalytic triad. Nat. Struct. Biol. 7, 560–564 (2000).
Stratmann, A. et al. Intermediates of rifamycin polyketide synthase produced by an Amycolatopsis mediterranei mutant with inactivated rifF gene. Microbiology 145(Pt 1), 3365–3375 (1999).
August, P. R. et al. Biosynthesis of the ansamycin antibiotic rifamycin: Deductions from the molecular analysis of the rif biosynthetic gene cluster of Amycolatopsis mediterranei S699. Chem. Biol. 5, 69–79 (1998).
Tyc, O., Song, C., Dickschat, J. S., Vos, M. & Garbeva, P. The ecological role of volatile and soluble secondary metabolites produced by soil bacteria. Trends Microbiol. 25, 280–292 (2017).
Keller, N. P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 17, 167–180 (2019).
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661 (2016).
Keller, N. P. Translating biosynthetic gene clusters into fungal armor and weaponry. Nat. Chem. Biol. 11, 671–677 (2015).
Glenn, A. E. et al. Two horizontally transferred xenobiotic resistance gene clusters associated with detoxification of benzoxazolinones by fusarium species. PLoS One 11, e0147486 (2016).
Jensen, P. R. Natural products and the gene cluster revolution. Trends Microbiol. 24, 968–977 (2016).
Anderton, M. C. et al. Characterization of the putative operon containing arylamine N-acetyltransferase (nat) in Mycobacterium bovis BCG. Mol. Microbiol. 59, 181–192 (2006).
Van der Geize, R. et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 104, 1947–1952 (2007).
Evangelopoulos, D. & Bhakta, S. Arylamine N-acetyltransferase in mycobacteria. In Arylamine N-acetyltransferases in Health and Disease (eds Laurieri, N. & Sim, E.) 303–324 (World Scientific, 2018). https://doi.org/10.1142/9789813232013_0012.
Glenn, A. E. & Bacon, C. W. FDB2 encodes a member of the arylamine N-acetyltransferase family and is necessary for biotransformation of benzoxazolinones by Fusarium verticillioides. J. Appl. Microbiol. 107, 657–671 (2009).
Hertweck, C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. Engl. 48, 4688–4716 (2009).
Walsh, C. T. & Fischbach, M. A. Natural products version 2.0: Connecting genes to molecules. J. Am. Chem. Soc. 132, 2469–2493 (2010).
Kawamura, A. et al. Eukaryotic arylamine N-acetyltransferase. Investigation of substrate specificity by high-throughput screening. Biochem. Pharmacol. 69, 347–359 (2005).
Lack, N. A. et al. Temperature stability of proteins essential for the intracellular survival of Mycobacterium tuberculosis. Biochem. J. 418, 369–378 (2009).
Tsirka, T. et al. Comparative analysis of xenobiotic metabolising N-acetyltransferases from ten non-human primates as in vitro models of human homologues. Sci. Rep. 8, 9759 (2018).
Karagianni, E.-P. et al. Fusarium verticillioides NAT1 (FDB2) N-malonyltransferase is structurally, functionally and phylogenetically distinct from its N-acetyltransferase (NAT) homologues. FEBS J. 290, 2412–2436 (2023).
Cronan, J. E. & Thomas, J. Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. Methods Enzymol. 459, 395–433 (2009).
Ziemert, N. et al. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. U. S. A. 111, E1130–E1139 (2014).
Vagena, E., Fakis, G. & Boukouvala, S. Arylamine N-acetyltransferases in prokaryotic and eukaryotic genomes: a survey of public databases. Curr. Drug Metab. 9, 628–660 (2008).
Glenn, A. E., Karagianni, E. P., Ulndreaj, A. & Boukouvala, S. Comparative genomic and phylogenetic investigation of the xenobiotic metabolizing arylamine N-acetyltransferase enzyme family. FEBS Lett. 584, 3158–3164 (2010).
Medema, M. H. et al. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339–W346 (2011).
Liu, G., Chater, K. F., Chandra, G., Niu, G. & Tan, H. Molecular regulation of antibiotic biosynthesis in streptomyces. Microbiol. Mol. Biol. Rev. 77, 112–143 (2013).
Bérdy, J. Bioactive microbial metabolites. J. Antibiot. (Tokyo) 58, 1–26 (2005).
Floss, H. G. & Yu, T. W. Lessons from the rifamycin biosynthetic gene cluster. Curr. Opin. Chem. Biol. 3, 592–597 (1999).
Kubiak, X. et al. Xenobiotic-metabolizing enzymes in Bacillus anthracis: Molecular and functional analysis of a truncated arylamine N-acetyltransferase isozyme. Br. J. Pharmacol. 174, 2174–2182 (2017).
Kubiak, X. et al. Structural and biochemical characterization of an active arylamine N-acetyltransferase possessing a non-canonical Cys-His-Glu catalytic triad. J. Biol. Chem. 288, 22493–22505 (2013).
Pluvinage, B. et al. Cloning and molecular characterization of three arylamine N-acetyltransferase genes from Bacillus anthracis: Identification of unusual enzymatic properties and their contribution to sulfamethoxazole resistance. Biochemistry 46, 7069–7078 (2007).
Mushtaq, A., Payton, M. & Sim, E. The COOH terminus of arylamine N-acetyltransferase from Salmonella typhimurium controls enzymic activity. J. Biol. Chem. 277, 12175–12181 (2002).
Sinclair, J. & Sim, E. A fragment consisting of the first 204 amino-terminal amino acids of human arylamine N-acetyltransferase one (NAT1) and the first transacetylation step of catalysis. Biochem. Pharmacol. 53, 11–16 (1997).
Helfrich, E. J. N., Lin, G.-M., Voigt, C. A. & Clardy, J. Bacterial terpene biosynthesis: Challenges and opportunities for pathway engineering. Beilstein J. Org. Chem. 15, 2889–2906 (2019).
Sim, E., Abuhammad, A. & Ryan, A. Arylamine N-acetyltransferases: from drug metabolism and pharmacogenetics to drug discovery. Br. J. Pharmacol. 171, 2705–2725 (2014).
Conway, L. P. et al. Unexpected acetylation of endogenous aliphatic amines by arylamine N-acetyltransferase NAT2. Angew. Chem. Int. Ed. Engl. 59, 14342–14346 (2020).
Nakazawa, T. et al. Overexpressing transcriptional regulator in Aspergillus oryzae activates a silent biosynthetic pathway to produce a novel polyketide. Chembiochem 13, 855–861 (2012).
Stanke, M. & Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467 (2005).
Galata, V., Fehlmann, T., Backes, C. & Keller, A. PLSDB: A resource of complete bacterial plasmids. Nucleic Acids Res. 47, D195–D202 (2019).
Frost, L. S., Leplae, R., Summers, A. O. & Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732 (2005).
Ruiz, B. et al. Production of microbial secondary metabolites: Regulation by the carbon source. Crit. Rev. Microbiol. 36, 146–167 (2010).
Top, E. M. & Springael, D. The role of mobile genetic elements in bacterial adaptation to xenobiotic organic compounds. Curr. Opin. Biotechnol. 14, 262–269 (2003).
Sohng, J. K., Oh, T. J., Lee, J. J. & Kim, C. G. Identification of a gene cluster of biosynthetic genes of rubradirin substructures in S. achromogenes var. rubradiris NRRL3061. Mol. Cells 7, 674–681 (1997).
Robinson, L. J., Verrett, J. N., Sorout, N. & Stavrinides, J. A broad-spectrum antibacterial natural product from the cystic fibrosis isolate, Pantoea agglomerans Tx10. Microbiol. Res. 237, 126479 (2020).
Schupp, T., Toupet, C., Engel, N. & Goff, S. Cloning and sequence analysis of the putative rifamycin polyketide synthase gene cluster from Amycolatopsis mediterranei. FEMS Microbiol. Lett. 159, 201–207 (1998).
Pompeo, F., Mushtaq, A. & Sim, E. Expression and purification of the rifamycin amide synthase, RifF, an enzyme homologous to the prokaryotic arylamine N-acetyltransferases. Protein Expr. Purif. 24, 138–151 (2002).
Terlouw, B. R. et al. MIBiG 3.0: A community-driven effort to annotate experimentally validated biosynthetic gene clusters. Nucleic Acids Res. 51, D603–D610 (2023).
Kim, T. K., Hewavitharana, A. K., Shaw, P. N. & Fuerst, J. A. Discovery of a new source of rifamycin antibiotics in marine sponge actinobacteria by phylogenetic prediction. Appl. Environ. Microbiol. 72, 2118–2125 (2006).
Wilson, M. C., Gulder, T. A. M., Mahmud, T. & Moore, B. S. Shared biosynthesis of the saliniketals and rifamycins in Salinispora arenicola is controlled by the sare1259-encoded cytochrome P450. J. Am. Chem. Soc. 132, 12757–12765 (2010).
Floss, H. G., Yu, T.-W. & Arakawa, K. The biosynthesis of 3-amino-5-hydroxybenzoic acid (AHBA), the precursor of mC7N units in ansamycin and mitomycin antibiotics: A review. J. Antibiot. (Tokyo) 64, 35–44 (2011).
Kang, Q., Shen, Y. & Bai, L. Biosynthesis of 3,5-AHBA-derived natural products. Nat. Prod. Rep. 29, 243–263 (2012).
Westwood, I. M. & Sim, E. Kinetic characterisation of arylamine N-acetyltransferase from Pseudomonas aeruginosa. BMC Biochem. 8, 3 (2007).
Eichner, S. et al. Broad substrate specificity of the amide synthase in S. hygroscopicus—new 20-membered macrolactones derived from geldanamycin. J. Am. Chem. Soc. 134, 1673–1679 (2012).
Yu, T. W. et al. Direct evidence that the rifamycin polyketide synthase assembles polyketide chains processively. Proc. Natl. Acad. Sci. U. S. A. 96, 9051–9056 (1999).
Wu, Y., Kang, Q., Shen, Y., Su, W. & Bai, L. Cloning and functional analysis of the naphthomycin biosynthetic gene cluster in Streptomyces sp. CS Mol. Biosyst. 7, 2459–2469 (2011).
Xu, Z. et al. Biosynthetic code for divergolide assembly in a bacterial mangrove endophyte. Chembiochem 15, 1274–1279 (2014).
Li, S. et al. Biosynthesis of hygrocins, antitumor naphthoquinone ansamycins produced by Streptomyces sp. LZ35. Chembiochem 15, 94–102 (2014).
Castro, J. F. et al. Identification and heterologous expression of the chaxamycin biosynthesis gene cluster from Streptomyces leeuwenhoekii. Appl. Environ. Microbiol. 81, 5820–5831 (2015).
**ao, Y. S. et al. Rifamorpholines A-E, potential antibiotics from locust-associated actinobacteria Amycolatopsis sp. Hca4. Org. Biomol. Chem. 15, 3909–3916 (2017).
Liu, Y. et al. Functional analysis of cytochrome P450s involved in Streptovaricin biosynthesis and generation of anti-MRSA analogues. ACS Chem. Biol. 12, 2589–2597 (2017).
Peek, J. et al. Rifamycin congeners kanglemycins are active against rifampicin-resistant bacteria via a distinct mechanism. Nat. Commun. 9, 4147 (2018).
Kim, C.-G. et al. Biosynthesis of rubradirin as an ansamycin antibiotic from Streptomyces achromogenes var. rubradiris NRRL3061. Arch. Microbiol. 189, 463–473 (2008).
Yu, T.-W. et al. The biosynthetic gene cluster of the maytansinoid antitumor agent ansamitocin from Actinosynnema pretiosum. Proc. Natl. Acad. Sci. U. S. A. 99, 7968–7973 (2002).
Ning, X., Wang, X., Wu, Y., Kang, Q. & Bai, L. Identification and engineering of post-PKS modification bottlenecks for ansamitocin P-3 titer improvement in Actinosynnema pretiosum subsp. pretiosum ATCC 31280. Biotechnol. J. 12, 1700484 (2017).
Zhang, M.-Q. et al. Optimizing natural products by biosynthetic engineering: Discovery of nonquinone Hsp90 inhibitors. J. Med. Chem. 51, 5494–5497 (2008).
Rascher, A. et al. Cloning and characterization of a gene cluster for geldanamycin production in Streptomyces hygroscopicus NRRL 3602. FEMS Microbiol. Lett. 218, 223–230 (2003).
Shin, J.-C. et al. Characterization of tailoring genes involved in the modification of geldanamycin polyketide in Streptomyces hygroscopicus JCM4427. J. Microbiol. Biotechnol. 18, 1101–1108 (2008).
He, W., Lei, J., Liu, Y. & Wang, Y. The LuxR family members GdmRI and GdmRII are positive regulators of geldanamycin biosynthesis in Streptomyces hygroscopicus 17997. Arch. Microbiol. 189, 501–510 (2008).
Wang, J., Li, W., Wang, H. & Lu, C. Pentaketide ansamycin microansamycins A-I from Micromonospora sp. reveal diverse post-PKS modifications. Org. Lett. 20, 1058–1061 (2018).
Li, X., Wu, X. & Shen, Y. Identification of the bacterial maytansinoid gene cluster asc provides insights into the post-PKS modifications of ansacarbamitocin biosynthesis. Org. Lett. 21, 5823–5826 (2019).
Rui, Z. et al. Biochemical and genetic insights into asukamycin biosynthesis. J. Biol. Chem. 285, 24915–24924 (2010).
Petříčková, K. et al. Biosynthesis of colabomycin E, a new manumycin-family metabolite, involves an unusual chain-length factor. Chembiochem 15, 1334–1345 (2014).
Dong, L.-B. et al. Biosynthesis of thiocarboxylic acid-containing natural products. Nat. Commun. 9, 2362 (2018).
Smanski, M. J. et al. Dedicated ent-kaurene and ent-atiserene synthases for platensimycin and platencin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 108, 13498–13503 (2011).
Zheng, C.-J. et al. PtmC catalyzes the final step of thioplatensimycin, thioplatencin, and thioplatensilin biosynthesis and expands the scope of arylamine N-acetyltransferases. ACS Chem. Biol. 16, 96–105 (2021).
Rodríguez Estévez, M., Myronovskyi, M., Gummerlich, N., Nadmid, S. & Luzhetskyy, A. Heterologous expression of the nybomycin gene cluster from the marine strain Streptomyces albus subsp. chlorinus NRRL B-24108. Mar. Drugs 16, 435 (2018).
Braesel, J., Lee, J.-H., Arnould, B., Murphy, B. T. & Eustáquio, A. S. Diazaquinomycin biosynthetic gene clusters from marine and freshwater actinomycetes. J. Nat. Prod. 82, 937–946 (2019).
Wu, X. et al. A comparative analysis of the sugar phosphate cyclase superfamily involved in primary and secondary metabolism. Chembiochem 8, 239–248 (2007).
Wu, X., Flatt, P. M., Xu, H. & Mahmud, T. Biosynthetic gene cluster of cetoniacytone A, an unusual aminocyclitol from the endosymbiotic bacterium Actinomyces sp. Lu 9419. Chembiochem 10, 304–314 (2009).
Wang, X. et al. Bioinformatics-guided connection of a biosynthetic gene cluster to the antitumor antibiotic gilvusmycin. Acta Biochim. Biophys. Sin. (Shanghai) 50, 516–518 (2018).
Sandmann, A., Sasse, F. & Müller, R. Identification and analysis of the core biosynthetic machinery of tubulysin, a potent cytotoxin with potential anticancer activity. Chem. Biol. 11, 1071–1079 (2004).
Tsutsumi, H. et al. Unprecedented cyclization catalyzed by a cytochrome P450 in benzastatin biosynthesis. J. Am. Chem. Soc. 140, 6631–6639 (2018).
Gould, S. J., Hong, S. T. & Carney, J. R. Cloning and heterologous expression of genes from the kinamycin biosynthetic pathway of Streptomyces murayamaensis. J. Antibiot. (Tokyo) 51, 50–57 (1998).
Gao, G. et al. Formation of an angular aromatic polyketide from a linear anthrene precursor via oxidative rearrangement. Cell Chem. Biol. 24, 881-891.e4 (2017).
Suzuki, H., Ohnishi, Y. & Horinouchi, S. Arylamine N-acetyltransferase responsible for acetylation of 2-aminophenols in Streptomyces griseus. J. Bacteriol. 189, 2155–2159 (2007).
Suzuki, H., Furusho, Y., Higashi, T., Ohnishi, Y. & Horinouchi, S. A novel o-aminophenol oxidase responsible for formation of the phenoxazinone chromophore of grixazone. J. Biol. Chem. 281, 824–833 (2006).
Hein, D. W., Boukouvala, S., Grant, D. M., Minchin, R. F. & Sim, E. Changes in consensus arylamine N-acetyltransferase gene nomenclature. Pharmacogenet. Genom. 18, 367–368 (2008).
Laurieri, N. & Sim, E. Arylamine N-Acetyltransferases in Health and Disease (World Scientific, 2018). https://doi.org/10.1142/10763.
Boukouvala, S. Arylamine N-acetyltransferase nomenclature. In Arylamine N-acetyltransferases in Health and Disease (eds Laurieri, N. & Sim, E.) (World Scientific, 2018). https://doi.org/10.1142/9789813232013_0016.
Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41, 95–98 (1999).
Okonechnikov, K., Golosova, O. & Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 28, 1166–1167 (2012).
Weber, T. et al. antiSMASH 3.0: A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243 (2015).
Blin, K. et al. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87 (2019).
Blin, K. et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 51, W46–W50 (2023).
Kautsar, S. A. et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 48, D454–D458 (2020).
Medema, M. H., Takano, E. & Breitling, R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol. Biol. Evol. 30, 1218–1223 (2013).
Veltri, D., Wight, M. M. & Crouch, J. A. SimpleSynteny: A web-based tool for visualization of microsynteny across multiple species. Nucleic Acids Res. 44, W41–W45 (2016).
Thompson, J. D., Gibson, T. J. & Higgins, D. G. Multiple sequence alignment using ClustalW and ClustalX. In Current Protocols in Bioinformatics, Chapter 2, Unit 2.3 (2002).
Stecher, G., Tamura, K. & Kumar, S. Molecular evolutionary genetics analysis (MEGA) for macOS. Mol. Biol. Evol. 37, 1237–1239 (2020).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Saitou, N. & Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987).
Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 17, 368–376 (1981).
Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39, 783–791 (1985).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz239 (2019).
Zallot, R., Oberg, N. & Gerlt, J. A. The EFI web resource for genomic enzymology tools: Leveraging protein, genome, and metagenome databases to discover novel enzymes and metabolic pathways. Biochemistry 58, 4169–4182 (2019).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Langille, M. G. I. & Brinkman, F. S. L. IslandViewer: An integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25, 664–665 (2009).
Bertelli, C. et al. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35 (2017).
Acknowledgements
The research project was partly supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “2nd Call for H.F.R.I. Research Projects to support Faculty Members & Researchers” (Project Number: 3712), providing funding to work performed by D.B., D.T., S.Z. and S.B. For her part of work, E.K. was recipient of a Ph.D. scholarship (2016-2019) co-financed by Greece and the European Union (European Social Fund-ESF) through Operational Program “Human Resources Development, Education and Lifelong Learning” in the context of project “Strengthening Human Resources Research Potential via Doctorate Research” (MIS-5000432), implemented by the State Scholarships Foundation (ΙΚΥ). We thank former students Marina Avramidou, Athina Eleftheraki, Christina Vagena-Pantoula, Maria-Giusy Papavergi, Charalampos Ioannidis and Vasiliki Garefalaki for assistance.
Author information
Authors and Affiliations
Contributions
S.B. conceptualized the study, supervised the team and wrote the manuscript with help from E.K. and other co-authors. E.K., I.O., D.P., D.T., K.A., A.M., M.A.T., D.B. and S.Z. implemented various aspects of the research with equal contributions, and they are featured in the chronological order of their participation in the project. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boukouvala, S., Kontomina, E., Olbasalis, I. et al. Insights into the genomic and functional divergence of NAT gene family to serve microbial secondary metabolism. Sci Rep 14, 14905 (2024). https://doi.org/10.1038/s41598-024-65342-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-65342-4
- Springer Nature Limited