
Abstract
Antimicrobial peptides (AMPs) are promising alternatives to classical antibiotics for the treatment of drug-resistant infections. Due to their versatility and unlimited sequence space, AMPs can be rationally designed by modulating physicochemical determinants to favor desired biological parameters and turned into novel therapeutics. In this study, we utilized key structural and physicochemical parameters, in combination with rational engineering, to design novel short α-helical hybrid peptides inspired by the well-known natural peptides, cathelicidin and aurein. By comparing homologous sequences and abstracting the conserved residue type, sequence templates of cathelicidin (P0) and aurein (A0) were obtained. Two peptide derivatives, P7 and A3, were generated by amino acid substitution based on their residue composition and distribution. In order to enhance antimicrobial activity, a hybrid analog of P7A3 was designed. The results demonstrated that P7A3 had higher antibacterial activity than the parental peptides with unexpectedly high hemolytic activity. Strikingly, C-terminal truncation of hybrid peptides containing only the α-helical segment (PA-18) and shorter derivatives confer potent antimicrobial activity with reduced hemolytic activity in a length‐dependent manner. Among all, PA-13, showed remarkable broad-spectrum antibacterial activity, especially against Pseudomonas aeruginosa with no toxicity. PA-13 maintained antimicrobial activity in the presence of physiological salts and displayed rapid binding and penetration activity which resulted in membrane depolarization and permeabilization. Moreover, PA-13 showed an anti-inflammatory response via lipopolysaccharide (LPS) neutralization with dose-dependent, inhibiting, LPS-mediated Toll-like receptor activation. This study revealed the therapeutic potency of a novel hybrid peptide, and supports the use of rational design in development of new antibacterial agents.
Similar content being viewed by others

Introduction
The increasing emergence and dissemination of antibiotic resistance among bacterial pathogens has become a global public health challenge1,2. Therefore, there is an urgent need to develop new antimicrobial agents to overcome this problem. Antimicrobial peptides (AMPs) are an essential component of the innate immune system produced as a first line of defense by all multicellular organisms3. With such exceptional properties as broad-spectrum antimicrobial activity, rapid action and infrequent development of resistance4,5, AMP-based pharmaceuticals provide excellent templates for a wide range of antimicrobial agents and biomedical applications.
In general, naturally occurring AMPs are between 12 and 50 amino acids in length, and often contain cationic and hydrophobic residues6. Previous studies contributing to the understanding of the structure-activity relationship of AMPs show that one important class of membrane-active AMPs infers an amphipathic α-helical conformation4,7,8. The initial electrostatic interaction between a positively-charged AMP and the negatively-charged microbial cell membrane, such as lipoteichoic acids in Gram-positive bacteria or lipopolysaccharides (LPSs) in Gram-negative bacteria, results in insertion of a hydrophobic segment into the lipid bilayer of the microbial membranes. The effect is to destabilize by pore formation and produce cell death, a process unlike that of conventional antibiotics which have specific modes of action such as inhibition of cell wall or nucleic acid synthesis9.
Cathelicidins, a prominent family of AMPs, play important roles in the innate immune system of practically all species of vertebrates10. The α-helical cathelicidins possess broad‐spectrum antimicrobial activity against bacteria, fungi and viruses, and of note also against antibiotic-resistant strains of pathogenic bacteria11. Besides their antimicrobial activity, cathelicidins bind and neutralize LPS, and protect against endotoxic shock in a murine model of septicemia12. Frog skin is a natural storehouse of active AMPs13. Upon contact with microorganisms, anuran skin peptides are produced in dermal serous glands and stored within granules for release onto the skin surface after stress or tissue injury as part of the immune system14. Amongst them, aurein peptides from the Australian southern bell frogs Litoria aurea and Litoria raniformis15, are a large family of peptides with prominent activity toward both bacteria and cancer cells16.
Despite the many desirable properties of AMPs, they also exhibit undesirable properties including hemolytic activity toward human red blood cells, sensitivity to protease, salt and serum, and high production cost which impede their development as therapeutic agents17. However, optimization of sequences or modification of AMPs can improve antimicrobial effects while reducing cytotoxicity and so overcome barriers and expedite development for clinical application7,18,8. The truncation of LL-37, based on the amino acid composition and 3D structure, retains its antimicrobial activity while losing its side effects21. The synthetic hybrid peptides of progetrin-1, bovine lactoferricin and cecropin A, LB-PG and CA-PG, exhibit broad antimicrobial activity against both Gram-positive and -negative bacteria along with reduced hemolytic activity8,27,38. We therefore modified parent peptide sequences by truncation of unstructured regions and amino acid substitution. Their derivatives, especially P7, showed perfect amphipathicity and higher hydrophobicity than the parental peptides with significantly improved antimicrobial activity. These results suggested that the unstructured region at positions 1 to 3 did not affect antimicrobial activity, but that both amphipathicity and hydrophobicity played crucial roles in antimicrobial activity. In addition, the potent antimicrobial activity of P7 may have resulted from the bulky side chain of tryptophan at position 7. Since this located on the hydrophobic/hydrophilic interface, the interaction of peptides with bacterial membranes is increased8.
Combinations of two or more native peptides, or hybrid peptide derivatives, have received great attention, as this can enhance their antimicrobial properties22. Since P7 showed much higher antimicrobial activity than A3, the sequence of P7 was maintained with its activity further improved by hybridizing with A3 at the C-terminus. P7A3 displayed higher antimicrobial activity against all tested bacteria when compared with P7 and A3 alone, however P7A3 also displayed the strongest hemolytic activity. Our results indicated that the high positive charge and hydrophobicity of P7A3 correlated with both its antimicrobial and hemolytic activities. To minimize toxicity, truncated derivatives were designed to identify the shortest amino acid sequence which retained the potent antimicrobial activity while reducing toxicity. It has been shown that the N-terminal α-helical domain of peptides such as PMAP-36 is its active region and has the ability to interact with and penetrate the bacterial membranes8. Moreover, an unstructured region (position 18 to 23) was observed in the C-terminus of the ribbon structure of P7A3. Therefore, the C-terminus of the hybrid peptide was truncated. The truncated derivatives of the hybrid analogue (PA-14 and -13) displayed high antimicrobial activity against both Gram-negative and -positive bacterial strains with relatively decreased toxicity. Our studies indicated that increasing amphipathicity (with suitable positive charges) and hydrophobicity improved antimicrobial activity and selectivity. Among truncated derivatives, the shortest peptide (PA-13) showed potent antimicrobial activity along with low toxicity against both hRBCs and L929 cells. Differences in amino acid sequence and position have been suggested to be important for antimicrobial activity. Importantly, the bulky side chain of tryptophan at the hydrophobic/hydrophilic interface of all truncated derivatives played an important role in facilitating the interaction of peptides with bacterial membranes. The antimicrobial activity and toxicity of AMPs are related to multiple physio-chemical properties, for instance the length, charge, hydrophobicity, amphipathicity and hydrophobic/hydrophilic angle. Thus, adjusting one can cause alterations to the others and so it is difficult to highlight the influence of a single factor for the activity4.
The therapeutic index is a widely employed parameter to indicate the specificity of AMPs. It is calculated as the ratio of MHC (hemolytic activity) and MIC (antimicrobial activity)27. Thus, larger TI values indicate greater antimicrobial specificity. Among all derivative peptides, P7 exhibited the highest TIs (8.78 and 2.06 against Gram-negative and -positive bacteria, respectively). The hybrid peptide P7A3 displayed high antimicrobial activity coupled with strong hemolytic activity. Hence its TI was low (0.09 and 0.31 for Gram-negative and -positive bacteria, respectively). PA-18, a truncated form containing the entire α-helical structure of the hybrid peptide, displayed comparable specificity with P7A3. Among truncated derivatives, PA-13 exhibited the highest TI against all tested bacterial strains suggesting that it had increased selectivity toward bacterial cells compared with hRBCs, and implying it had a wider therapeutic window. Our studies demonstrated that an increase of TI mainly resulted from a reduction of hemolytic activity, and indicated that truncation at the C-terminus of peptides affected their hemolytic activity. Therefore, the potent antimicrobial activity with low host toxicity of PA-13 reflects an attractive direction for AMP development.
It has been suggested that the propensity of AMPs to form an amphipathic α-helix in membrane-mimetic environments is the key to their membrane-disruptive activity27. In the current study, the secondary structure of PA-13 in water, membrane-mimicking environments and LPS was analyzed using CD spectra. The results indicated that PA-13 exhibited a random coil in aqueous environments. In 30 mM SDS, 50% TFE and 0.05% LPS of P. aeruginosa, PA-13 exhibited a typical α-helical structure, with more helical content in 30 mM SDS than in 50% TFE and 0.05% LPS. This result was consistent with previous studies indicating that the structural change of peptides from a random coil to an α-helix structure might be one of the most important factors determining peptide function at bacterial cell membranes, including antimicrobial activity39.
Previous studies have demonstrated that a positive charge facilitates antimicrobial peptide binding to the negatively-charged bacterial membranes, and/or LPS, via electrostatic interactions40. However, other positively charged molecules, including salts, can weaken this electrostatic interaction. The antibacterial activity of PA-13 in the presence of different physiological salts was investigated. PA-13 retained its antimicrobial activity in the presence of NH4+, Zn2+, Fe3+, Ca2+ and K+, suggesting that cation valence [monovalent (NH+, K+), divalent (Zn2+, Ca2+) and trivalent (Fe3+)] had little or no effect on the strength of PA-13’s antimicrobial activity. This might be due to the bulky side chain of tryptophan, which could enhance the affinity of this antimicrobial peptide for the bacterial membrane and contribute to its strong antibacterial activity in the presence of salts8. However, there was a marked decrease in the antibacterial activity of PA-13 against P. aeruginosa in the presence of Na+ and Mg2+. Mg2+ compromised the antimicrobial activity, possibly due to competition with cationic peptide for binding with LPS molecules on bacterial outer membranes. The presence of Na+ could hinder electrostatic interactions and decrease the binding efficacy of the peptide. This might be attributed to the first step in antimicrobial activity, ultimately compromising the killing efficiency of a peptide41,42. However, this is in consistent with the results of other studies43,44. While F4 peptide retains killing activity in the presence of salts at physiologic concentrations, except Na+ and Mg2+, it still displays effective antimicrobial potency in the mouse model45.
Although the exact mechanism of action of AMPs has not been established, it has been proposed that the cytoplasmic membranes of bacteria are the main target of these peptides via their disruption or the formation of pore/ion channels35. To assess membrane-penetrating activity and localization, TAMRA-labelled PA-13 was incubated with P. aeruginosa and the bacteria investigated by flow cytometry and confocal microscopy. The results indicated that PA-13 exhibited time-dependent penetrating activity. PA-13 rapidly punctured the bacterial membranes (within 5 min at 1 × MIC). The results were strongly in accordance with localization studies. That is, TAMRA-labelled PA-13 clearly localized on P. aeruginosa membranes and then accumulated in bacterial cytoplasm. After inserting into the cytoplasmic membrane, PA-13 potentially disrupted the membrane integrity in dose- and time-dependent manner. This appeared concordant with the collapse of membrane electrical potential as observed by flow cytometry. The results of SEM and TEM further confirmed the killing mechanisms of PA-13 being membrane rupture and pore formation, leading to the leakage of intracellular contents and cell death. The fluorophore-PEN conjugates displayed altered modes of membrane interaction with increased insertion into the core of the model cell membrane and exert membrane-thinning effects without effecting membrane-penetrating ability46. This agrees with our results, showing the penetrating ability of TAMRA-labelled PA-13 in a time- dependent manner and that the unlabeled PA-13 behaved similarly to the labeled compound determined by circular dichroism and flow cytometry (data not shown).
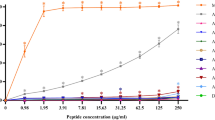
Lipopolysaccharide from bacterial endotoxin, a major component of the outer membrane of all Gram-negative bacteria, can cause inflammation, sepsis and shock47. Previous research demonstrated that the peptide-LPS interaction strongly promotes bacterial cell death and reduces inflammation. Anti-inflammatory activity of AMPs can occur as a result of neutralization of biological inflammatory cytokines or inhibition of their production48. Our studies revealed that PA-13 was able to bind and neutralize LPS, the negatively charged molecules constituting an important barrier in the membranes of Gram-negative bacteria, in a dose-dependent manner. Furthermore, PA-13 showed dose-dependent inhibition of LPS-mediated, Toll-like receptor activation, which inhibited the release of pro-inflammatory cytokines and down-regulated further severe inflammation. Therefore, PA-13 appears to be a very promising anti-inflammatory agent.
There are several reports on the relationship between in vitro activity and in vivo efficacy and safety of antimicrobial peptides49,50,51,52,53,54. A novel peptide, BSN-37, exhibits strong antibacterial activity against S. Typhimurium and significantly inhibits the proliferation of intracellular S. Typhimurium with no associated toxicity to the eukaryotic cells49. A broad-spectrum antimicrobial peptide, GL13K, with low cytotoxicity in vitro exhibits a low level of hemolysis and anti-inflammatory activity in vivo in a mouse model of inactivated LPS-induced sepsis50,51. Although less peptide binding is observed, GL13K is still active in physiological salts conditions52. The in vivo protective effect of buforin II in an experimental rat model of A. baumannii sepsis confirms the in vitro effectiveness of this peptide53. The proline-rich, bovine innate immune peptide, Bac5, is able to kill some mycobacterial species and supports macrophage activation in vitro in synergy with Mycobacterium marinum. Peptide Bac5 is able to slow M. marinum infections of zebrafish54. The in vitro characterization of antimicrobial peptides may be applicable as predictors of in vivo efficacy and safety. Our in vitro data may, similarly, predict the in vivo efficacy and safety of PA-13. To continue the clinical development of this peptide, studies of efficacy and safety in animal models will be conducted.
In conclusion, among rationally designed, short α-helical hybrid peptides inspired by cathelicidin and aurein, PA-13 exhibited potent antibacterial activity against P. aeruginosa, including multidrug resistant strains, via penetration through the bacteria’s membrane and causing depolarization, permeabilization and rupture of membranes, and subsequent leakage of intracellular contents and cell death. The peptide displayed low hemolytic and cytotoxic activities against mammalian cells suggesting a therapeutic potential. Furthermore, PA-13 showed anti-inflammatory activity via LPS neutralization. Our research supports the usefulness of rational design of hybrid peptides as promising candidates for development of novel antimicrobial agents.
References
Rossolini, G. M., Arena, F., Pecile, P. & Pollini, S. Update on the antibiotic resistance crisis. Curr Opin Pharmacol 18, 56–60, https://doi.org/10.1016/j.coph.2014.09.006 (2014).
World Health Organization. Antimicrobial Resistance: Global Report on Surveillance 2014. (2014) Available at, http://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf (Date of access: 24/02/2018).
Zhang, L. J. & Gallo, R. L. Antimicrobial peptides. Curr Biol 26, R14–19, https://doi.org/10.1016/j.cub.2015.11.017 (2016).
Huang, Y., Huang, J. & Chen, Y. Alpha-helical cationic antimicrobial peptides: relationships of structure and function. Protein Cell 1, 143–152, https://doi.org/10.1007/s13238-010-0004-3 (2010).
Waghu, F. H. et al. Designing Antibacterial Peptides with Enhanced Killing Kinetics. Front Microbiol 9, 325, https://doi.org/10.3389/fmicb.2018.00325 (2018).
Li, J. et al. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front Neurosci 11, 73, https://doi.org/10.3389/fnins.2017.00073 (2017).
Zelezetsky, I. & Tossi, A. Alpha-helical antimicrobial peptides–using a sequence template to guide structure-activity relationship studies. Biochim Biophys Acta 1758, 1436–1449, https://doi.org/10.1016/j.bbamem.2006.03.021 (2006).
Lv, Y. et al. Antimicrobial properties and membrane-active mechanism of a potential alpha-helical antimicrobial derived from cathelicidin PMAP-36. Plos One 9, e86364, https://doi.org/10.1371/journal.pone.0086364 (2014).
Hall, K. & Aguilar, M. I. Surface plasmon resonance spectroscopy for studying the membrane binding of antimicrobial peptides. Methods Mol Biol 627, 213–223, https://doi.org/10.1007/978-1-60761-670-2_14 (2010).
Yu, H. et al. Identification and polymorphism discovery of the cathelicidins, Lf-CATHs in ranid amphibian (Limnonectes fragilis). Febs j 280, 6022–6032, https://doi.org/10.1111/febs.12521 (2013).
Kosciuczuk, E. M. et al. Cathelicidins: family of antimicrobial peptides. A review. Mol Biol Rep 39, 10957–10970, https://doi.org/10.1007/s11033-012-1997-x (2012).
Bals, R., Weiner, D. J., Moscioni, A. D., Meegalla, R. L. & Wilson, J. M. Augmentation of innate host defense by expression of a cathelicidin antimicrobial peptide. Infect Immun 67, 6084–6089 (1999).
Conlon, J. M. Structural diversity and species distribution of host-defense peptides in frog skin secretions. Cell Mol Life Sci 68, 2303–2315, https://doi.org/10.1007/s00018-011-0720-8 (2011).
Konig, E., Bininda-Emonds, O. R. & Shaw, C. The diversity and evolution of anuran skin peptides. Peptides 63, 96–117, https://doi.org/10.1016/j.peptides.2014.11.003 (2015).
Pan, Y. L. et al. Characterization of the structure and membrane interaction of the antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys J 92, 2854–2864, https://doi.org/10.1529/biophysj.106.097238 (2007).
Rozek, T. et al. The antibiotic and anticancer active aurein peptides from the Australian Bell Frogs Litoria aurea and Litoria raniformis the solution structure of aurein 1.2. Eur J Biochem 267, 5330–5341, https://doi.org/10.1046/j.1432-1327.2000.01536.x (2000).
Aoki, W. & Ueda, M. Characterization of Antimicrobial Peptides toward the Development of Novel Antibiotics. Pharmaceuticals (Basel) 6, 1055–1081, https://doi.org/10.3390/ph6081055 (2013).
Hilpert, K. et al. Sequence requirements and an optimization strategy for short antimicrobial peptides. Chem Biol 13, 1101–1107, https://doi.org/10.1016/j.chembiol.2006.08.014 (2006).
Liu, Y., **a, X., Xu, L. & Wang, Y. Design of hybrid beta-hairpin peptides with enhanced cell specificity and potent anti-inflammatory activity. Biomaterials 34, 237–250, https://doi.org/10.1016/j.biomaterials.2012.09.032 (2013).
Ong, Z. Y., Wiradharma, N. & Yang, Y. Y. Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Adv Drug Deliv Rev 78, 28–45, https://doi.org/10.1016/j.addr.2014.10.013 (2014).
Kiattiburut, W. et al. Antimicrobial peptide LL-37 and its truncated forms, GI-20 and GF-17, exert spermicidal effects and microbicidal activity against Neisseria gonorrhoeae. Hum Reprod 33, 2175–2183, https://doi.org/10.1093/humrep/dey315 (2018).
Xu, W., Zhu, X., Tan, T., Li, W. & Shan, A. Design of embedded-hybrid antimicrobial peptides with enhanced cell selectivity and anti-biofilm activity. Plos One 9, e98935, https://doi.org/10.1371/journal.pone.0098935 (2014).
Fox, M. A., Thwaite, J. E., Ulaeto, D. O., Atkins, T. P. & Atkins, H. S. Design and characterization of novel hybrid antimicrobial peptides based on cecropin A, LL-37 and magainin II. Peptides 33, 197–205, https://doi.org/10.1016/j.peptides.2012.01.013 (2012).
Steinberg, D. A. et al. Protegrin-1: a broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob Agents Chemother 41, 1738–1742 (1997).
Andrews, J. M. Determination of minimum inhibitory concentrations. J Antimicrob Chemother 48 Suppl 1, 5–16, https://doi.org/10.1093/jac/48.suppl_1.5 (2001) MBC.
Stark, M., Liu, L. P. & Deber, C. M. Cationic hydrophobic peptides with antimicrobial activity. Antimicrob Agents Chemother 46, 3585–3590, https://doi.org/10.1128/aac.46.11.3585-3590.2002 (2002).
Zhang, S. K. et al. Design of an alpha-helical antimicrobial peptide with improved cell-selective and potent anti-biofilm activity. Sci Rep 6, 27394, https://doi.org/10.1038/srep27394 (2016).
**dal, H. M. et al. Antimicrobial Activity of Novel Synthetic Peptides Derived from Indolicidin and Ranalexin against Streptococcus pneumoniae. Plos One 10, e0128532, https://doi.org/10.1371/journal.pone.0128532 (2015).
Rabanal, F. et al. A bioinspired peptide scaffold with high antibiotic activity and low in vivo toxicity. Sci Rep 5, 10558, https://doi.org/10.1038/srep10558 (2015).
Torcato, I. M. et al. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-negative and Gram-positive bacteria. Biochim Biophys Acta 1828, 944–955, https://doi.org/10.1016/j.bbamem.2012.12.002 (2013).
Taylor, P. K., Yeung, A. T. & Hancock, R. E. Antibiotic resistance in Pseudomonas aeruginosa biofilms: towards the development of novel anti-biofilm therapies. J Biotechnol 191, 121–130, https://doi.org/10.1016/j.jbiotec.2014.09.003 (2014).
Breidenstein, E. B., de la Fuente-Nunez, C. & Hancock, R. E. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19, 419–426, https://doi.org/10.1016/j.tim.2011.04.005 (2011).
Ventola, C. L. The antibiotic resistance crisis: part 1: causes and threats. P t 40, 277–283 (2015).
Boman, H. G. Antibacterial peptides: basic facts and emerging concepts. J Intern Med 254, 197–215, https://doi.org/10.1046/j.1365-2796.2003.01228.x (2003).
Li, Y., **ang, Q., Zhang, Q., Huang, Y. & Su, Z. Overview on the recent study of antimicrobial peptides: origins, functions, relative mechanisms and application. Peptides 37, 207–215, https://doi.org/10.1016/j.peptides.2012.07.001 (2012).
Kumar, P., Kizhakkedathu, J. N. & Straus, S. K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the cctivity and biocompatibility in vivo. Biomolecules 8, https://doi.org/10.3390/biom8010004 (2018).
Boland, M. P. & Separovic, F. Membrane interactions of antimicrobial peptides from Australian tree frogs. Biochim Biophys Acta 1758, 1178–1183, https://doi.org/10.1016/j.bbamem.2006.02.010 (2006).
Qu, P. et al. The central hinge link truncation of the antimicrobial peptide Fowlicidin-3 enhances its cell selectivity without antibacterial activity loss. Antimicrob Agents Chemother 60, 2798–2806, https://doi.org/10.1128/aac.02351-15 (2016).
Takahashi, D., Shukla, S. K., Prakash, O. & Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 92, 1236–1241, https://doi.org/10.1016/j.biochi.2010.02.023 (2010).
Teixeira, V., Feio, M. J. & Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog Lipid Res 51, 149–177, https://doi.org/10.1016/j.plipres.2011.12.005 (2012).
Huang, J. et al. Inhibitory effects and mechanisms of physiological conditions on the activity of enantiomeric forms of an alpha-helical antibacterial peptide against bacteria. Peptides 32, 1488–1495, https://doi.org/10.1016/j.peptides.2011.05.023 (2011).
Aquila, M., Benedusi, M., Koch, K. W., Dell’Orco, D. & Rispoli, G. Divalent cations modulate membrane binding and pore formation of a potent antibiotic peptide analog of alamethicin. Cell Calcium 53, 180–186, https://doi.org/10.1016/j.ceca.2012.11.012 (2013).
Yu, H.-Y. et al. Easy strategy to increase salt resistance of antimicrobial peptides. Antimicrob Agents Chemother 55, 4918–4921, https://doi.org/10.1128/AAC.00202-11 (2011).
Chou, S. et al. Short, symmetric-helical peptides have narrow-spectrum activity with low resistance potential and high selectivity. Biomater. Sci 7, 2394–2409, https://doi.org/10.1039/C9BM00044E (2019).
Chou, S. et al. Short, symmetric-helical peptides have narrow-spectrum activity with low resistance potential and high selectivity. Biomater. Sci 7, 2394–2409, https://doi.org/10.1039/C9BM00044E (2019).
Hedegaard, S. F. et al. Fluorophore labeling of a cell-penetrating peptide significantly alters the mode and degree of biomembrane interaction. Sci. Rep 8, 6327–6327, https://doi.org/10.1038/s41598-018-24154-z (2018).
Erridge, C., Bennett-Guerrero, E. & Poxton, I. R. Structure and function of lipopolysaccharides. Microbes Infect 4, 837–851 (2002).
Sun, Y. & Shang, D. Inhibitory effects of antimicrobial peptides on lipopolysaccharide-induced inflammation. Mediators Inflamm 2015, 167572, https://doi.org/10.1155/2015/167572 (2015).
Yang, L. et al. Antibacterial Peptide BSN-37 Kills Extra- and Intra-Cellular Salmonella enterica Serovar Typhimurium by a Nonlytic Mode of Action. Front Microbiol 11, https://doi.org/10.3389/fmicb.2020.00174 (2020).
Hirt, H. & Gorr, S.-U. Antimicrobial peptide GL13K is effective in reducing biofilms of Pseudomonas aeruginosa. Antimicrob Agents Chemother 57, 4903–4910, https://doi.org/10.1128/AAC.00311-13 (2013).
Abdolhosseini, M., Nandula, S. R., Song, J., Hirt, H. & Gorr, S. U. Lysine substitutions convert a bacterial-agglutinating peptide into a bactericidal peptide that retains anti-lipopolysaccharide activity and low hemolytic activity. Peptides 35, 231–238, https://doi.org/10.1016/j.peptides.2012.03.017 (2012).
Balhara, V., Schmidt, R., Gorr, S.-U. & DeWolf, C. Membrane selectivity and biophysical studies of the antimicrobial peptide GL13K. Biochim. Biophys. Acta 1828, 2193–2203, https://doi.org/10.1016/j.bbamem.2013.05.027 (2013).
Cirioni, O. et al. Therapeutic efficacy of buforin II and rifampin in a rat model of Acinetobacter baumannii sepsis. Crit Care Med 37, 1403–1407, https://doi.org/10.1097/CCM.0b013e31819c3e22 (2009).
Price, R. L. et al. In vitro and in vivo properties of the bovine antimicrobial peptide, Bactenecin 5. Plos One 14, e0210508, https://doi.org/10.1371/journal.pone.0210508 (2019).
Acknowledgements
The authors gratefully acknowledge the financial support provided by Thai Goverment Research Fund Contract No. 38/2561 and 21/2562 for fiscal year 2018-2019. This work was partially supported by Research Unit in Antimicrobial Agent and Application, Thammasat University. Ratchaneewan Aunpad was supported by TRF Research Career Development Grant (RSA6080033). Natthaporn Klubthawee was supported by the Thailand Research Fund under the Royal Golden Jubilee Ph.D. Programme (PHD/0015/2558, 5.L.TU/58/I.1). We would like to thank Dr. Arthur Brown, Faculty of Medical Technology, Mahidol University, Thailand, for providing language assistance.
Author information
Authors and Affiliations
Contributions
Natthaporn Klubthawee: Investigation, Methodology, Writing - original draft; Poom Adisakwattana: Resources, Supervision; Warunee Hanpithakpong: Investigation, Validation; Sangdao Somsri: Methodology; Ratchaneewan Aunpad: Conceptualization, Funding acquisition, Investigation, Validation, Writing - original draft, Writing - review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klubthawee, N., Adisakwattana, P., Hanpithakpong, W. et al. A novel, rationally designed, hybrid antimicrobial peptide, inspired by cathelicidin and aurein, exhibits membrane-active mechanisms against Pseudomonas aeruginosa. Sci Rep 10, 9117 (2020). https://doi.org/10.1038/s41598-020-65688-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65688-5
- Springer Nature Limited
This article is cited by
-
Dual-function antimicrobial-antibiofilm peptide hybrid to tackle biofilm-forming Staphylococcus epidermidis
Annals of Clinical Microbiology and Antimicrobials (2024)
-
One-pot synthesis of alginate-antimicrobial peptide nanogel
Photochemical & Photobiological Sciences (2024)
-
LL-37_Renalexin hybrid peptide exhibits antimicrobial activity at lower MICs than its counterpart single peptides
Applied Microbiology and Biotechnology (2024)
-
In vitro and in vivo antibiofilm activity of the synthetic antimicrobial peptide WLBU2 against multiple drug resistant Pseudomonas aeruginosa strains
BMC Microbiology (2023)
-
Molecular hybridization strategy for tuning bioactive peptide function
Communications Biology (2023)