
Abstract
A new structure of mesoporous spherical nanocomposites was designed and easily prepared from the reaction between NiCuFe2O4 nanoparticles and mesoporous silica in three steps. The prepared multi-yolk@shell NiCuFe2O4@mSiO2 mesoporous sphere was characterized by using FT-IR, XRD, VSM, EDX, BET, FE-SEM and HR-TEM techniques. This unique mesoporous nanocomposite sphere as a heterogeneous nanocatalyst has demonstrated highly catalytic activity for the green synthesis of tetrahydronaphthalene derivatives in 92–98% yields at reaction times of 60–75 min. This process was carried out through multi-component reaction of the cyclic ketone, malononitrile and aromatic aldehyde under solvent-free conditions. Furthermore, the procedure was optimized on the basis of catalyst loading amounts, various solvents and temperature conditions. This novel methodology exposes obvious benefits such as; catalyst reusability, easy reaction procedure, simplicity of work-up, excellent product yields and short reaction times.
Similar content being viewed by others


Introduction
Recently, nanotechnology and nanoscale materials, especially magnetic nanoparticles (MNPs) have attracted a lot of attention and shown many applications in research and industrial fields in the chemical, medical and environmental sectors. For example, the application of magnetic nanoparticles in removal of pollutants or reducing toxicity is promising1. The properties of MNPs can be related to the physical characteristics of nanoparticles such as; shape, chemical composition, size, crystal structure morphology and surface area effects. These MNPs are usually very different from the properties of bulk materials2 and coated with a shell to show the core–shell nanoparticles (CSNPs)3,4,5. The core–shell nanoparticles are often created by the formation of a dense layer surrounding center. The properties of CSNPs are mostly determined by the type of core and the contained shell, as well as the sizes of both core and shell and the combination of both. Although CSNPs offer several improved features and advantages over traditional MNPs such as; narrow size distribution, higher chemical stability and magnetic moment can be tuned with the nanoparticle cluster size6. However, these particles still have some limitations such as; a dense shell coating that restricts the native surface of MNPs and the accessibility of reagents to the catalytic active sites of MNPs7. Therefore, to remove these limitations, a type of core–shell nanoparticles was considered.
Yolk–shell nanoparticles (YSNPs) are a novel class of unique core–shell nanoparticles that are commonly referred to as hollow shells with a void area between the solid particle core and shell, allowing the core to move freely within the shell8,9. Because of their unique optical, chemical, mechanical, and electrical properties, the YSNPs have recently received a number of attention from researchers, and they have a lot of potential in catalysis10,11,12, lithium-ion batteries13,14,15, biosensors16,17, bioimaging18,19,20, surface-enhanced Raman scattering21,22,23, and biomedicine applications24,25,26.
Multicomponent reactions (MCRs) are one-pot reactions using more than two substrate materials, where most of the atoms of the starting materials are incorporated in the final product. These reactions are economical and environmentally friendly methods for organic synthesis27,28.
One of the multicomponent reactions is the synthesis of tetrahydronaphtalenes. Tetralin or tetrahydronaphthalene is obtained from the hydrogenation of naphthalene29. This compound is a colorless liquid with a chemical formula, C10H12 that is used as a hydrogenation solvent30. Its laboratory synthesis was first prepared by Darnz. The ortho-aminocarbonitriles with two chiral centers that exhibit optical activity are also crucial intermediates in the synthesis of a number of heterocyclic compounds. Several interesting organic chemistry works related to the ortho-aminocarbonitrile tetrahydronaphthalene derivatives has led to the development of numerous techniques in recent years31.
Recently years, the different catalysts are used for synthesis of tetralin compound such as; Fe3O4@SiO2@mSiO2-Pd32, Pd/S-1@ZSM-5 core–shell33, CaMg@ mesoporous silica yolk shell34, CaMgFe2O4 magnetic nanocatalyst35 and NaOH catalyst36. However, these catalysts have disadvantages such as; long reaction times, low reaction yields and high loading catalyst. While, in this work, by using the prepared nanocatalyst, the tetrahydronaphthalene derivatives are easily produced in excellent yields, low catalyst amounts and low reaction times.
In continuation of ongoing our team work on the basis of the catalytic reactions37,38, the purpose of this research was to create a new catalyst with high activity through simple procedure. Therefore, the multi-yolk@shell NiCuFe2O4@mSiO2 sphere was fabricated and used for multi-component reaction of cyclic ketone, malononitrile and aromatic aldehyde under solvent-free conditions. In this methodology, the tetrahydronaphthalenes were purely obtained in highly efficiency and short reaction times.
Experimental
Materials and apparatus
All chemicals and reagents, namely calcium nitrate trihydrate (99%), nickel nitrate hexahydrate (99%), iron nitrate nonanehydrate (99%), tetraethyl orthosilicate, glucose, barbituric acid, 1-naphthylamine and the respective benzaldehyde derivatives, were purchased from Merck Chemical Company. 1H NMR spectra were taken with 400 MHz NMR spectrometer for solution NMR analysis. FT-IR spectra were obtained with Shimadzu’s instrument. Electrothermal programmable digital melting point apparatus was used to measure the melting points. BGMN/Profex/AutoQuan software reported the results of the X-ray diffraction studies. The nanoparticles were examined using a field emission scanning electron microscope (FE-SEM), on the Zeiss that run at an accelerating voltage of 15 kV. The magnetic properties of nanostructure was measured by a vibrating sample magnetometer (VSM) with PPMS-9T at 300 K. The Brunauer–Emmett–Teller (BET) specific area and pore volume was measured via the nitrogen adsorption–desorption isotherms method using BELSORP MINI II. The high resolution transmission electron microscopy (HR-TEM) was prepared by a FEI TECNAI F20 at 200 kV instrument.
General procedure for preparation of NiCuFe2O4@mSiO2
In accordance to the previously reported procedure39, in the first step, to a glucose (4.95 mg) dissolved in 25 mL of distilled water, the Fe(NO3)3.9H2O (0.8 mg), Ni(NO3)2⋅6H2O (0.14 mg), and Cu(NO3)2⋅3H2O (0.12 mg) salts were dispersed in 10 mL distilled water. After being stirred for about 30 min, the mixture was transferred into a Teflon-lined stainless steel autoclave. The autoclave was maintained at 180 °C for 24 h and then naturally cooled to ambient temperature. The precipitate was filtered, washed with water and ethanol, then dried at 100 °C and NiCuFe2O4@C was obtained. In the second step, NiCuFe2O4@C (0.1 g), CTAB (0.15 g), and NH3⋅H2O (580 µL) were dispersed into the mixture of 40 mL H2O and ethanol on the magnetic stirring for 30 min. Then, 150 µL TEOS added to mixture reaction and the reaction maintained for 6 h at room temperature. The black solid was obtained, washed with water and ethanol; then dried in the oven at 70 °C and then, calcination for 3 h under air atmosphere at 600 °C. The final, multi yolk@shell NiCuFe2O4@mSiO2 sphere was made.
A general procedure for the tetrahydronaphthalene derivatives
In this section, cyclohexanon derivatives (1 mmol), malononitrile (2 mmol), aromatic aldehyde (1 mmol), and 1.0 mg NiCuFe2O4@mSiO2 as a catalyst are mixed under stirring at 50 °C. The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion of the reaction, the ethanol (3 mL) was added and the catalyst was separated by an external magnet. The reaction mixture filtered and washed with ethanol (3 mL), then the pure products are obtained.
2-Amino-4-phenyl-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbonitrile (4a)
White solid; m.p.: 255–257 °C (Lit.40 m.p 255–258 °C); IR (KBr) ʋ = 3419, 3339, 2933, 2856, 2210, 1648, 1451, 1276, 1037, 784, 580 cm−1; 1H NMR (400 MHz, DMSO-d6): δ = 7.37–7.60 (m, 7H, NH2, CH), 5.71 (s, 1H), 3.52 (d, J = 12.0 Hz, 1H), 2.76–2.83 (m, 1H), 2.00–2.20 (m, 2H), 1.64 (t, J = 12.0 Hz, 1H), 1.39–1.48 (m, 2H), 0.79–0.89 (m, 1H).
2-Amino-4-(4-bromophenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbonitrile (4b)
White solid; m.p.: 244–246 °C (Lit.40 m.p 243–245 °C); IR (KBr) ʋ = 3428, 3346, 2946, 2210, 1642, 1522, 1348, 1276, 1113, 807 cm−1; 1H NMR (400 MHz, DMSO-d6): δ = 8.32 (t, J = 8.0 Hz, 2H), 7.91 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.43 (s, 2H, NH2), 5.74 (s, 1H), 3.87 (d, J = 12.0 Hz, 1H), 2.84–2.91 (m, 1H), 2.02–2.20 (m, 2H), 1.67 (d, J = 12.0 Hz, 1H), 1.39–1.48 (m, 2H), 0.81–0.90 (m, 1H).
2-Amino-4-(4-nitrophenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbonitrile (4c)
Yellow solid; m.p.: 264–266 °C (Lit.40 m.p 263–266 °C); IR (KBr) ʋ = 3419, 3341, 2947, 2856, 2215, 1646, 1604, 1455, 1256, 1025, 833, 570 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 8.33 (t, J = 12.0 Hz, 2H), 7.91 (d, J = 8.0 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.43 (s, 2H, NH2), 5.74 (s, 1H), 3.90 (d, J = 12.0 Hz, 1H), 2.84–2.91 (m, 1H), 2.00–2.21 (m, 2H), 1.66–1.70 (m, 1H), 1.39–1.48 (m, 2H), 0.81–0.90 (m, 1H).
2-Amino-4-(4-(dimethylamino)phenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbo-nitrile (4d)
Yellow solid; m.p.: 263–265 °C (Lit.40 m.p 264–265 °C); IR (KBr) ʋ = 3427, 3341, 2921, 2203, 1614, 1518, 1379, 1184, 814 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 7.17–7.34 (m, 4H, NH2, CH), 6.60–6.80 (m, 2H), 5.08 (s, 1H), 3.31 (d, J = 12.0 Hz, 1H), 2.92 (s, 6H, OCH3), 2.66–2.75 (m, 1H), 2.49 (s, H), 2.00–2.19 (m, H), 1.64–1.70 (m, 1H), 1.41–1.53 (m, 2H), 0.77–0.86 (m, 1H).
2-Amino-4-(4-fluorophenyl)-4a, 5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbonitrile (4e)
White solid; m.p.: 262–264 °C (Lit.40 m.p 263–267 °C); IR (KBr) ʋ = 3418, 3378, 2951, 2869, 2211, 1621, 1511, 1449, 1390, 1231, 1163, 844 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 7.63 (s, H), 7.48 (s, H), 7.21–7.37 (m, 4H, NH2, CH), 5.71 (s, 1H), 3.62 (d, J = 12.0 Hz, 1H), 2.75–2.82 (m, 1H), 2.05–2.20 (m, 2H), 1.66–1.76 (m, 1H), 1.40–1.45 (m, 2H), 0.82–0.88 (m, 1H) ppm.
2-Amino-4-(4-chlorophenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbonitrile (4f)
White solid; m.p.: 248–250 °C (Lit.40 m.p 246–249 °C); IR (KBr) ʋ = 3443, 3339, 3033, 2923, 2225, 1643, 1583, 1289, 1093, 1007, 825, 748, 501 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 7.46–7.61 (m, 4H), 7.38 (s, 2H, NH2), 5.71 (s, 1H), 3.63 (d, J = 12.0 Hz, 1H), 2.75–2.83 (m, 1H), 2.01–2.20 (m, 2H), 1.65–1.69 (m, 1H), 1.42–1.46 (m, 2H), 0.79–0.89 (m, 1H) ppm.
2-Amino-4-(4-methoxyphenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbonitrile (4g)
White solid; m.p.: 261–262 °C (Lit.40 m.p 258–261 °C); IR (KBr) ʋ = 3421, 3334, 2953, 2213, 1649, 1600, 1451, 1390, 1160, 1037, 706, 594 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 6.95–750 (m, 6H, NH2, CH), 5.70 (s, 1H), 3.78 (s, 3H, OCH3), 3.46 (d, J = 12.0 Hz, 1H), 2.70–2.78 (m, 1H), 2.00–2.19 (m, 2H), 1.66–1.70 (m, 1H), 1.43–1.49 (m, 2H), 0.77–0.87 (m, 1H) ppm.
2-Amino-4-(4-methylphenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbonitrile (4h)
White solid; m.p.: 235–237 °C (Lit.41 m.p 234–236 °C); IR (KBr) ʋ = 3437, 3341, 2956, 2210, 1645, 1599, 1425, 1264, 1044, 818, 513 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 7.17–7.47 (m, 6H, NH2, CH), 5.70 (s, 1H), 3.47 (d, J = 12.0 Hz, 1H), 2.73–2.79 (m, 1H), 2.49 (s, 3H, CH3), 2.02–2.19 (m, 2H), 1.64–1.68 (m, 1H), 1.41–1.49 (m, 2H), 0.77–0.87 (m, 1H) ppm.
2-Amino-4-(3-bromophenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbonitrile (4i)
White solid; m.p.: 250–253 °C (Lit.40 m.p 248–253 °C); IR (KBr) ʋ = 3417, 3339, 2940, 2859, 2210, 1650, 1568, 1476, 1339, 1212, 1076, 887, 796, 688, 578 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 7.39–7.77 (m, 6H, NH2, CH), 5.72 (s, 1H), 3.64 (d, J = 12.0 Hz, 1H), 2.78–2.86 (m, 1H), 2.00–2.20 (m, 2H), 1.66–1.72 (m, 1H), 1.43–1.53 (m, 2H), 0.82–0.92 (m, 1H) ppm.
2-Amino-5-methyl-4-phenyl-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbonitrile (4j)
White solid; m.p.: 260–262 °C; IR (KBr) ʋ = 3417, 3357, 2940, 2859, 2210, 1650, 1568, 1476, 1393, 1278, 1076, 578 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 7.37–7.60 (m, 7H, NH2, CH), 5.71(s, 1H), 3.53 (d, J = 8.0 Hz, 1H), 2.82–2.88 (m, 1H), 1.65–1.74 (m, H), 1.30–1.40 (m, 1H), 0.89–0.94 (m, H), 0.81 (d, J = 8.0 Hz, 3H, CH3), 0.60–0.66 (m, 1H) ppm.
2-Amino-5-methyl-4-(4-bromophenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbo-nitrile (4k)
White solid; m.p.: 253–257 °C; IR (KBr) ʋ = 3442, 3345, 2946, 2859, 2209, 1644, 1600, 1488, 1391, 1268, 1074, 1009, 563 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 7.58–7.67 (m, 2H), 7.45–748 (m, 1H), 7.16–7.28 (m, 1H), 6.05(s, 1H), 4.84 (s, 2H, NH2), 3.07 (d, J = 12.0 Hz, 1H), 2.87–2.97 (m, 1H), 2.33–2.40 (m, 1H), 1.71–1.81 (m, 2H), 1.52–1.60 (m, 1H), 0.92 (d, J = 8.0 Hz, 3H), 0.68–0.75 (m, 1H) ppm.
2-Amino-5-methyl-4-(4-hydroxyphenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbo nitrile (4l)
White solid; m.p.: 256–257 °C; IR (KBr) ʋ = 3443, 3344, 2947, 2866, 2209, 1646, 1518, 1449, 1384, 1271, 1170, 831, 575 cm−1, 1H NMR (400 MHz, DMSO-d6): δ = 9.66 (s, H, OH), 7.17–7.39 (m, 4H, NH2, CH), 6.76–6.87 (m, 2H), 5.68 (s, 1H), 3.39 (s, 1H), 2.76–2.83 (m, 1H), 2.21–2.28 (m, 1H), 1.67 (d, J = 12.0 Hz, 2H), 1.38–1.45 (m, 1H), 0.81 (d, J = 2.0 Hz, 3H), 0.59–0.64 (m, 1H) ppm.
2-Amino-5-methyl-4-(4-chlorophenyl)-4a,5,6,7-tetrahydronaphthalene-1,3,3(4H)-tricarbo-nitrile (4m)
White solid; m.p.: 236–240 °C; IR (KBr) ʋ = 3423, 3345, 2947, 2856, 2211, 1643, 1493, 1452, 1391, 1268, 1093, 825, 511 cm-1; 1H NMR (400 MHz, DMSO-d6): δ = 7.27–7.53 (m, 4H), 6.06 (s, 1H), 4.93 (s, 2H, NH2), 2.90–2.96 (m, 1H), 2.33–2.41 (m, 1H), 2.19 (s, 1H), 1.73–1.80 (m, 1H), 1.53–1.61 (m, 1H), 0.91–0.93 (d, J = 8.0 Hz, 3H), 0.72–0.79 (m, 1H) ppm.
Results and discussions
Preparation and characterization of catalyst
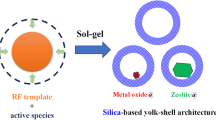
The multi-yolk@shell NiCuFe2O4@mSiO2 has been designed and easily prepared in a triple step procedure in accordance to the literature42 that completely described in “General procedure for preparation of NiCuFe2O4@mSiO2” section and shown in Fig. 1.

Preparation of multi yolk@shell NiCuFe2O4@mSiO2 spheres.
The prepared catalyst was characterized by FT-IR, XRD, FE-SEM, EDX, HR-TEM, VSM and BET techniques. FT-IR spectra of the created samples in the region of 400 to 4000 cm−1 are shown in Fig. 2. This Figure shows the FT-IR spectra of hollow catalyst before and after the calcination. Figure 2a is shown the spectrum of NiCuFe2O4@C@SiO2 catalyst that was appropriated before calcination. The peaks of 3422 and 1619 cm−1 are shown the stretching vibration of the O–H bond and H–O–H bending vibration. The peaks of the 2922, and 2853 cm−1 can be appropriated to the stretching vibration of the C–H bond. The peaks of 1473 and 1382 cm−1 identify the bending vibration of the C–H bond and the stretching vibration of the C–O bond which shown the template carbon is formed and the CTAB is in the NiCuFe2O4@C@SiO2 catalyst. The peaks that appeared in 460 and 794 cm−1 can be related to the Ni–O, Cu–O, and Fe–O bonds. The stretching vibration of the Si–O bond is appeared in 1079 cm-1. Figure 2b is shown the spectrum of NiCuFe2O4@C@SiO2 catalyst after calcination. In this step, the absence of the peaks at 2922, 2853, 1473 and 1382 cm−1 indicated that the calcination is completely done, the catalyst voided and the NiCuFe2O4@mSiO2 is formed regularly.

FT-IR spectra of the multi-yolk@shell NiCuFe2O4 spheres, (a) before, and (b) after calcination.
The XRD pattern of multi-yolk@shell NiCuFe2O4 is shown in Fig. 3 which the related peaks are shown. Figure 3a shows the catalyst before calcination that it is irregularly indicated. The presence of the peaks at 2Ɵ = 20.2, 33, and 36.6 in Fig. 3b can be appropriated to the catalyst before calcination that shows the catalyst is regularly made. Figure 3c shows the XRD low angle of catalyst and the presence of the sharp peak at 2Ɵ indicates that multi-yolk@shell NiCuFe2O4 mesoporous silica catalyst is made.

XRD pattern of the NiCuFe2O4@mSiO2 spheres (a) before, (b) after calcination and (c) XRD low angle of the NiCuFe2O4@mSiO2 spheres.
Figure 4 shows the field emission scanning electron microscopy (FE-SEM) images for hollow multi-shell NiCuFe2O4@mSiO2 spheres catalyst. The FE-SEM images (Fig. 4) is indicated that the catalyst is made up of uniform, spheres, and hollow.

FE-SEM images of the multi-yolk@shell NiCuFe2O4@mSiO2 nanospheres.
Furthermore, Fig. 5 is demonstrated the EDX analysis of the hollow multi shell NiCuFe2O4@mSiO2 spheres. This result is indicated that the nanospheres included the Ni, Cu, O, Si, and Fe elements. This finding is quite following the FT-IR results.

EDX spectrum of the multi-yolk@shell NiCuFe2O4@mSiO2 spheres.
Also, the results of elemental map** analysis is indicated the Fe, Cu, Ni, O, and Si elements (Fig. 6). This result is quite following the EDX analysis.

Elemental map** analysis: for (a) Fe, (b) Cu, (c) Ni, (d) O, (e) Si.
The high resolution TEM (HR-TEM) of the multi-yolk@shell NiCuFe2O4@mSiO2 spheres indicates the structure of the catalyst in the Fig. 7. These images show that the catalyst is made in a sphere uniform, multi-shell, yolk–shell and is multi-metal.

High resolution TEM (HR-TEM) of the multi-yolk@shell NiCuFe2O4@mSiO2.
A vibrating sample magnetometer (VSM) has been used to explore the magnetic properties of various materials for NiCuFe2O4@mSiO2 spheres magnetization pattern are shown in Fig. 8. The multi-yolk@shell NiCuFe2O4@mSiO2 spheres have a 28.1 emu per g magnetic valence.

VSM analysis of the multi-yolk@shell NiCuFe2O4@mSiO2 spheres.
The nitrogen adsorption–desorption isotherm of the hollow multi-shell NiCuFe2O4@mSiO2 and corresponding BJH pore size distribution is shown in Fig. 9. The BJH is shown an average pore size of 4.6 nm which indicated the catalyst is mesoporous (Fig. 9b). Due to the mesoporous silica, the available surface area is 180.51 m2/g that higher than the catalyst reported without mesoporous silica (Fig. 9a)42.

The nitrogen adsorption–desorption isotherm (a) and corresponding BJH pore size (b) of multi-yolk@shell NiCuF2O4@mSiO2.
Investigation of catalytic activity
In order to assess its ability as just a catalyst for the synthesis of tetrahydronaphtalene, multi-yolk@shell NiCuF2O4@mSiO2 was investigated (Table 1). The reaction of cyclohexanone, aromatic aldehyde and the 4-Br-benzaldehyde was selected as a model reaction. So, some studies of the influence of heat, suitability of solvent, and nanocatalyst amounts loading on the reaction speed was conducted (Table 1). This Table shows the low product yields when using the ethanol, water, ethanol–water and, methanol as solvent (Table 1, entries 1–4). In this research, the best conditions for the model reaction at 50 °C to create the tetrahydronaphtalene derivative are reported to be solvent-free (Table 1, entry 5).
Several kinds of catalysts for this reaction were examined once the reaction parameters were optimized (Table 2). When the present catalyst was compared with the other catalysts, such as; NiFe2O4, HCl and H2SO4, it was found that the reaction in the presence of multi-yolk@shell NiCuFe2O4@mSiO2 spheres as a catalyst, obtained the highest product yield. As a result, the multi-yolk@shell NiCuFe2O4@mSiO2 spheres are preferable to other catalysts because of short reaction time, low catalyst loading and highest product yield.
Following the adjustment of the reaction conditions including temperature, catalyst loading, and different solvents, the development and aim of our methodology for the production of various tetrahydronaphtalenes were carried out. This study looked at how different benzaldehydes reacted with cyclohexanone derivatives, malononitrile, and the corresponding results are summarized in Table 3. As can be seen in this Table, the malononitrile effectively reacted with a variety of cyclohexanones and benzaldehydes including electron-withdrawing and electron-donating substituents. The results are shown that the necessary tetrahydronaphtalenes, including different substituents, are synthesized in the majority of cases in high yields and short reaction times. So that the present reaction was absolutely uniform.
Furthermore, the comparison of tetrahydronaphtalenes synthesis using the present nanocatalyst with the previously reported catalysts are reported in Table 4. As indicated in this Table, the works using different catalysts have disadvantages than the using multi-yolk@shell NiCuFe2O4@mSiO2 catalyst based on yields, reaction times and catalyst loading amounts (Table 4, entries 1–5 vs 6).
Proposed reaction mechanism
Scheme 1 shows a plausible reaction mechanism for the formation of tetrahydronaphtalen. The mesoporous silica can be lead to highly large surface area in the catalyst and coordinating agent with the carbonyl group of aldehyde to give the product in higher isolated yield. In this reaction, at the first time, the catalyst as a Lewis acid activated the benzaldehyde, then the activated benzaldehyde reacted with malononitrile and created an intermediate I. Then, the cyclohexanone reacted with another malononitrile molecule, an intermediate II is produced. Subsequently, the reaction between I and II carried out to obtain the intermediate III. The cyclization reaction of III through intramolecular nucleophilic addition of carbanion into cyano group to form IV. Finally, the isomerization of IV to give V compound as a target product.

The proposed reaction mechanism for the formation of tetrahydronaphtalene.
In order to study the reusability of catalyst, after completion of the reaction, the catalyst is separated by an external magnet, then, washed with ethanol and dried. The benzaldehyde with cyclohexanone derivative and malononitrile was chosen and reacted together as a model reaction to measure the catalyst reusability. Then, Fig. 10 is shown the multi-yolk@shell NiCuFe2O4@mSiO2 nanospheres can be reused in the reaction after 8 times without loss activity.

Reusability of the multi-yolk@shell NiCuFe2O4@mSiO2 spheres after 8 times used in the reaction.
Conclusion
In this research, the multi-yolk@shell NiCuFe2O4@mSiO2 spheres was designed and prepared. Then, it was used as a reactive nanocatalyst for synthesis of tetrahydronaphtalene derivatives under solvent free conditions. The target products were purely obtained in highly yields and short reaction times. The XRD and EDX are indicated the existence of metals in the catalyst. HR-TEM of the catalyst is shown the confirmation of multi-yolk@shell and multi metallic structure. It was found that according to VSM analysis of catalyst, it has high magnetic property. Therefore, the catalyst was easily separated from the reaction by an external magnet and reused in the reaction 8 times without any decrease the catalytic reactivity.
Data availability
All data generated or analysed during this study are included in this published article [and its Supplementary Information files].
References
Grześkowiak, B. F. et al. Nanomagnetic activation as a way to control the efficacy of nucleic acid delivery. Pharm. Res. 32, 103–121 (2015).
Majewski, P. & Thierry, B. Functionalized magnetite nanoparticles—Synthesis, properties, and bio-applications. Crit. Rev. Solid State Mater. Sci. 32, 203–215 (2007).
Ghosh Chaudhuri, R. & Paria, S. Core/shell nanoparticles: Classes, properties, synthesis mechanisms, characterization, and applications. Chem. Rev. 112, 2373–2433 (2012).
Chiu, Y.-H. et al. Hollow Au nanosphere-Cu2O core–shell nanostructures with controllable core surface morphology. J. Phys. Chem. C 124, 11333–11339 (2020).
Kuo, M.-Y. et al. Au@Cu2O core@shell nanocrystals as dual-functional catalysts for sustainable environmental applications. Appl. Catal. B Environ. 242, 499–506 (2019).
Kralj, S. et al. Effect of surface charge on the cellular uptake of fluorescent magnetic nanoparticles. J. Nanopart. Res. 14, 1151–1165 (2012).
Liu, W., Miroshnichenko, A. E., Neshev, D. N. & Kivshar, Y. S. Broadband unidirectional scattering by magneto-electric core-shell nanoparticles. ACS Nano 6, 5489–5497 (2012).
Li, A., Zhu, W., Li, C., Wang, T. & Gong, J. Rational design of yolk–shell nanostructures for photocatalysis. Chem. Soc. Rev. 48, 1874–1907 (2019).
Purbia, R. & Paria, S. Yolk/shell nanoparticles: Classifications, synthesis, properties, and applications. Nanoscale 7, 19789–19873 (2015).
Sun, J., Hu, J., Han, J., Yuan, G. & Guo, R. Dumbbell-like Pt-Fe3O4 nanoparticles encapsulated in N-doped carbon hollow nanospheres as a novel yolk@shell nanostructure toward high-performance nanocatalysis. Langmuir 35, 12704–12710 (2019).
Lin, F. H. & Doong, R. A. Catalytic nanoreactors of Au@Fe3O4 yolk-shell nanostructures with various Au sizes for efficient nitroarene reduction. J. Phys. Chem. C 121, 7844–7853 (2017).
Wang, G. H. et al. Scalable one-pot synthesis of yolk–shell carbon nanospheres with yolk-supported Pd nanoparticles for size-selective catalysis. Chem. Mater. 30, 2483–2487 (2018).
Liu, N. et al. A yolk-shell design for stabilized and scalable Li-ion battery alloy anodes. Nano Lett. 12, 3315–3321 (2012).
Zhang, J. et al. Beyond yolk–shell nanoparticles: Fe3O4@Fe3C Core@Shell nanoparticles as yolks and carbon nanospindles as shells for efficient lithium ion storage. ACS Nano 9, 3369–3376 (2015).
Liu, R. et al. Carbon nanotube-connected yolk–shell carbon nanopolyhedras with cobalt and nitrogen do** as sulfur immobilizers for high-performance lithium–sulfur batteries. ACS Appl. Energy Mater. 1, 6487–6496 (2018).
Patel, S. K. S. et al. Fe2O3 yolk–shell particle-based laccase biosensor for efficient detection of 2,6-dimethoxyphenol. Biochem. Eng. J. 132, 1–8 (2018).
Lu, N. et al. Multifunctional yolk–shell nanostructure as a superquencher for fluorescent analysis of potassium ion using guanine-rich oligonucleotides. ACS Appl. Mater. Interfaces 9, 30406–30413 (2017).
Gao, J. et al. Multifunctional yolk–shell nanoparticles: A potential MRI contrast and anticancer agent. J. Am. Chem. Soc. 130, 11828–11833 (2008).
Xu, J. et al. Yolk-structured upconversion nanoparticles with biodegradable silica shell for FRET sensing of drug release and imaging-guided chemotherapy. Chem. Mater. 29, 7615–7628 (2017).
Pahari, S. K., Olszakier, S., Kahn, I. & Amirav, L. Magneto-fluorescent yolk–shell nanoparticles. Chem. Mater. 30, 775–780 (2018).
Kang, T. et al. Fabrication of Ag nanoaggregates/SiO2 yolk–shell nanoprobes for surface-enhanced Raman scattering. J. Ind. Eng. Chem. 32, 34–38 (2015).
Wan, G. et al. The preparation of Au@TiO2 yolk–shell nanostructure and its applications for degradation and detection of methylene blue. Nanoscale Res. Lett. 12, 535 (2017).
Liu, K. et al. Porous Au–Ag nanospheres with high-density and highly accessible hotspots for SERS analysis. Nano Lett. 16, 3675–3681 (2016).
Deng, X. et al. Yolk–shell structured Au nanostar@metal–organic framework for synergistic chemo-photothermal therapy in the second near-infrared window. Nano Lett. 19, 6772–6780 (2019).
Niu, D. et al. Extraction-induced fabrication of yolk–shell-structured nanoparticles with deformable micellar cores and mesoporous silica shells for multidrug delivery. ACS Appl. Bio Mater. 2, 5707–5716 (2019).
Wang, Y., Wang, G., **ao, Y., Yang, Y. & Tang, R. Yolk–shell nanostructured Fe3O4@NiSiO3 for selective affinity and magnetic separation of his-tagged proteins. ACS Appl. Mater. Interfaces 6, 19092–19099 (2014).
Khan, A. T., Lal, M. & Khan, M. M. Synthesis of highly functionalized piperidines by one-pot multicomponent reaction using tetrabutylammonium tribromide (TBATB). Tetrahedron Lett. 51, 4419–4424 (2010).
Paprocki, D., Madej, A., Koszelewski, D., Brodzka, A. & Ostaszewski, R. Multicomponent reactions accelerated by aqueous micelles. Front. Chem. 6, 502–604 (2018).
Maleki, B., Rooky, R., Rezaei-Seresht, E. & Tayebee, R. One-pot synthesis of bicyclic ortho-aminocarbonitrile and multisubstituted cyclohexa-1,3-dienamine derivatives. Org. Prep. Proced. Int. 49, 557–567 (2017).
Collin, G., Höke, H. & Greim, H. Naphthalene and hydronaphthalenes. In Ullmann’s Encyclopedia of Industrial Chemistry (eds Collin, G. et al.) 661–669 (Wiley, 2003).
Khorasani, M., Naeimi, H. & Zahraei, Z. Sonochemical synthesis of ortho-amino-carbonitrile tetrahydronaphthalenes using mesoporous yolk–shell nanocomposites as a recyclable heterogeneous catalyst and evaluation of their in-vitro antimicrobial activities. Appl. Organomet. Chem. 1, e7305 (2023).
Yonghui, Y. et al. Magnetically separable mesoporous silica-supported palladium nanoparticle catalyzed selective hydrogenation of naphthalene to tetralin. Appl. Organomet. Chem. 33, 5204–5212 (2019).
Lu, N., Zhao, J., Dong, Q., Zhao, Y. & Fan, B. Supported noble metal catalyst with a core-shell structure for enhancing hydrogenation performance. Mol. Catal. 506, 111543 (2021).
Khorasani, M. & Naeimi, H. Fabrication and characterization of mesoporous yolk–shell nanocomposites as an effective reusable heterogeneous base catalyst for the synthesis of ortho-aminocarbonitrile tetrahydronaphthalenes. RSC Adv. 13, 18690–18699 (2023).
Naeimi, H. & Mohammadi, S. Synthesis of 1 H-isochromenes, 4 H-chromenes and orthoaminocarbonitrile tetrahydronaphthalenes by CaMgFe2O4 base nanocatalyst. ChemistrySelect 5, 2627–2633 (2020).
Rong, L., Han, H., Jiang, H. & Tu, S. Efficient NaOH-catalyzed reaction of aromatic aldehyde, cyclic ketones, and malononitrile under solvent-free conditions using a grinding method. Synth. Commun. 38, 3530–3542 (2008).
Mohammadi, S. & Naeimi, H. A bifunctional yolk–shell nanocatalyst with Lewis and organic functional base for the synthesis of spirooxindoles. Appl. Catal. A Gen. 602, 117720–117750 (2020).
Naeimi, H., Salimi, F. & Rabiei, K. Mild and convenient one pot synthesis of Schiff bases in the presence of P2O5/Al2O3 as new catalyst under solvent-free conditions. J. Mol. Catal. A Chem. 260, 100–104 (2006).
Yao, T., Cui, T., Fang, X., Cui, F. & Wu, J. Preparation of yolk–shell FexOy/Pd@mesoporous SiO2 composites with high stability and their application in catalytic reduction of 4-nitrophenol. Nanoscale 5, 5896–5904 (2013).
Khorasani, M. & Naeimi, H. Synthesis of orthoaminocarbonitrile tetrahydronaphthalenes catalyzed by butyl-3-methylimidazolium hexafluorophosphate ionic liquid base catalyst. Synth. Commun. 52, 19–29 (2022).
Wan, Y. et al. Tandem synthesis of bicyclic ortho-aminocarbonitrile derivatives in ionic liquids. J. Heterocycl. Chem. 52, 623–627 (2015).
Rajabzadeh, M., Khalifeh, R., Eshghi, H. & Bakavoli, M. A facile hydrothermal synthesis of novel hollow triple-shell CuNiFe2O4 nanospheres with robust catalytic performance in the Suzuki–Miyaura coupling reaction. J. Catal. 360, 261–269 (2018).
Moshtaghi, Z. A., Eskandari, I. & Notash, B. An efficient and green procedure for the synthesis of highly substituted polyhydronaphthalene derivatives via a one-pot, multi-component reaction in aqueous media. Curr. Chem. Lett. 4, 85–92 (2015).
Wang, J. et al. Primary 1,2-diamine catalysis III: An unexpected domino reaction for the synthesis of multisubstituted cyclohexa-1,3-dienamines. Org. Biomol. Chem. 8, 4240–4242 (2010).
Maleki, B. & Veisi, H. Facile and efficient synthesis of bicyclic ortho-aminocarbonitrile derivatives using nanostructured diphosphate Na2CaP2O7. Org. Prep. Proced. Int. 52, 232–237 (2020).
Acknowledgements
The authors are grateful to University of Kashan for supporting this work by Grant No. 159148/89.
Author information
Authors and Affiliations
Contributions
S.K. and H.N. wrote the main manuscript text and S.K. prepared all of the figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kazempour, S., Naeimi, H. Design, fabrication and characterization of mesoporous yolk–shell nanocomposites as a sustainable heterogeneous nanocatalyst for synthesis of ortho-aminocarbonitrile tetrahydronaphthalenes. Sci Rep 13, 22464 (2023). https://doi.org/10.1038/s41598-023-50021-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-50021-7
- Springer Nature Limited