
Abstract
Climate changes and anthropogenic pressures are causing a biodiversity decline in terms of species number and genetic diversity, reducing the adaptability and evolvability of natural communities. Transitional water ecosystems are more sensitive to habitat reduction and degradation and, thus, are more exposed to biodiversity declines requiring biodiversity monitoring programs for their conservation. Environmental DNA (eDNA) metabarcoding represents a high-throughput tool for biodiversity assessment that is facilitating data collection for biodiversity monitoring. In this study, we applied, for the first time, eDNA metabarcoding in a Mediterranean coastal lagoon to assess the ecological features of eukaryotic phytoplankton communities. We sampled water in seven different lagoon sites and amplified the extracted DNA with primers targeting the variable region 4 (V4) of the 18S rRNA gene marker. The results demonstrated the validity of eDNA studies to provide insights into lagoon phytoplankton composition, establish the structure and spatial variation of phytoplankton communities, and evaluate its correlation to abiotic factors. Finally, the genetic distances analysis suggests that the different spatial distribution of OTUs, at least for the Tetraselmis genus, reflects the genetic background.
Similar content being viewed by others


Introduction
Climate changes and anthropogenic pressures are the main drivers of a biodiversity decline characterized by an unprecedented rate of species extinction1,2. In addition to a decrease in species composition, habitat reduction and degradation are also causing a loss of species’ genetic diversity3,4. The genetic diversity of natural populations and communities ensures adaptive potential and evolvability5, reducing the risk of extinction6. To address these problems, monitoring actions and conservation strategies are essential. The inter-governmental group on Earth Observations and Biodiversity Observation Network (GEO BON) has developed the Essential Biodiversity Variables (EBVs), which are a set of variables aiming to capture different levels of biodiversity and are divided into 6 classes: genetic composition, species populations, species traits, community composition, ecosystem function, ecosystem structure. Specifically, the class of genetic composition includes genetic diversity and genetic differentiation or genetic distance; the class of species populations includes species distribution and abundances; the class of community composition includes community abundance and taxonomic/phylogenetic diversity7.
The advent of environmental DNA (eDNA) metabarcoding as a high-throughput tool for biodiversity assessment is certainly facilitating data collection for biodiversity monitoring.
Environmental DNA (eDNA) refers to DNA from an environmental sample and it is possible to identify two main ways to obtain this genetic material8,9. The first method involves DNA to be extracted from a bulk sample of organisms. The bulk sampling method has been applied to small planktonic organisms, but also to larger and multicellular species10. The second method uses traces of DNA collected by filtering water or using soil or sediment samples11. The sources of this DNA include cells, faeces, mucous, gametes, degrading cells or tissues and fragments resulting from predation9.
eDNA metabarcoding as a biodiversity assessment tool has already been applied in a variety of environments indicating its capability to unveil the macro-guilds used as ecological indicators as well as ecological communities structure12,12,Statistical OTU diversity estimation OTU richness has been calculated as the total number of OTUs per sampling site. OTU occurrence has been calculated as the percentage of OTUs occurring in the same number of sampling sites. All calculations have been done through the software Microsoft Excel. Bray–Curtis distance-based Redundancy Analysis (db-RDA) was performed by using the R Package Vegan. Response variables are represented by the occurrence of OTUs in each sampling site, while explanatory variables are represented by the mean abiotic parameter values registered in the Aquatina lagoon during routine surveys, specifically during the sampling period (Salinity, Temperature, Dissolved Oxygen, and pH). Two-way ANOVA was performed for significance values. The results are visualised as an RDA plot obtained using the R Package ggplot2. OTUs consensus sequences were multiple aligned using Clustal Omega 1.2.2 through the Geneious Prime alignment options, with settings automatically adjusted based on the number of sequences. The obtained multiple sequence alignment was used to construct the Neighbour-Joining tree. For the tree construction, Resampling Method was set on Bootstrap with 674,476 random seeds and 1000 replicates. The genetic distance model was set on Jukes-Cantor, with no outgroup. Tree Build Method was set on Neighbour-Joining.Neighbour-joining phylogenetic analysis
Results
eDNA detects phytoplankton biodiversity in the lagoon
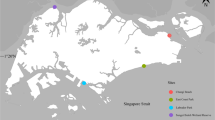
To analyse the biodiversity of phytoplankton communities characterizing the study area, we identified seven sampling sites along the entire lagoon. DNA was extracted from water samples of the selected sites, and a region of the 18S rRNA gene marker was amplified with primers spanning the V4 region of the gene. Illumina sequencing generated a total of about 3,000,000 raw 18S rRNA reads with an average of about 420,000 sequences per sample, of which 41% were retained after bioinformatic processing (Table S1). The Good’s coverage and Shannon Index rarefaction curve of filtered sequences indicated that the samples reached a plateau (Figs. S1, S2), and therefore sequencing depth was appropriate in all the samples.
Following taxonomic assignment, only the OTUs annotated as phytoplankton taxa and classified beyond the D2 level of the SILVA 138 database were retained, resulting in a total of 2293 phytoplankton OTUs. This eDNA metabarcoding experiment detected a large fraction of phytoplankton groups (Table S2), of which 48.8% were assigned up to genus level and 9% up to species level. Analysing the biodiversity of phytoplankton OTUs, 47% of OTUs belong to the Ochrophyta phylum, 37% of OTUs belong to the Myzozoa phylum, 7% and 6% of OTUs belong to Cryptophyta and Chlorophyta phyla, respectively (Fig. 2). eDNA analysis detected OTUs annotated as nano- and pico-phytoplanktonic genera such as Ostreococcus, Micromonas and Bolidomonas.

Total percentage of Phytoplankton OTUs at phyla level.
Interestingly, eDNA from water samples, in our experimental conditions, was able to detect a wide fraction of phytoplankton taxa ranging over different phytoplankton size classes. In addition, both the total and relative numbers of OTUs belonging to the identified phyla present a specific and different distribution in the seven selected sites of the lagoon (Fig. 3).

Number of OTUs at phyla level in each of the seven sampling sites.
Phytoplankton OTU richness and occurrence across lagoon sites
To quantify the differences in phytoplankton OTU richness and composition, we compiled the OTU presence/absence matrix (Table S3). Based on this matrix, OTU richness and OTU occurrence were calculated.
OTU richness represents the number of phytoplankton OTUs observed per sampling site, revealing that sites A (n. of OTUs 1771), E (n. of OTUs 1599), and C (n. of OTUs 1417) host most of the OTU richness, site AF (n. of OTUs 496) appears to be the poorest, and sites B (n. of OTUs 899), D (n. of OTUs 909), and F (n. of OTUs 959) present intermediate values.
The OTU occurrence has been calculated as the percentage of phytoplankton OTUs occurring in the same number of sampling sites and visualised as a bar plot (Fig. 4). The majority of OTUs (26.72%) occur in two sampling sites, while the minority of OTUs (3.46%) occur in all seven sampling sites.

OTUs occurrence analysis calculated as the percentage of OTUs occurring in the same number of sampling sites.
Abiotic parameters explain occurrence heterogeneity among lagoon sites
To infer the dissimilarity between sampling sites and correlate it to the main abiotic drivers that characterize the environment under study through a constrained ordination, we performed a Bray–Curtis distance-based Redundancy Analysis (db-RDA). All loading and score values are summarised in Table S4. The most commonly routinely surveyed environmental parameters have been used as explanatory variables (independent variables): Temperature (T), Salinity (PSU), Dissolved Oxygen (DO) and pH (Fig. 5). Specifically, these environmental parameters are particularly relevant for transitional water ecosystems, in which they describe their heterogeneity. The explained variables (dependent variables) are represented by the OTUs composition in each sampling site. According to the p-values calculated for each of the abiotic parameters, it results that salinity (p < 0.01) and temperature (p < 0.01) are the main explanatory variables for the phytoplankton OTUs distribution across the sampling sites, while pH (p < 0.05) and especially DO (p < 0.1) do not seem to affect it.

Bray–Curtis distance-based Redundancy Analysis (db-RDA) calculated to explain the differences in the phytoplankton OTUs distribution across the Aquatina di Frigole lagoon. PSU = Salinity (PSU); T = Temperature (°C); DO = Dissolved oxygen (mg/L). Black dots and letters indicate the seven sampling sites.
The biplot of this analysis displays sites A, C, and D grouped, while sites AF, B, E, and F do not show any grou** pattern. This result reflects the similar environmental conditions occurring in sites A, C, and D, where comparable levels of the considered environmental variables have been registered. The proximity of these three sites in the biplot also indicates similarities in the phytoplankton OTUs composition across these sites. At the same time, the four sites far apart in the biplot represent areas with distinct environmental differences, influencing the phytoplankton OTUs distribution in a way that separates them apart and from the other three sites.
Exploring genetic diversity across the lagoon: the case of Tetraselmis genus
The analysis of the presence/absence matrix of OTUs highlighted that different OTUs assigned to the same taxon exhibited a different distribution across sampling sites. Specifically, we focused on multiple OTUs assigned to the Tetraselmis genus because the relative and different OTUs presented a clearly different distribution among sampling sites. The bioinformatics analysis recovered 8 OTUs assigned to the Tetraselmis genus highly differently occurring across the sampling sites (Table S5). 2 OTUs occurred only in the marine site AF, 4 OTUs presented wide occurrence across sampling sites, and 2 OTUs presented a peculiar occurrence in two sampling sites. We analysed the genetic distance, calculated as nucleotide substitutions in a phylogenetic tree, to inspect if the differences in the spatial distribution reflect genetic variations (Fig. 6).

Neighbour-joining phylogenetic tree (Jukes-Cantor genetic distance model with 674,476 random seed and 1000 bootstrap replicates) of OTUs of Tetraselmis genus for V4 region of 18S marker. The Clustal omega multiple sequence alignment consists of about 390 bp region. Branch labels indicate substitutions per site.
Interestingly, the OTUs present only in the marine site AF (Tetraselmis 6 and 8) clustered and separated from OTUs widely distributed across the sampling sites, especially Tetraselmis 1, 2, 3, but significantly also from Tetraselmis 5.
This analysis suggests that the different spatial distribution of OTUs, at least for the Tetraselmis genus, presents a relation with the genetic background.
Discussion
Coastal lagoons provide valuable ecosystem services and are suitable areas for biodiversity conservation24. However, they are sensitive to climatic and anthropogenic pressures, which are compromising their ecological integrity25, 26. Among the ecological indicators, eukaryotic phytoplankton is known to exhibit remarkable species richness in transitional waters27. However, molecular phytoplankton surveys are still in their infancy in these environments, where they are only limited to the detection of harmful algal blooms28. In this research, we conducted eDNA metabarcoding with the 18S rRNA gene marker in a protected Mediterranean coastal lagoon. We selected 18S as a gene marker because it is a largely represented barcode gene for phytoplankton in Mediterranean ecosystems29. We assessed an experimental plan aimed to overcome specific issues related to lagoon ecosystems such as water turbidity and abundance of humic substances. We extracted the DNA present in the water column, recovering phytoplankton biodiversity ranging over all the phytoplankton size classes. In general, for planktonic organisms has been applied the bulk sample approach. We used the eDNA metabarcoding method and the results highlight the identification of a wide fraction of nanophytoplankton and microphytoplankton, representing about 80% of OTUs. The high percentage of OTUs belonging to these phytoplanktonic size classes could reflect the different coverage of phytoplankton species in the DNA barcode reference databases.
In this study, eDNA also detected significant correlations between phytoplankton OTUs occurrence and lagoon abiotic components. Specifically, the significant environmental explanatory variables were salinity and temperature (p-values < 0.01), displaying a high difference between site AF, located at the interface between the sea and the lagoon, characterized by high salinity and low temperature, site F, located in proximity to the freshwater input, characterized by low salinity and higher temperature, and the other sites, characterized by a greater intermediate homogeneity. The specific pattern observed in the db-RDA biplot shows that similar environmental conditions correspond to similarities in the phytoplankton community composition, while distinct environmental differences reflect in separated communities. These results confirm that environmental heterogeneity can represent the spatial factor playing a key role in influencing the phytoplankton community structure in transitional water ecosystems30.
Different OTUs were taxonomically assigned to the Tetraselmis genus. The absence of species assignment is probably related to the absence of 18S DNA barcodes for Mediterranean Tetraselmis species in the reference databases. OTUs belonging to the Tetraselmis genus and highly differently distributed across the sampling sites were analysed for the genetic distances. The results outlined that genetic backgrounds correlate with specific spatial distribution. Although the analysis is limited to a short fragment of a conserved gene, the results suggest that sequencing data from eDNA amplicons could be useful to inspect directional genetic variations.
A high genetic variation is important for the ability of phytoplanktonic assemblages to be extremely viable in highly heterogeneous environments such as coastal lagoons, which are subjected to strong variations driven by extrinsic factors such as climate and tides, other than by intrinsic variability given by abiotic heterogeneity.
Overall, this work demonstrates that eDNA metabarcoding is a valuable tool for rapidly surveying the phytoplanktonic communities and describing their spatial distribution in transitional water ecosystems.
Data availability
The FASTQ files are deposited in GenBank (SRA) of NCBI with identification number PRJNA880489.
References
Boyce, D. G., Lewis, M. R. & Worm, B. Global phytoplankton decline over the past century. Nature 466(7306), 591–596 (2010).
Johnson, C. N. et al. Biodiversity losses and conservation responses in the Anthropocene. Science 56(6335), 270–275 (2017).
Exposito-Alonso, M. et al. Genetic diversity loss in the Anthropocene. Science 377(6613), 1431–1435 (2022).
Leigh, D. M., Hendry, A. P., Vázquez-Domínguez, E. & Friesen, V. L. Estimated six per cent loss of genetic variation in wild populations since the industrial revolution. Evol. Appl. 12(8), 1505–1512 (2019).
Blows, M. W. & Hoffmann, A. A. A reassessment of genetic limits to evolutionary change. Ecology 86, 1371–1384 (2005).
Hoffmann, A. & Sgrò, C. Climate change and evolutionary adaptation. Nature 470, 479–485 (2011).
Geijzendorffer, I. R. et al. Bridging the gap between biodiversity data and policyreporting needs: An essential biodiversity variables perspective. J. Appl. Ecol. 53, 1341–1350 (2016).
Suter, L., Polanowski, A. M., Clarke, L. J., Kitchener, J. A. & Deagle, B. E. Capturing open ocean biodiversity: Comparing environmental DNA metabarcoding to the continuous plankton recorder. Mol. Ecol. 30(13), 3140–3157 (2021).
Taberlet, P., Coissac, E., Hajibabaei, M. & Rieseberg, L. H. Environmental DNA. Mol. Ecol. 21(8), 1789–1793 (2012).
Hirai, J., Kuriyama, M., Ichikawa, T., Hidaka, K. & Tsuda, A. A metagenetic approach for revealing community structure of marine planktonic copepods. Mol. Ecol. Resour. 15(1), 68–80 (2015).
Jeunen, G. J. et al. Environmental DNA (eDNA) metabarcoding reveals strong discrimination among diverse marine habitats connected by water movement. Mol. Ecol. Resour. 19(2), 426–438 (2019).
Pawlowski, J. et al. The future of biotic indices in the ecogenomic era: Integrating (e)DNA metabarcoding in biological assessment of aquatic ecosystems. Sci. Total Environ. 637, 1295–1310 (2018).
Specchia, V., Saccomanno, B., Zangaro, F., Tzafesta, E. & Pinna, M. Exploring the biodiversity of a European Natura 2000 Mediterranean lagoon through eDNA metabarcoding. Diversity 14, 991 (2022).
**e, R. et al. eDNA metabarcoding revealed differential structures of aquatic communities in a dynamic freshwater ecosystem shaped by habitat heterogeneity. Environ. Res. 201, 111602 (2021).
Apothéloz-Perret-Gentil, L. et al. Monitoring the ecological status of rivers with diatom eDNA metabarcoding: A comparison of taxonomic markers and analytical approaches for the inference of a molecular diatom index. Mol. Ecol. 30, 2959–2968 (2021).
Kermarrec, L. et al. A next-generation sequencing approach to river biomonitoring using benthic diatoms. Freshw. Sci. 33, 349–363 (2014).
Rimet, F., Pinseel, E., Bouchez, A., Japoshvili, B. & Mumladze, L. Diatom endemism and taxonomic turnover: Assessment in high-altitude alpine lakes covering a large geographical range. Sci. Total Environ. 871, 161970 (2023).
Rivera, S. F. et al. Metabarcoding of lake benthic diatoms: From structure assemblages to ecological assessment. Hydrobiologia 807, 37–51 (2018).
Lowery, C. M., Bown, P. B., Fraass, A. J. & Hull, P. M. Ecological response of plankton to environmental change: Thresholds for extinction. Annu. Rev. Earth Planet. Sci. 48, 403–429 (2020).
Moe, J., Hobæk, A., Persson, J., Skjelbred, B. & Løvik, J. Shifted dynamics of plankton communities in a restored lake: Exploring the effects of climate change on phenology through four decades. Clim. Res. 86, 125–143 (2021).
Raven, J. A. & Beardall, J. Influence of global environmental change on plankton. J. Plankton Res. 43(6), 779–800 (2021).
Winder, M. & Sommer, U. Phytoplankton response to a changing climate. Hydrobiologia 698(1), 5–16 (2012).
Stoeck, T. et al. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19, 21–31 (2010).
Newton, A. et al. Assessing, quantifying and valuing the ecosystem services of coastal lagoons. J. Nat. Conserv. 44, 50–65 (2018).
Domingues, R. B., Guerra, C. C., Galvão, H. M., Brotas, V. & Barbosa, A. B. Short-term interactive effects of ultraviolet radiation, carbon dioxide and nutrient enrichment on phytoplankton in a shallow coastal lagoon. Aquat. Ecol. 51(1), 91–105 (2017).
Pérez-Ruzafa, A., Marcos, C. & Pérez-Ruzafa, I. M. Mediterranean coastal lagoons in an ecosystem and aquatic resources management context. Phys. Chem. Earth 36(5–6), 160–166 (2011).
Bazin, P., Jouenne, F., Deton-Cabanillas, A. F., Pérez-Ruzafa, Á. & Véron, B. Complex patterns in phytoplankton and microeukaryote diversity along the estuarine continuum. Hydrobiologia 726(1), 155–178 (2014).
Suarez-Menendez, M., Planes, S., Garcia-Vazquez, E. & Ardura, A. Early alert of biological risk in a coastal lagoon through eDNA metabarcoding. Front. Ecol. Evol. 8, 9 (2020).
Tzafesta, E. et al. DNA barcode gap analysis for multiple marker genes for phytoplankton species biodiversity in Mediterranean aquatic ecosystems. Biology 11(9), 1277 (2022).
Nogueira, I. D. S., Nabout, J. C., Ibañez, M. D. S. R. & Bourgoin, L. M. Determinants of beta diversity: The relative importance of environmental and spatial processes in structuring phytoplankton communities in an Amazonian floodplain. Acta Limnol. Bras. 22, 247–256 (2010).
Funding
This research was supported by the ex-60% fund from the Italian Ministry of University and Research, by Funding of Basic Research Activities (FFABR 2017) from the Italian Ministry of University and Research (MUR) awarded to M. Pinna and V. Specchia and by RIPARTI project (Code a467414b) funded by Apulia Region, Italy awarded to V. Specchia and supporting F. Zangaro.
Author information
Authors and Affiliations
Contributions
Conceptualization, V.S. and M.P.; Data curation, F.Z., V.S. and M.P.; Formal analysis, V.S., F.Z., E.T., B.S., M.V. and M.P.; Funding acquisition, V.S. and M.P.; Methodology, F.Z., E.T.; Resources, V.S., M.P.; Supervision, V.S. and M.P.; Writing—original draft, V.S., F.Z. and M.P.; Writing—review and editing, V.S., F.Z., B.S., E.T., M.V. and M.P.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Specchia, V., Zangaro, F., Tzafesta, E. et al. Environmental DNA detects biodiversity and ecological features of phytoplankton communities in Mediterranean transitional waters. Sci Rep 13, 15192 (2023). https://doi.org/10.1038/s41598-023-42389-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42389-3
- Springer Nature Limited