
Abstract
Phlebotomine sand flies are remarkable vectors of several etiologic agents (virus, bacterial, trypanosomatid Leishmania), posing a heavy health burden for human populations mainly located at develo** countries. Their intestinal microbiota is involved in a wide range of biological and physiological processes, and could exclude or facilitate such transmission of pathogens. In this study, we investigated the Eubacterial microbiome from digestive tracts of Lu. evansi adults structure using 16S rRNA gene sequence amplicon high throughput sequencing (Illumina MiSeq) obtained from digestive tracts of Lu. evansi adults. The samples were collected at two locations with high incidence of the disease in humans: peri-urban and forest ecosystems from the department of Sucre, Colombia. 289,068 quality-filtered reads of V4 region of 16S rRNA gene were obtained and clustered into 1,762 operational taxonomic units (OTUs) with 97% similarity. Regarding eubacterial diversity, 14 bacterial phyla and 2 new candidate phyla were found to be consistently associated with the gut microbiome content. Proteobacteria, Firmicutes, and Bacteroidetes were the most abundant phyla in all the samples and the core microbiome was particularly dominated by Methylobacterium genus. Methylobacterium species, are known to have mutualistic relationships with some plants and are involved in sha** the microbial community in the phyllosphere. As a remarkable feature, OTUs classified as Wolbachia spp. were found abundant on peri-urban ecosystem samples, in adult male (OTUs n = 776) and unfed female (OTUs n = 324). Furthermore, our results provide evidence of OTUs classified as Cardinium endosymbiont in relative abundance, notably higher with respect to Wolbachia. The variation in insect gut microbiota may be determined by the environment as also for the type of feeding. Our findings increase the richness of the microbiota associated with Lu. evansi. In this study, OTUs of Methylobacterium found in Lu. evansi was higher in engorged females, suggesting that there are interactions between microbes from plant sources, blood nutrients and the parasites they transmit during the blood intake.
Similar content being viewed by others

Introduction
The current situation of leishmaniasis and difficulties for its control1, requires integral explorations of various aspects of the vector and parasite lifestyle, nutrient sources, diversity and interplay between insect, feeding regimes and resources, microbiomes, niches, modes of transmission, urban adaptation and influence of human built environments, among others2. One initial step to tackle this issue is to describe the microbial diversity associated with the intestine of insect vectors recognized for the transmission of parasites3,4,5,6. Such knowledge may contribute to understand and to design new alternative control methods, as the ones currently available are evidently insufficient7. In Colombia, there are around 14 species of Lutzomyia (Lu.) reported as vectors of Leishmania sp8,9,10. Only the intestinal microbiota of a wild population of Lu. longipalpis, and Lu. evansi from caribbean coast have been recently reported in other studies8,11.
Lu. evansi, is recognized because to the vectorial competence that has to transmit parasites causing visceral leishmaniasis in rural and urban environments of the Caribbean coast of Colombia11,12,13,14. Its abundance and epidemiological importance also makes it an attractive biological model for studies exploring the influence of intestinal microbiota on the fitness, survival, reproduction, parasite transmission and infection establishment in the mammalian host15. Therefore, microbiome-based approaches to control vector transmission of Leishmania infantum could provide new alternatives and solutions.
Given the environmental plasticity of Lu. evansi, the corresponding intestinal microbial communities of this species may require further characterization to see if a similar complex, shifting and diverse composition is observed or if there is a core microbiome with patterns stably maintained between individual specimens. In this context, microbiome studies of insect vectors largely relied on culture techniques used for microbial growth of species aerobic mainly and use to identify associations symbiotic, pathogenic and vectoring. Nevertheless, this approach addresses a limited proportion of the bacterial representation that can be grown on standard conditions, restricting our current knowledge about the microbial communities in insect guts3,16,17.
Bacterial diversity analysis by culture-independent means in wild Lutzomyia spp. are still scarce. In America, there are reports for sand flies phlebotomine, considering sequences of the 16S rRNA gene, DGGE and NGS with 16S rDNA amplicons8,18,19. Gut bacterial types detected on these species are mainly belonging to Serratia, Enterobacter, Acinetobacter, and Pseudomonas genera8,18,19, probably representing a partial or modified microbiome diversity of the original wild insect population, as studies referred were made in laboratory colonies.
The bacterial diversity of Lu. intermedia sand fly from an endemic area in Brazil using metagenomics sequencing was also studied by Monteiro et al. 2016, indicating the presence of genera as Ochrobactrum and Bradyrhizobium across all the groups5. Recently, the bacterial gut composition from Lu. evansi using cultured and uncultured approaches was described, being similar to the reported above11. Ochrobactrum, Shinella, Paenibacillus, Lysobacter, Microbacterium, Streptomyces, Bacillus, Rummeliibacillus, Staphylococcus, Brevibacterium, and Pantoea were also reported, including microbiota analyses immature states11.
Recently, alternative options to the chemical control of insect vectors as sand flies include advances in insect genomics and transformation technology providing new strategies for the control of insect-borne pathogen transmission and insect pest management19,20 such as the genetic modification of insects with genes that block pathogen development. Another strategy consist of suppressing insect populations by releasing either sterile males or males carrying female-specific dominant lethal genes into the environment. Considering that arthropods harbor a rich microbiota, its potential egestion after bites may be a shared mechanism that contributes to severity of vector-borne disease, thus, in-depth knowledge of sand flies gut microbiota could help to design new strategies for disease transmission21.
Recent studies have highlighted the capacity of endogenous bacteria to modulate viral and parasitic infections in mosquito (Ae. Aegypti, An. gambiae) and sand flies vectors (Lu. longipalpis, P. papatasi) by activating their immune responses (Toll pathway) or directly inhibiting pathogen development22, β-diversity analysis of microbial communities associated with the groups of guts established of Lu. evansi. (a) Constrained Analysis of Principal Coordinates (CAP) of 16S rRNA data (b) Principal Coordinate Analysis (PCoA) of Bray-Curtis dissimilarities of 16S rRNA data. Statistical significance of the CAP and PCoA was assessed by Permanova (P < 0.005).
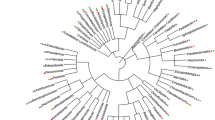
The principal components analysis (PCA) showed the composing the core microbiome (Figs 4, 8). Fed and unfed females of Lu. evansi from peri-urban and forest biotypes has closer associations mainly with S. marcenscens, M. organiphilum (Three different OTUs), Unclassified renibacterium, Unclassified intrasporangiaceae. Unlike the samples of Lu. evansi appearing closely related to OTUs of Unclassified Bacillus, Unclassified EC94, Unclassified Pseudomonadaceae y Unclassified Enterobacteriaceae (Fig. 8).

Principal Component Analysis (PCA) showing the distribution of the core microbiome (B) associated with the groups of guts established of Lu. evansi. The PCA revealed mainly a cluster matching with fed and unfed females of both locations. Also revealed a cluster with samples of males and unfed. Nomenclature of gut pools: COM: males from Colosó; COFF: fed females from Coloso; COUF: Unfed females from Coloso; OVM: males from Ovejas; OVFF: fed females from Ovejas; OVUF: unfed females from Ovejas.
Discussion
Lu. evansi is an important vector insect in the transmission cycle of visceral leishmaniasis in the Caribbean coast of Colombia, its control is a health priority. For this reason, a complete analysis of their intestinal bacterial microbiome was developed in this work, in order to find possible alternatives for its biological control. A detailed understanding about how pathogens interact with their vectors and the resident microbes can lead to the discovery of new tools to block disease transmission35. The first and crucial step in this approach is the identification of suitable commensal bacteria in the vector.
Previous reports of gut microbiome in female sandflies from South America have been performed applying classic techniques of molecular ecology such as DGGE and band sequencing of 16S rRNA gene fragments, and some bacterial types (Enterobacter, Bacillus, Serratia) are usually found in plants and soils, including some sequences of potential plant pathogens or species with entomopathogenic potential8,11,35. In this study, we investigated the Eubacterial microbiome from digestive tracts of Lu. evansi adults structure using 16S rRNA gene sequence amplicon high throughput sequencing (Illumina MiSeq).
Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes were the most abundant phyla in all Lu. evansi samples. This result is in good agreement with a previous study on the gut microbiota of 81 insects, showing that Proteobacteria and Firmicutes were the predominant phyla, comprising 57.4% and 21.7% of sequences, respectively36,37. It is also consistent with microbiota reported to be associated to different species of Lutzomyia and Phlebotomus genus, as well of mosquitoes (Aedes and Anopheles) although, Bacteroidetes and Actinobacteria phylum showed lower abundance with respect to our study3,5,19,38.
Gut microbiomes on Lu. evansi clearly show such abundant phyla shared across all the samples, but also a large fraction of phyla with quite distinct and variable composition even between similar sample groups. The dynamic variation in insect gut microbiota could be explained due to the influence of microhabitats, gut morphology and physicochemical conditions, such as pH, oxygen availability in the insect gut, as well as type of diet39. These diverse gut conditions may cause variation in host-specific gut microbiota in insects, influencing nutritional and immune system status as well as reproductive fitness15.
Regarding the presence of constant OTUs shared among all Lu. evansi samples, notably most dominant Methylobacterium, a common cosmopolitan inhabitant of phyllospheres of plants (endophytic, transporting and rhizosphere tissues), thus suggesting the constant feeding from plants in specimens collected40,41. Recently, the family Methylobacteriaceae was reported with low representation in (gravid and fed females) Lu. intermedia, in an endemic area of leishmaniasis in Brazil5,38. These findings together allowed us to suggest that Methylobacteriaceae have the capabilities to remain into the midgut during physiological processes carried out during the blood intake, proposal backed up by experimental evidence of the study published by Kelly et al., 2017, showing the continued presence or even increase in abundance of Methylobacteriaceae after the sixth day of feeding and infection with Leishmania infantum parasites in Lu. longipalpis19.
The relative abundance of Methylobacterium is significant in salivary glands and midgut of mosquitoes infected and non-infected with P. falciparum, which suggests that interactions occur between microbes and parasites42. The above indicates that Methylobacterium can be important in several physiological and metabolic processes in Lu. evansi, which suggests that interactions could occur with Leishmania parasite. More studies are needed to deepen in these interactions.
Studies on distribution of Wolbachia among its arthropod hosts are important both for better understanding why this bacterium is so common43,44, as well as for its potential use as a biological control agent. In America, Wolbachia infection has been observed previously in Lu. cruciata, Lu. shannoni, Lu. vespertilionis, Lu. trapidoi, Lu. longipalpis, Lu. whitmani, and Lu. intermedia19,23,45. One of the main objectives of this study was to confirm, in a new set of samples and with a higher resolution experimental approach the natural presence of Wolbachia in the microbiome of Lu. evansi specimens wild populations collected in Colombia (department of Sucre). Wolbachia endosymbiont was detected in adult male and unfed female specimens, distinctly found only in samples coming from Ovejas. A high proportion of Wolbachia was determined, which allows to confirm the preliminary results by conventional PCR46, supporting the need of further studies to evaluate the natural infection by Leishmania and the presence of this symbiont simultaneously.
Recently, in groups of females fed from Lu. intermedia, the family Rickettsiaceae was the most prevalent, with 46.7% of the group represented by Wolbachia5. This group of females midgut microbiomes was also analyzed by Illumina 16S rRNA gene amplicon sequencing. Wolbachia is the microbiota genus most enhanced by the presence of blood meal in the midgut5. This is in stark contrast with our study of Lu. evansi and for Kelly et al. 2017 about Lu. longipalpis, in which Wolbachia spp., were identified only in sucrose-fed flies and not in blood-fed or infected flies19,46.
Despite several reports about the distribution of intracellular symbionts that regulate host reproduction, such as Wolbachia, there are few studies addressing the broad distribution of other specific symbionts on insect hosts47,48. Among these symbionts, we would like to remark the case of Cardinium, which is reported for the first time for Lu. evansi, and not reported so far in any other insect vector in Colombia. It is showing a variability between similar groups (ex: Cardinium present in one group of OVM, but not in the other one). Wild populations of Lu. longipalpis from Brazil had been reported as hosts of this bacterial type37. ‘Candidatus Cardinium’ is a recently described bacterium from the Bacteroidetes group involved in diverse reproduction alterations of its arthropod hosts (including cytoplasmic incompatibility, parthenogenesis, and feminization) similar to Wolbachia49. Therefore, this finding could have potential and significance for biological control design. Cardinium is also vertically transmitted to progeny but rarely show co-speciation with the host. In sap-feeding insects, as phlebotomine sandflies, plant tissues have been proposed as alternative horizontal routes of interspecific transmission, but experimental evidence is limited50.
This remarks the potential of these intracellular bacteria to be studied and considered as possible controlling agents51,52; and allows setting new platforms and perspectives to further expand the observations to correlate its presence and abundance in a larger sampled population where Leishmania content is also defined on each specimen. Given the recent reports by Kelly et al. (2017) and Louradour et al. (2017), that providing experimental evidence about the requirement of a gut microbiome linked to the survival of L. infantum in Lu. longipalpis and of L. major in P. duboscqi sand flies, respectively19,53, this kind of conditioning and controlling of parasite hosting, mediated by the insect gut microbiome, could also may be happening in wild populations of Lu. evansi. Further efforts are underway to continue exploring these possibilities.
The presence of OTUs of Oxalabacteraceae and Staphylococcus in Lu. evansi, is also very remarkable, mainly in males and unfed females. Oxalobacteraceae is a family within the order Burkholderiales in the subclass Betaproteobacteria that contains 13 genera. Possibly these OTUs were acquired by Lu. evansi during the larval stage and remained in the intestine until adult. These results correlate with the previous finding of Burkholderia cenocepacia in larvae11.
It is notable that Staphylococcus genera has been associated only to the intestinal microbiota of sandflies from Old World (Phlebotomus argentipes, Phlebotomus chiniensis, Phlebotomus papatasi)6, but with significant representation of OTUs, which is similar to the results obtained in Lu. evansi. Staphylococcus has a variable consistency between hosts, and is mainly related to plants and sap suggesting that they are acquired through sugar feeding39.
Microbiota can vary between sexes, feeding status, origin, and status of Leishmania infection. Sequencing by Illumina MiSeq platform, allowed us to define the composition of the intestinal microbiota of phlebotomines in the wild54. The results presented here open up a promising path to continue the studies on these and other vector species of Leishmania in aspects such as enzymatic and antimicrobial activities, influence of microbiota on the resistance to insecticides, and especially, the recalcitrant or favorable effects that specific microbiome components may exert on vector competence.
