
Abstract
Epigenetic dysregulation is a common mechanism shared by molecularly and clinically heterogenous neurodegenerative diseases (NDs). Histone acetylation homeostasis, maintained by the antagonistic activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs), is necessary for appropriate gene expression and neuronal function. Disruption of neural acetylation homeostasis has been implicated in multiple types of NDs including Alzheimer’s disease (AD), yet mechanisms underlying alterations remain unclear. We show that like AD, disruption of Tip60 HAT/HDAC2 balance with concomitant epigenetic repression of common Tip60 target neuroplasticity genes occurs early in multiple types of Drosophila ND models such as Parkinson’s Disease (PD), Huntington’s Disease (HD) and Amyotrophic Lateral Sclerosis (ALS). Repressed neuroplasticity genes show reduced enrichment of Tip60 and epigentic acetylation signatures at all gene loci examined with certain genes showing inappropriate HDAC2 repressor enrichment. Functional neuronal consequences for these disease conditions are reminiscent of human pathology and include locomotion, synapse morphology, and short-term memory deficits. Increasing Tip60 HAT levels specifically in the mushroom body learning and memory center in the Drosophila brain protects against locomotion and short-term memory function deficits in multiple NDs. Together, our results support a model by which Tip60 protects against neurological impairments in different NDs via similar modes of action.
Similar content being viewed by others


Introduction
Neurodegenerative diseases (NDs) are a heterogeneous group of disorders marked by progressive loss of neuronal cells in the brain and/or the spinal cord. Some common NDs include Alzheimer’s Disease (AD), Huntington’s. Depending on the specific subset of neurons and/or the area of nervous system severely affected, NDs vary with respect to the molecular pathways underlying the disease and the subsequent clinical manifestations1. Despite exhibiting a wide range of molecular and phenotypic heterogeneity, NDs share some general characteristics, such as protein misfolding and aggregation, mitochondrial dysfunction, dysregulation of similar molecular/cellular pathways and loss of chromatin dynamics2,3,4. Together, these similarities support convergence of heterogeneous NDs, and better insight into these common links will help target multiple NDs with potential unifying therapeutics3,4.
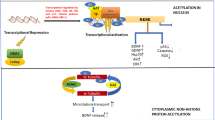
Over the past decade, histone acetylation mediated epigenetic mechanisms have been implicated in a variety of neural processes required for brain function that include neuronal differentiation and maintenance of activity-dependent synaptic plasticity in mature neurons5,6. Acetylation homeostasis in the brain is maintained by the antagonistic activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs), and is essential for proper neuronal gene expression and concomitant neural health4,7. Compelling evidence demonstrates that histone acetylation dysregulation contributes to age related cognitive impairment and age associated neurodegenerative diseases, such as HD, PD, ALS and AD5,7,8,9,10. For instance, the polyglutamate repeat region in HD was shown to sequester HATs CREB-binding protein (CBP) and p300/CBP associated factor (PCAF), resulting in global reduction of H3 and H4 acetylation levels and altered gene expression11. In PD, associated α-synuclein can inhibit H3 histone acetylation by CBP, p300 and PCAF, causing apoptosis in human neuroblastoma cells12 and mitochondrial dysfunction13. In a mouse model of ALS, loss of CBP HAT activity was observed14. Additionally, impaired expression levels of class I, II and IV HDACs and Sirtuins have been observed in human postmortem ALS brain and spinal cord15,16. In line with these studies, we previously reported decreased levels of the HAT, Tat interactive protein 60 kDa (Tip60), and concomitant loss of Tip60 associated specific histone acetylation marks in an AD Drosophila model and in human postmortem AD brain. We further demonstrated that increasing Tip60 levels in the AD Drosophila brain protects against alteration of Tip60 HAT/HDAC2 balance, epigenetic-mediated neuroplasticity gene repression and functional cognitive deficits that occur during early stages of neurodegenerative progression17. These findings raise the possibility that Tip60 HAT/HDAC2 imbalance may be an early common event that contributes to epigenetic mediated gene misregulation and cognition deficits in other NDs as well.
Here, we test the hypothesis that Tip60 mediated histone acetylation homeostasis is disrupted during early stages of neurodegeneration in multiple types of NDs. We show that disruption of Tip60 HAT/HDAC2 balance with concomitant epigenetic repression of common neuroplasticity Tip60 target genes is an early event in multiple types of Drosophila ND models that include Parkinson’s Disease (PD), Huntington’s Disease (HD) and Amyotrophic Lateral Sclerosis (ALS). Functional neuronal consequences of each of these disease conditions are consistent with human pathology and include defects in locomotion, synapse morphology, and short-term memory deficits. Increasing Tip60 HAT levels in the mushroom body learning and memory center in the Drosophila brain protects against these neural deficits. Our findings are the first to demonstrate that disruption of Tip60 HAT/HDAC2 homeostasis and concomitant Tip60 gene control is a critical initial mechanism common to multiple neurodegenerative disorders.
Results
Common Tip60 target neuroplasticity genes are repressed during early stages of multiple types of neurodegenerative disorders
Transcriptional misregulation of critical neuronal genes in the brain are central late stage features of most neurodegenerative disorders including AD18, PD19, HD53. Moreover, dsh is a downstream effector of the Wnt signaling, which has been shown to facilitate LTP expression54. Consistent with these functions, and in general agreement with prior reports39, here we observed functional deficits in locomotor ability in all three ALS, PD and HD ND models examined and concomitant alterations in NMJ synaptic morphology in ALS and PD. Although no morphological defects in the NMJ were observed in our HD model, previous literature shows Htt(128Q) expression has functional consequences on synaptic transmission, such as reduction in evoked transmission and quantal size55. Further, we observed short term memory deficits in both the HD and PD models with the HD fly model also exhibiting defects in olfactory learning and the ALS fly model exhibiting a non-significant yet downward trend in STM. Thus, early alterations in Tip60 epigenetic mediated control of target neuroplasticity genes may cause the subtle synaptic defects and mild cognitive impairment (MCI) believed to represent a transitional period before full pathophysiology of these NDs appears56,57. Our results are in general agreement with recent findings that report early alterations in epigenetic gene control in an HD and AD17,58,59,60,61, and modest but still early transcriptome changes in ALS and PD62,63,64. We propose that epigenetic Tip60/HDAC2 imbalance occurs early in multiple NDs and triggers certain critical synaptic gene expression alterations that persist and/or initiate additional gene disruptions during later stages of the disease that negatively impact synaptic plasticity and LTP.
We previously demonstrated that increasing Tip60 levels in the AD larval brain restores Tip60 HAT/HDAC2 homeostatic balance at least in part, by decreasing HDAC2 levels. Consequently, we observed Tip60 mediated protection against alterations in neuroepigenetic acetylation signatures, repression of neuroplasticity gene expression profiles and functional deficits in brain morphology and cognition. Here, we show that similar to AD, increased Tip60 levels in the fly brain protects against locomotion deficits in all three HD, PD and ALS ND models tested. In addition, increasing Tip60 levels in the brain protected against the significant STM defects observed in the PD and non-significant deficit trend in the ALS and HD larvae. Our results that Tip60 not significantly rescue STM in the HD model is consistent with our observation that Tip60 was not reduced in the HD larval brain. Together, our results reveal that functional neural deficits common in Drosophila models of multiple NDs are alleviated by increasing Tip60 HAT levels in the brain, strengthening the concept that fine-tuning of HAT/HDAC balance is critical for neural function (Fig. 7).

Tip60 promotes neuronal health by protecting against early Tip60 HAT/HDAC imbalance in multiple neurodegenerative diseases. Our results support a model for synaptic defects and cognitive decline in preclinical stages of multiple NDs due to early disruption of Tip60/HDAC homeostasis. This imbalance results in reduction of Tip60 mediated histone acetylation at synaptic gene loci with concomitant gene repression. Increasing cellular levels of specific neural HATs such as Tip60 protects against altered acetylation homeostasis in the brain to maintain appropriate neuroplasticity gene expression profiles and neural health.
During the last decade, there has been extensive research focusing on restoration of histone acetylation homeostasis in several animal models of NDs. As such, a number of HDAC inhibitors (HDACi) have risen as a promising new strategy for treatment of neurodegenerative conditions that include cognitive deficits, motor impairments and other neuropathological phenotypes65,66,67,68,69. However, many HDACi are non-specific and act by increasing global acetylation levels, resulting in unwanted side effects that raise concerns about their therapeutic applicability7,70,71,72. Here, we show that early disruption of HAT/HDAC2 balance is a common feature in multiple NDs, supporting the concept of targeting HAT activity in a combinatorial HAT/HDAC therapeutic approach. To this end, we previously demonstrated that HDACi treatments are not sufficient for full rescue of certain neural gene expression patterns, as they may still require recruitment of Tip60 for site-specific acetylation and/or for interaction with additional transcriptional factors44. Further, many HATs have non-redundant functions and studies have shown success in restoring histone acetylation homeostasis in the brain and concomitant neurodegenerative phenotypes by increasing specific HAT levels7,17,73,74. For example, overexpression of p300 HAT, but not HDAC inhibition, restored acetylation levels of H3 histone and p53, aiding axonal regeneration in mature retinal ganglion cells after optic nerve injury75. Accordingly, HAT activators have emerged as novel potential alternatives to HDACi for neuroprotection76,77. In support of this concept, here we show protection against nervous system related locomotion deficits and short term memory loss in multiple ND Drosophila models solely by increasing Tip60 HAT levels specifically in the brain. Moreover, our observations that Tip60 HAT/HDAC2 imbalance and concomitant transcriptional dysregulation occur early during neurodegenerative progression, such HAT based therapeutics may be effective for early and general neurodegenerative disease protection. Our findings lay the groundwork for discovering a potentially broad neuroprotective role for Tip60 HAT action in multiple neurological disorders, providing opportunity for future therapeutic intervention.
Materials and methods
Fly strains and crosses
All fly lines were raised under standard conditions at 22 °C on standard yeasted Drosophila media (Applied Scientific Jazz Mix Drosophila Food, Thermo Fischer Scientific). The pan-neuronal driver elavC155, MB-specific driver 201Y, UAS-Htt(128Q), UAS-Hsap/SNCAA30P and UAS-Vap-33-1, Cyo/Sco, and TM3/TM6 were all obtained from Bloomington Drosophila Stock Center (Bloomington, IN). For controls for all experiments, the respective UAS or GAL4 driver lines were used alone and yielded identical phenotypes. All experimental crosses were carried out at normal physiological temperature of 25 °C with 12 h light/dark cycle. Controls for all experiments.
Generation of 201Y; Tip60 fly strain
Generation of the double transgenic 201Y-Gal4; UAS-Tip60 line expressing Tip60WT in the mushroom body of the brain followed standard genetic techniques. Balancers with visible phenotypes on the 2nd chromosome (Cyo/Sco) and 3rd chromosome (TM3/TM6), as well as eye phenotype associated with mini white gene in 201Y-Gal4 and UAS-Tip60 insertions were used to select for desired genotypes. Briefly, flies containing homozygous 201Y-Gal4 (201Y) on 2nd chromosome were crossed with TM3/TM6 whereas flies containing homozygous UAS-Tip60 (Tip60) on 3rd chromosome were crossed with Cyo/Sco. These crosses resulted in the F1 progeny being heterozygous for the balancer as well as 201Y or Tip60. These flies were then crossed with their balancer counterpart (i.e. TM3 with TM6 and Cyo with Sco), and F2 progeny were selected for 201Y/201Y; TM3/TM6 or CyO/Sco; Tip60/Tip60. Next, F2 progeny flies were crossed together, and flies were selected for 201Y/CyO; Tip60/TM3 in the next generation. A final self-cross was executed and flies homozygous for 201Y and Tip60 (that is, 201Y; Tip60) were selected for experiments. Tip60 overexpression in the larval brains was confirmed by RT-qPCR using a mixture of exogenous and endogenous Tip60 primers. Flies containing an extra ectopic copy of UAS-GFP (201Y-Gal4; UAS-GFP) which is the same genetic background as the 201Y-Gal4; UAS-Tip60 used to test Tip60 rescue, showed no significant difference in line-crossing then third instar larvae expressing either Htt(128Q), SNCAA30P or Vap-33-1 under the control of mushroom body (MB)-specific 201Y-Gal4 driver alone. These results confirmed that the Tip60 mediated rescue we observed is not due to GAL4 dilution caused by the extra Tip60 construct.
Immunohistochemistry
Antibody staining and imaging was performed as previously described78. UAS-Htt (128Q), UAS-Hsap/SNCAA30P and UAS-Vap-33-1 were crossed with elavC155-Gal4 for expression of the respective transgene in all neurons. elavC155-Gal4 crossed to w1118 served as wild type control. Briefly, wandering third instar larvae were dissected in calcium-free Haemolymph-like saline (HL3) saline on a sylgard dish. Fillet preparations were fixed in 4% paraformaldehyde for 30 min, followed by 2 washes in 1X PBS and one with 1 × PBS with 0.2% Triton (PBS-T). Samples were blocked with 5% goat serum for 30 min and then incubated overnight in conjugated primary antibodies at 4 °C. After 2 washes in 1X PBS-T and one wash in 1X PBS, samples were mounted on Vectashield mounting media (Vector laboratories, Burlingame CA). Slides were imaged within 24 h of mounting. Goat anti-HRP primary antibody conjugated with Alexa488 was obtained from Jackson Immunoresearch Laboratories Inc. and used at a dilution of 1:25. Rhodamine phalloidin was obtained from Molecular Probes, Invitrogen and used at a dilution of 1:40.
Chromatin immunoprecipitation (ChIP)
Chromatin was extracted and sheared from ~ 200third instar larval heads per experiment. To obtain larval heads, the first 1/3 of the larvae (anterior head region) was isolated. Remaining fat bodies were carefully dissected and discarded. All larval heads were inspected visually to ensure that the entire CNS was intact. Because we use a GAL4-inducible system to target gene expression exclusively in the nervous system of the larvae, this method ensures virtually no variability in gene expression in the samples used. For ChIPs, we used truChIP Chromatin Shearing Kit from Covaris following the manufacturer’s instructions. Briefly, protein–DNA cross-links were made at RT for 5 min with 1% formaldehyde and tissue was pulverized using the CryoPrep from Covaris. Cells were lysed and nuclei were prepared using Covaris lysis buffer. Sonication of DNA was performed using a Covaris E220 Ultrasonicator for 13 min. The sheared chromatin was immunoprecipitated using the EZ-Magna ChIP A Chromatin Immunoprecipitation Kit (Millipore) following the manufacturer's instructions. Briefly, ChIP was performed with 30 μg of sheared chromatin using anti-Rpd3 (Abcam, ab1767), anti-Tip60 (Abcam, ab23886), anti-H4K5ac (Active Motif, 39699), anti-H4K12ac (Active Motif, 39166), anti-H4K16ac (Active Motif, 39168), and Normal Mouse IgG Polyclonal Antibody control (Millipore) antibodies. The eluted material from the immunoprecipitation was then purified using a QIAquick PCR purification kit (QIAGEN) and used directly for qPCR. Primer sets were designed by NCBI/Primer-BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast/). Fold enrichment for all the respective genes was calculated relative to the non-specific Mouse IgG Polyclonal Antibody control.
Imaging and analysis
Larval NMJs were imaged using Olympus FV1000 Fluoview laser scanning confocal microscope. Muscle segmentation was used to locate the NMJ synapses present exactly between muscles 6/7 in the segment A3 of larval body wall. All images were captured using constant confocal gain settings. NMJs were optically sectioned with the help of Olympus FV1000 software and images were acquired as a Z-stack. Later, a 2D projection at maximum intensity was rendered with the help of ImageJ software (NIH). Quantitative analysis of boutons per NMJ, area of NMJ and area of muscle was performed using ImageJ. In order to minimize variation across larval dissection preparations between samples, NMJ area was normalized to the area covered by muscles 6/7 in segment A3 for each sample.
Larval line crossing assay
Larval locomotion was analyzed as previously described79. The line crossing apparatus consisted of a petri plate containing 1% agar positioned on a 0.2 cm2 grid paper. A single third instar larva was placed on the petri plate and allowed to acclimate to the plate for 5 min. After initial acclimation, the number of grid lines passed by the head of the larva in 30 s were recorded. This was performed a total of three times for each larva and averaged. Data from thirty larvae were collected.
Olfactory learning and memory assay
Third instar larvae were trained and tested for olfactory learning and memory performance following guidelines in Honjo and Furukubo-Tokunaga 46. Freshly prepared 2.5% agar plates with 2 ml 1 M SUC or DW (for control) spread over the agar were used to train the third instar larvae. 10 µl of undiluted odor Linalool (LIN) (Nacalai, Tokyo, Japan) was spotted on a filter disk and placed on the lid of the petri dish (Fig. 6A). 50 to 100 larvae were transferred to the plate and kept for 30 min. After training, the larvae were rinsed with distilled water and transferred to the test plate. The test plate contained odor (2.5 µl) on one side and none on the other side. The plate was observed for 3 min and responsive index was determined based on the number of larvae (RI) within the 3 cm semicircular area. RI = (number of larvae in the odor area – number of larvae in the control area)/total number for larvae counted. ∆RI was calculated as the difference in RI of LIN/SUC and control LIN/DW. For memory performance test, the larvae were trained as described above, and kept on 2.5% agar plate for the indicated time until the olfactory test was performed. Naïve olfactory and gustatory response test was performed as described in Honjo and Furukubo-Tokunaga46. To determine the speed of the larvae from different genotypes, third instar larvae were placed on 2.5% agar plates, allowed to crawl for one minute and video was recorded using a Sony DCR-SR47 Handycam with Carl Zeiss optics. The recorded path length was quantified, and locomotion speed was calculated using the software Tracker. RIs of all the genotype were normalized using their respective locomotion speed.
RT-qPCR analysis
Total RNA was isolated from 40 staged third instar larval brains using the RNeasy Plus Mini Kit (QIAGEN). Complementary DNA (cDNA) was prepared using SuperScript II reverse transcriptase kit (Invitrogen) according to the manufacturer’s instructions with 1 µg total RNA and 0.2 µg/ml random hexamer primers (Roche Applied Science). PCRs were performed in a 20 µl reaction volume containing cDNA, 1 µM Power SYBR Green PCR Master Mix (Applied Biosystems), and 10 µM of both forward and reverse primers. Primer sequences are available in the Supplementary Table 2. The fold change for each dataset is calculated using the standard ∆∆Ct method using the reference Drosophila RP49 gene as the internal control. For the heterochromatin region, 4 µl of 2.5 µM primer set (Active Motif, 71,028) was added to 20ul reaction volume. PCR was performed using an ABI 7500 Real-Time PCR system (Applied Biosystems) following the manufacturer’s instructions. Fold change in mRNA expression was determined by the ∆∆Ct method.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Przedborski, S., Vila, M. & Jackson-Lewis, V. Series introduction: neurodegeneration: what is it and where are we?. J. Clin. Investig. 111, 3–10 (2003).
Sheikh, S., Haque, E. & Mir, S. S. Neurodegenerative diseases: multifactorial conformational diseases and their therapeutic interventions. J. Neurodegener. Dis. https://doi.org/10.1155/2013/563481 (2013).
Habib, R., Noureen, N. & Nadeem, N. Decoding common features of neurodegenerative disorders: from differentially expressed genes to pathways. Curr. Genom. 19, 300–312 (2018).
Berson, A., Nativio, R., Berger, S. L. & Bonini, N. M. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 41, 587–598 (2018).
Lardenoije, R. et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 131, 21–64 (2015).
Sharma, S. K. Protein acetylation in synaptic plasticity and memory. Neurosci. Biobehav. Rev. 34, 1234–1240 (2010).
Saha, R. & Pahan, K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 13, 539 (2006).
Feng, Y., Jankovic, J. & Wu, Y. C. Epigenetic mechanisms in Parkinson’s disease. J. Neurol. Sci. 349, 3–9. https://doi.org/10.1016/j.jns.2014.12.017 (2015).
Konsoula, Z. & Barile, F. A. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J. Pharmacol. Toxicol. Methods 66, 215–220 (2012).
Rossaert, E. et al. Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model. Acta Neuropathol. Commun. 7, 107 (2019).
Steffan, J. S. et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413, 739–743. https://doi.org/10.1038/35099568 (2001).
Kontopoulos, E., Parvin, J. D. & Feany, M. B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15, 3012–3023. https://doi.org/10.1093/hmg/ddl243 (2006).
Siddiqui, A. et al. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson’s disease. Free Radic. Biol. Med. 53, 993–1003. https://doi.org/10.1016/j.freeradbiomed.2012.05.024 (2012).
Rouaux, C. et al. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 22, 6537–6549 (2003).
Janssen, C. et al. Differential histone deacetylase mRNA expression patterns in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 573–581. https://doi.org/10.1097/NEN.0b013e3181ddd404 (2010).
Korner, S. et al. Differential sirtuin expression patterns in amyotrophic lateral sclerosis (ALS) postmortem tissue: neuroprotective or neurotoxic properties of sirtuins in ALS?. Neurodegener. Dis. 11, 141–152. https://doi.org/10.1159/000338048 (2013).
Panikker, P. et al. Restoring Tip60 HAT/HDAC2 balance in the neurodegenerative brain relieves epigenetic transcriptional repression and reinstates cognition. J. Neurosci. 38, 4569–4583. https://doi.org/10.1523/JNEUROSCI.2840-17.2018 (2018).
Blalock, E. M. et al. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. USA 101, 2173–2178 (2004).
Elstner, M. et al. Expression analysis of dopaminergic neurons in Parkinson’s disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol. 122, 75 (2011).
**ang, C., Zhang, S., Dong, X., Ma, S. & Cong, S. Transcriptional dysregulation and post-translational modifications in polyglutamine diseases: from pathogenesis to potential therapeutic strategies. Front. Mol. Neurosci. 11, 153. https://doi.org/10.3389/fnmol.2018.00153 (2018).
Riva, N. et al. Unraveling gene expression profiles in peripheral motor nerve from amyotrophic lateral sclerosis patients: insights into pathogenesis. Sci. Rep. 6, 39297 (2016).
Romero, E. et al. Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron 57, 27–40 (2008).
Feany, M. B. & Bender, W. W. A Drosophila model of Parkinson’s disease. Nature 404, 394 (2000).
Sanhueza, M., Zechini, L., Gillespie, T. & Pennetta, G. Gain-of-function mutations in the ALS8 causative gene VAPB have detrimental effects on neurons and muscles. Biol. Open 3, 59–71 (2014).
Roos, J., Hummel, T., Ng, N., Klämbt, C. & Davis, G. W. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron 26, 371–382 (2000).
Noordermeer, J., Klingensmith, J., Perrimon, N. & Nusse, R. dishevelled and armadillo act in the wingless signalling pathway in Drosophila. Nature 367, 80 (1994).
Jan, L. Y., Papazian, D. M., Timpe, L., O’Farrell, P. & Jan, Y. N. Application of drosophila molecular genetics in the study of neural function—studies of the shaker locus for a potassium channel. Trends Neurosci. 8, 234–238 (1985).
Budnik, V. et al. Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron 17, 627–640 (1996).
Guan, J. S. et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. https://doi.org/10.1038/nature07925 (2009).
Tea, J. S., Chihara, T. & Luo, L. Histone deacetylase Rpd3 regulates olfactory projection neuron dendrite targeting via the transcription factor Prospero. J Neurosci 30, 9939–9946. https://doi.org/10.1523/JNEUROSCI.1643-10.2010 (2010).
Fitzsimons, H. L. & Scott, M. J. Genetic modulation of Rpd3 expression impairs long-term courtship memory in Drosophila. PLoS ONE 6, e29171. https://doi.org/10.1371/journal.pone.0029171 (2011).
Keshishian, H., Broadie, K., Chiba, A. & Bate, M. The Drosophila neuromuscular junction: a model system for studying synaptic development and function. Annu. Rev. Neurosci. 19, 545–575 (1996).
Jan, L. Y. & Jan, Y. N. Antibodies to horseradish peroxidase as specific neuronal markers in Drosophila and in grasshopper embryos. Proc. Natl. Acad. Sci. USA 79, 2700–2704 (1982).
Ruiz-Cañada, C. & Budnik, V. Introduction on the use of the Drosophila embryonic/larval neuromuscular junction as a model system to study synapse development and function, and a brief summary of pathfinding and target recognition. Int. Rev. Neurobiol. 75, 1–31 (2006).
Menon, K. P., Carrillo, R. A. & Zinn, K. Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip. Rev. 2, 647–670 (2013).
Prokop, A. Organization of the efferent system and structure of neuromuscular junctions in Drosophila. Int. Rev. Neurobiol. 75, 71–90 (2006).
Kurdyak, P., Atwood, H., Stewart, B. & Wu, C. F. Differential physiology and morphology of motor axons to ventral longitudinal muscles in larval Drosophila. J. Comp. Neurol. 350, 463–472 (1994).
Slater, C. The structure of human neuromuscular junctions: some unanswered molecular questions. Int. J. Mol. Sci. 18, 2183 (2017).
Pennetta, G., Hiesinger, P. R., Fabian-Fine, R., Meinertzhagen, I. A. & Bellen, H. J. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron 35, 291–306 (2002).
Lloyd, T. E. & Taylor, J. P. Flightless flies: Drosophila models of neuromuscular disease. Ann. N. Y. Acad. Sci. 1184, e1-20 (2010).
Lee, W.-C.M., Yoshihara, M. & Littleton, J. T. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 101, 3224–3229 (2004).
Heisenberg, M. What do the mushroom bodies do for the insect brain? An introduction. Learn. Mem. 5, 1–10 (1998).
Zars, T. Behavioral functions of the insect mushroom bodies. Curr. Opin. Neurobiol. 10, 790–795 (2000).
Johnson, A. A., Sarthi, J., Pirooznia, S. K., Reube, W. & Elefant, F. Increasing Tip60 HAT levels rescues axonal transport defects and associated behavioral phenotypes in a Drosophila Alzheimer’s disease model. J. Neurosci. 33, 7535–7547. https://doi.org/10.1523/JNEUROSCI.3739-12.2013 (2013).
Xu, S. et al. Epigenetic control of learning and memory in Drosophila by Tip60 HAT action. Genetics 198, 1571–1586. https://doi.org/10.1534/genetics.114.171660 (2014).
Honjo, K. & Furukubo-Tokunaga, K. Induction of cAMP response element-binding protein-dependent medium-term memory by appetitive gustatory reinforcement in Drosophila larvae. J. Neurosci. 25, 7905–7913. https://doi.org/10.1523/JNEUROSCI.2135-05.2005 (2005).
McFarland, K. N. et al. Genome-wide histone acetylation is altered in a transgenic mouse model of Huntington’s disease. PLoS ONE 7, e41423 (2012).
Yakhine-Diop, S. M. et al. The paradigm of protein acetylation in Parkinson’s disease. Neural Regener. Res. 14, 975 (2019).
Francis, Y. I. et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 18, 131–139 (2009).
Yakhine-Diop, S. M. et al. Impaired mitophagy and protein acetylation levels in fibroblasts from Parkinson’s disease patients. Mol. Neurobiol. 56, 2466–2481 (2019).
Tan, Y. et al. Upregulation of histone deacetylase 2 in laser capture nigral microglia in Parkinson’s disease. Neurobiol. Aging 68, 134–141 (2018).
Graff, J. et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226. https://doi.org/10.1038/nature10849 (2012).
Ruiz-Canada, C. et al. New synaptic bouton formation is disrupted by misregulation of microtubule stability in aPKC mutants. Neuron 42, 567–580 (2004).
Tang, S. J. Synaptic activity-regulated Wnt signaling in synaptic plasticity, glial function and chronic pain. CNS Neurol. Disord. Drug Targets 13, 737–744 (2014).
Steinert, J. R. et al. Rab11 rescues synaptic dysfunction and behavioural deficits in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 21, 2912–2922 (2012).
Petersen, R. C. et al. Mild cognitive impairment: ten years later. Arch. Neurol. 66, 1447–1455. https://doi.org/10.1001/archneurol.2009.266 (2009).
Minhas, S., Khanum, A., Riaz, F., Khan, S. & Alvi, A. Predicting progression from mild cognitive impairment to Alzheimer’s disease using autoregressive modelling of longitudinal and multimodal biomarkers. IEEE J. Biomed. Health Inform. https://doi.org/10.1109/JBHI.2017.2703918 (2017).
Bossers, K. et al. Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer’s disease. Brain 133, 3699–3723 (2010).
Hervás-Corpión, I. et al. Early alteration of epigenetic-related transcription in Huntington’s disease mouse models. Sci. Rep. 8, 9925 (2018).
Luthi-Carter, R. et al. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 9, 1259–1271 (2000).
Scherzer, C. R., Jensen, R. V., Gullans, S. R. & Feany, M. B. Gene expression changes presage neurodegeneration in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 12, 2457–2466 (2003).
Yacoubian, T. A. et al. Transcriptional dysregulation in a transgenic model of Parkinson disease. Neurobiol. Dis. 29, 515–528 (2008).
Barham, C. et al. RNA-Seq analysis of spinal cord tissues from hPFN1 G118V transgenic mouse model of ALS at pre-symptomatic and end-stages of disease. Sci. Rep. 8, 13737 (2018).
Bandyopadhyay, U. et al. RNA-Seq profiling of spinal cord motor neurons from a presymptomatic SOD1 ALS mouse. PLoS ONE 8, e53575 (2013).
Hockly, E. et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 100, 2041–2046. https://doi.org/10.1073/pnas.0437870100 (2003).
Thomas, E. A. et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 105, 15564–15569. https://doi.org/10.1073/pnas.0804249105 (2008).
Monti, B. et al. Valproic acid is neuroprotective in the rotenone rat model of Parkinson’s disease: involvement of alpha-synuclein. Neurotox. Res. 17, 130–141. https://doi.org/10.1007/s12640-009-9090-5 (2010).
Ryu, H. et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 93, 1087–1098. https://doi.org/10.1111/j.1471-4159.2005.03077.x (2005).
Gräff, J. & Tsai, L.-H. The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev. Pharmacol. Toxicol. 53, 311–330 (2013).
Chuang, D.-M., Leng, Y., Marinova, Z., Kim, H.-J. & Chiu, C.-T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32, 591–601 (2009).
Boutillier, A.-L., Tzeplaeff, L. & Dupuis, L. The dark side of HDAC inhibition in ALS. EBioMedicine 41, 38 (2019).
Didonna, A. & Opal, P. The promise and perils of HDAC inhibitors in neurodegeneration. Ann. Clin. Transl. Neurol. 2, 79–101 (2015).
Pirooznia, S. K. & Elefant, F. Targeting specific HATs for neurodegenerative disease treatment: translating basic biology to therapeutic possibilities. Front. Cel. Neurosci. 7, 30 (2013).
Caccamo, A., Maldonado, M. A., Bokov, A. F., Majumder, S. & Oddo, S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 107, 22687–22692 (2010).
Gaub, P. et al. The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain 134, 2134–2148. https://doi.org/10.1093/brain/awr142 (2011).
Schneider, A. et al. Acetyltransferases (HATs) as targets for neurological therapeutics. Neurotherapeutics 10, 568–588 (2013).
Selvi, B. R., Cassel, J.-C., Kundu, T. K. & Boutillier, A.-L. Tuning acetylation levels with HAT activators: therapeutic strategy in neurodegenerative diseases. Biochim. Biophys. Acta 1799, 840–853 (2010).
Sarthi, J. & Elefant, F. dTip60 HAT activity controls synaptic bouton expansion at the Drosophila neuromuscular junction. PLoS ONE 6, e26202 (2011).
Nichols, C. D., Becnel, J. & Pandey, U. B. Methods to assay Drosophila behavior. JoVE 61, e3795 (2012).
Acknowledgements
This work was supported by the National Institutes of Health Grant R01HD057939 to F.E.
Author information
Authors and Affiliations
Contributions
M.B., A.B. prepared Figs. 3,4,6,7, wrote manuscript text, carried out experiments, analyzed results and performed statistics. H.Z., P.P., R.S. prepared Figs. 1,2,5 carried out experiments, analyzed results and performed statistics. V.P., G.V., N.B., S.A. helped with experiments. F.E. wrote manuscript text and oversaw all experiments and interpretations.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beaver, M., Bhatnagar, A., Panikker, P. et al. Disruption of Tip60 HAT mediated neural histone acetylation homeostasis is an early common event in neurodegenerative diseases. Sci Rep 10, 18265 (2020). https://doi.org/10.1038/s41598-020-75035-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75035-3
- Springer Nature Limited
This article is cited by
-
Small molecule modulators of chromatin remodeling: from neurodevelopment to neurodegeneration
Cell & Bioscience (2023)
-
Dissecting the Relationship Between Neuropsychiatric and Neurodegenerative Disorders
Molecular Neurobiology (2023)
-
NAA60 (HAT4): the newly discovered bi-functional Golgi member of the acetyltransferase family
Clinical Epigenetics (2022)
-
The role of histone modifications: from neurodevelopment to neurodiseases
Signal Transduction and Targeted Therapy (2022)