
Abstract
Alzheimer’s disease (AD), the most common age-related neurodegenerative disorder, is currently conceptualized as a disease of synaptic failure. Synaptic impairments are robust within the AD brain and better correlate with dementia severity when compared with other pathological features of the disease. Nevertheless, the series of events that promote synaptic failure still remain under debate, as potential triggers such as β-amyloid (Aβ) can vary in size, configuration and cellular location, challenging data interpretation in causation studies. Here we present data obtained using adeno-associated viral (AAV) constructs that drive the expression of oligomeric Aβ either intra or extracellularly. We observed that expression of Aβ in both cellular compartments affect learning and memory, reduce the number of synapses and the expression of synaptic-related proteins, and disrupt chemical long-term potentiation (cLTP). Together, these findings indicate that during the progression AD the early accumulation of Aβ inside neurons is sufficient to promote morphological and functional cellular toxicity, a phenomenon that can be exacerbated by the buildup of Aβ in the brain parenchyma. Moreover, our AAV constructs represent a valuable tool in the investigation of the pathological properties of Aβ oligomers both in vivo and in vitro.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the most common age-related neurodegenerative disorder, afflicting approximately 5.5 million individuals in the USA and with a new case develo** every 66 seconds1. The pathophysiologic mechanisms of AD begin long before symptom onset provides an opportunity to apply advanced imaging and biomarker methods to diagnose individuals during the preclinical stages of neurodegeneration. All AD brains are characterized by two main hallmarks: plaques, consisting of extracellular deposits of β-amyloid (Aβ), and tangles, aggregates of hyperphosphorylated tau2. A fundamental mechanism in AD pathogenesis is synapse failure3,4, and several studies suggest that Aβ oligomers can interact with different proteins, ultimately leading to synapse toxicity, alterations in long-term potentiation (LTP), and cognitive impairments5,6,7,8. Also, significant synaptic loss is already observed in patients with mild cognitive impairment (MCI), and progressive loss is the most robust hallmark that correlates with cognitive decline in AD9,10. However, the underlying molecular mechanisms that impair the synaptic function, and cause their loss at early stages of the disease, are poorly understood. Dendritic spines are specialized anatomical structures in neuronal cells that serve as the postsynaptic component for the vast majority of CNS synapses, and are major sites of processing and storage of information in the brain2. Structural changes at dendritic spines underlie learning and memory processes in the brain, and alterations in spine structure and function might lead to cognitive impairments4.
A continuing debate is whether intra- or extracellular Aβ is more deleterious to the brain. It is known that intraneuronal Aβ accumulation occurs early in adult life in basal forebrain cholinergic neurons, increasing with both aging and AD in the presence of intermediate and large oligomeric states11. The early intraneuronal accumulation has also been observed in transgenic rats McGill-R-Thy1-APP, which harbor the human APP751 transgene with the Swedish and Indiana mutations under the control of the murine Thy1.2 promoter12. In these animals, the intraneuronal pathology was due to a mixture of APP, CTFs and a considerable amount of Aβ, leading to deleterious effects in the CNS before amyloid plaques develop. Alternatively, additional studies suggest that intraneuronal Aβ precedes plaques in brain areas affected early in the disease13, is associated with synaptic deficits14, and can be secreted in a prion-like manner15. There is evidence of the accumulation of extracellular Aβ in an age-dependent manner16. Exogenous oligomers accumulate particularly at synaptic spines, and presynaptic sites can also be targeted17. However, as plaques correlate poorly with AD-related dementia and little is known about the effects of intracellular Aβ on cognitive function, we asked whether these two distinct accumulation sites could have different effects on the development of pathology.
Several animal models of AD employ an approach based on the overexpression of mutant human amyloid precursor protein (APP) or presenilin to increase Aβ expression18. Although these have been very useful and are still fundamental to the understanding of AD development, increasing the expression of Aβ42 via AAV-mediated gene transfer has been demonstrated useful in develo** AD animal models19,20. In this study, we used this method to overexpress Aβ42 oligomers in mouse hippocampus and neuronal cultures and to analyze their effect in synaptic function and cognitive impairments. Overall, our findings reveal a faster yet physiologically relevant model that show that synaptic impairment and cognitive decline initiates as soon as Aβ accumulates in the intracellular compartment in AD.
Results
Characterization of Aβ expression following incubation and injection of AAV-BRI-Aβ42 and AAV-UBI-Aβ42 vectors
The detailed description of generating AAV constructs used here was reported previously19,20,21. AAV vectors encoding BRI-Aβ cDNAs, fusions between human Aβ peptides and the BRI protein (known to be associated with amyloid deposition in British familial dementia), are able to promote high-level expression of Aβ peptide in the absence of APP overexpression19. AAV-BRI-Aβ42 and AAV-UBI-Aβ42 were created to facilitate the expression of extracellular and intracellular Aβ, respectively22.
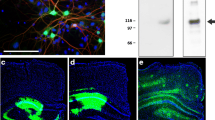
Prior to in vivo testing, we incubated hippocampal neuronal cell cultures with AAV constructs encoding BRI-Aβ42 or UBI-Aβ42 fusion proteins in order to determine the optimal concentration and efficacy for each construct. Exogenous synthetic oligomers of Aβ were used as a positive control. We detected significant levels of Aβ42 in the culture medium using three concentrations of BRI-Aβ42, in contrast to the UBI-Aβ42 and EGFP control AAV constructs (Fig. 1a). We also measured the expression of Aβ oligomers in cell media and lysate of cell cultures. There is no significant increase in the cell media after incubation of either AAV constructs, even though there is an increased trend in the AAV-BRI-Aβ42 in accordance with what has been observed in Fig. 1a. We observed an increase in the levels of oligomers in the cell lysate after incubation with AAV-BRI-Aβ42 compared to EGFP (Fig. 1c). With these results we confirmed the ability of these constructs to promote the overexpression of Aβ peptides.

Expression of Aβ by AAV-BRI-Aβ42 and AAV-UBI-Aβ42 in hippocampal neuronal cell cultures. (a) Total Aβ42 levels in the media after AAV constructs incubation. AAV-BRI-Aβ42: [1] 4.7 × 1010 genome particles/ml, [2] 9.3 × 1010 genome particles/ml, and [3] 2.8 × 1011 genome particles/ml. AAV-UBI-Aβ42: [1] 1.5 × 1010 genome particles/ml, [2] 7.7 × 1010 genome particles/ml, [3] 4.6 × 1011 genome particles/ml. AAV-BRI-Aβ42 [1] and [2] promoted a 37-fold and 465-fold increase and AAV-UBI-Aβ42 [3] promoted an 18-fold increase in Aβ42 levels when compared to those elicited by EGFP. In comparison to AAV-BRI-Aβ42 [2], the concentration [3] promoted a 3-fold decrease in the levels of Aβ42. Compared to EGFP, Aβ 10 mM elicited a 669-fold increase (*p < 0.0001). (b) No significant change was observed in Aβ42 oligomers in the cell media after incubation of either AAV constructs. (c) AAV-BRI-Aβ42 promoted a significant increase in the levels of Aβ42 oligomers in the cell lysate compared to EGFP (*p < 0.0001).
Next, we tested the AAV-BRI-Aβ42 and AAV-UBI-Aβ42 expression in vivo by determining the Aβ42 relative levels in the soluble and insoluble fractions of mice hippocampus after AAVs injection. Mice were divided into 3 cohorts: AAV-BRI-Aβ42-treated, AAV-UBI-Aβ42-treated or AAV-EGFP-treated. Each subject received bilateral intrahippocampal injection of a single specific AAV construct, and the brains were collected and analyzed 3 months later. The overexpression of BRI-Aβ42 construct resulted in higher expression of both soluble and insoluble Aβ42 as compared to the UBI-Aβ42 construct, while there was no detectable Aβ following EGFP incubation (Fig. 2a,b). Immunostaining for 6E10 in the BRI-Aβ42 construct demonstrated a high amyloid deposition in the hippocampus. However, animals that received the UBI-Aβ42 presented distinct neuronal processes staining with mild intraneuronal accumulation of Aβ (Fig. 2c), without accumulation of Aβ deposits23.

Expression of Aβ by AAV-BRI-Aβ42 and AAV-UBI-Aβ42 in mice hippocampus. (a,b) Both soluble and insoluble fractions of the mouse hippocampus show an increase in Aβ42 levels after AAV-BRI-Aβ42 or AAV-UBI-Aβ42 transfection (*p < 0.0001). (c) Light microscopy images of the hippocampus (CA1 region) immunostained with anti-Aβ antibody (6E10) of ntg mice treated with EGFP, AAV-BRI-Aβ42 or AAV-UBI-Aβ42.
Hippocampal Aβ expression promoted by AAV constructs leads to impaired cognition
Animals treated with both Aβ AAV constructs presented significant cognitive impairment, measured by performance in the Morris Water maze test (Fig. 3). In this evaluation, both groups took more time to find the hidden platform – as demonstrated by latency (Fig. 3b) – and also crossed the platform fewer times (Fig. 3c). The injection of both vectors did not affect motor skills, as demonstrated by velocity and distance evaluation (Fig. 3d,e). These results are significant and demonstrate the importance of both intra- and extracellular Aβ in the development of spatial cognitive impairments. There are no significant changes with contextual fear conditioning (Fig. 3f).

Aβ AAV-mediated gene transfer impair cognition and LTP in ntg mice. (a) Mice were trained on the spatial reference version of the Morris water maze at 8–9 months of age. Acquisition curves for the 5 days of training show significant differences in groups treated with AAV-BRI-Aβ42 and AAV-UBI-Aβ42 compared to EGFP (*p < 0.0001). (b) Animals in the AAV-BRI-Aβ42 and AAV-UBI-Aβ42 groups had an increase in the latency to reach the hidden platform compared to EGFP group (175.82% ± 20.86 and 156.00% ± 20.13, respectively; *p = 0.0127). (c) A significant decrease in frequency was observed in the AAV-BRI-Aβ42 and AAV-UBI-Aβ42 groups (30.22% ± 9.49 and 36.06% ± 3.58, respectively; *p = 0.0016). (d) No differences were observed between groups in distance or velocity behavior. Values represent the mean ± S.E.M (n = 10 per group).
In vitro synaptic function is altered by both BRI-Aβ42 and UBI-Aβ42
As Aβ is known to cause dendritic spine density and synaptic function changes24, we next examined if the cognitive impairments observed in our animals were associated with functional or structural changes in dendritic spines or specific synaptic markers. To determine possible mechanisms by which the AAV-driven expression of Aβ42 could impair cognition, we evaluated the effect of BRI-Aβ42 and UBI-Aβ42 expression on long-term potentiation (LTP), the best-known cellular correlate of memory25. We used FASS-LTP, a flow cytometry-based method that quantifies surface GluA1 expression in isolated synaptosomes following chemical LTP (cLTP)26,27,28. Synaptosomal cLTP is based on application of the NMDA receptor co-agonist glycine, which facilitates NMDA receptor activation29. By flow-cytometry, FASS-LTP selectively and reliably identifies synaptosomal particles (subset of particles of ~1.0 μm) using calibrated beads (Fig. 4a,b), and quantifies potentiated synaptosomes by extracellular labeling of the AMPA receptor subunit GluA1 and neurexin-1β (Nrx1β)26,27, a presynaptic adhesion molecule stabilized at the membrane surface by synaptic activity30. GluA1 and Nrx1β double-labeling ensures the analysis of intact synaptosomes that contain both pre- and postsynaptic elements. In synaptosomes of mice treated with the control AAV-EGFP vector, cLTP stimulation increased the proportion of synaptosomes expressing both GluA1 and Nrx1β at the surface (GluA1 + Nrx1β+), relative to non-stimulated synaptosomes (basal condition, Fig. 4d). In contrast, the levels of potentiated GluA1 + Nrx1β+ synaptosomes were similar in the basal and cLTP conditions in the BRI-Aβ42 and UBI-Aβ42 groups (P > 0.05, Fig. 3d), thereby indicating that both BRI-Aβ42 and UBI-Aβ42 impair LTP mechanisms directly at the synapse.

AAV-BRI-Aβ42 and AAV-UBI-Aβ42 induce functional, morphological, and structural synaptic alterations. (a) Flow cytometry FASS-LTP identifies synaptosomes by size. Based on calibrated beads, we set a threshold in the forward scatter (FSC-H) channel, as well as a gate region. We excluded small particles having a size equivalent to 0.5 µm calibrated beads (red particles), while selecting particles of ~ 1.0 µm calibrated beads (blue particles). (b) Forward-Side (FSC-SSC) profile of particles in the synaptosomal P2 fraction isolated from the hippocampus. The inside rectangle (gate) selects putative synaptosomes according with to size (~1.0 µm = size-gated synaptosomes). (c) In size-gated synaptosomes, FASS-LTP identifies potentiated synapses by tracking GluA1 and Nrx1β surface staining. To induce cLTP in samples from each experimental group (EGFP, AAV-BRI-Aβ42 and AAV-UBI-Aβ42 samples were run in parallel), synaptosomal P2 fractions maintained in Mg2+ -free external solution were sequentially stimulated using 500 µM glycine (15 min) and 37 mM KCl (30 min). As controls, equivalent volumes of external solution were added to a parallel set of synaptosomal fractions maintained in external solution (basal). Representative two-color parameter plots show GluA1 (x-axis) and Nrx1β (y-axis) surface levels in basal and cLTP conditions. Thresholds for endogenous/non-specific fluorescence for each marker were set by staining with secondary antibodies only. (d) Values normalized to the basal state in each experimental group, mean ± SEM. Basal vs cLTP: EGFP, *p = 0.041 (n = 6); AAV- BRI-Aβ42, P = 0.244 (n = 7); AAV-UBI-Aβ42, P = 0.461 (n = 6).
Both BRI-Aβ42 and UBI-Aβ42 drive synapse loss in vivo
Consistent with the suppression of synaptosomal cLTP by BRI-Aβ42 and UBI-Aβ42, we found that our AAV constructs reduced the total number of spines (Fig. 5a), as well as that of stable mushroom spines (Fig. 5b). The BRI-Aβ42 construct also significantly reduced stubby (Fig. 5c) and filopodia-like spines (Fig. 5d), whereas the UBI-Aβ42 construct did not affect the stubby spines, i.e. there was a higher number of these immature spines in this group. In addition, we observed reduced protein levels of the pre- and postsynaptic markers PSD-95 and synaptophysin, respectively (Fig. 5f,g), as well as of profilin-1 (Fig. 5h), an actin cytoskeleton protein, suggesting that impairments in synaptic function and strength could be related to intra- and extracellular Aβ.

Impairments in synaptic number and synaptic proteins are related to Aβ overexpression via AAV-mediated gene transfer. (a–d) Stereological quantification showed a significant decrease in total spines for the AAV-BRI- Aβ42 and AAV-UBI- Aβ42 groups (67.65% ± 5.06; 43.02% ± 4.93; *p < 0.0001) when compared to AAV-EGFP-transfected mice. There was also a significant decrease in mushroom (BRI-Aβ42: 44.09% ± 4.83; UBI-Aβ42: 40.13% ± 9.78; *p = 0.0021), stubby (BRI-Aβ42: 62.34% ± 4.20, *p = 0.0015) and filopodia-like spines (BRI-Aβ42: 76.72% ± 0.59; UBI-Aβ42: 62.53% ± 8.42; *p < 0.0001) compared to the number observed in EGFP-transfected mice. Notably, there was no significant difference in stubby spines in the AAV-UBI-Aβ42 group. The values represent the mean ± SEM (n = 5 per group). (e) Light microscopic 3D reconstruction images of dendritic spines in the CA1 subfield in AAV-EGFP, AAV-BRI- Aβ42 and AAV-UBI- Aβ42 mice. (f–i) Immunoblot analysis of hippocampal homogenates of 8–9 month-old mice, normalized to GAPDH and expressed as arbitrary units, showing a significant reduction in the expression of PSD-95 (BRI-Aβ42: 37.20% ± 9.19; UBI-Aβ42: 44.80% ± 5.35; *p = 0.0028), synaptophysin (BRI-Aβ42: 43.69% ± 9.75; UBI-Aβ42: 39.82% ± 6.46; *p = 0.0030) and profilin-1 (BRI-Aβ42: 51.87% ± 8.00; UBI-Aβ42: 42.04% ± 11.13; *p = 0.0075) compared to the EGFP group. Quantification of western blots was performed via densitometric analysis and is presented as arbitrary units, normalized to GAPDH. Colored circles represent the groups tested: white - EGFP; yellow - BRI-Aβ42; blue - UBI-Aβ42. Values represent the mean ± SEM (n = 4 per group).
Discussion
The findings of this study imply that intra- and extracellular Aβ accumulation mediated by AAV-gene transfer can promote deleterious effects on synaptic and cognitive functions. Our study indicates that these AAV constructs of Aβ42 induce memory impairments, alongside reductions in the number of spines and of proteins related to synaptic function and LTP. These results are significant as they demonstrate that mild intraneuronal accumulation of Aβ42 (UBI-42) is as potent as combined intraneuronal and extracellular accumulation (BRI-42), significantly impacting our understanding of the detrimental aspects of AD.
The view that insoluble Aβ fibrils are the major factor in AD pathogenesis was firmly held until prefibrillar soluble Aβ oligomers were shown to be more detrimental in some experimental settings31,32,33,34,35. These oligomers can interact with glutamate receptors, dysregulating calcium influx and also altering spine morphology and density36. Moreover, when extracted directly from AD brains and injected into rodent brains, they can inhibit LTP, enhance long-term depression (LTD), and reduce dendritic spine numbers37. Although the potential role of extracellular Aβ oligomers is more well-established, there is also evidence that intracellular Aβ and its deposition precede the formation of plaques in animal models and human brains38. We observed a significant increase in both soluble and insoluble fractions of Aβ, particularly with the BRI-Aβ42 construct. Moreover, this construct also induced the formation of plaques as well as intraneuronal accumulation of Aβ in the CA1 area of the hippocampus.
The first reports describing the existence of intracellular Aβ appeared shortly after the original identification of Aβ as the main component of plaques. Since then, other studies have provided evidence for intracellular Aβ accumulation in transgenic mouse brain and in post-mortem brain samples from AD and Down syndrome patients. A rat model with a doubly mutated APP, driven by a Thy1.2 promoter highly expressed in neurons, triggered the accumulation of intraneuronal human Aβ in 2–3-month-old rats, coinciding with cognitive impairments and pre-plaque generation39. Further evidence linking intraneuronal Aβ accumulation to cognitive deficits and synaptic dysfunction came from transgenic mouse models38,40,41,42. However, the overexpression of specific genes and the subsequent formation of plaques and/or tau pathology in such models make it hard to attribute the associated cognitive impairments they promote exclusively to the presence of intracellular Aβ. Our results are significant because, through the promotion of both intra- and extracellular Aβ, we can conclude that the accumulation of Aβ peptides, even in young mice, is sufficient to promote behavioral and synaptic impairments.
Synaptic deficits and loss are the pathological hallmarks that best correlate with the progressive cognitive decline observed in AD patients43,44,45. Here, we have demonstrated that synaptic function as measured by FASS-LTP is affected very early. We used FASS-LTP, an approach to evaluate chemically-induced LTP directly in isolated synaptosomes. FASS-LTP identifies the subset of synaptosomes that are double-labeled for surface GluA1 and Nrx1β (GluA1 + Nrx1β +). Importantly, the increase in surface GluA1 + Nrx1β+ levels after cLTP stimulation is sustained, and mechanistically parallels the facilitation of synaptic transmission following electrically induced LTP (e.g., dependence on NMDAR and CaMKII), as previously shown26,27. Specifically, the generation of cLTP was reduced significantly in the presence of either intra- and extracellular Aβ. Our results are in accordance with recent findings, whereas LTP was blocked by the intracellular injection of Aβ42 into hippocampal pyramidal cells46, and by extracellular Aβ at the CA3-CA1 synapses of APP-knockout mice47. Moreover, the Aβ42-induced impairment of glutamatergic synaptic function is dependent on its internalization and intracellular accumulation48.
Information storage underlying learning and memory, as well as signal transduction at excitatory synapses, develop at the postsynaptic density (PSD)49,50. Within PSDs, the most abundant scaffolding protein is PSD-95, which is known to play key roles in synaptic plasticity51,52. In the pre-synapse, the loss of synaptophysin is one of the best brain correlates of cognitive decline in AD53, occurring early in the development of disease and accompanied by increased APP and hyperphosphaorylated tau expression in neurons of the hippocampus and entorhinal cortex54. Interestingly, the reduction in synaptic density is more pronounced within immature and mature plaques55. Moreover, the actin cytoskeleton and its dynamics play pivotal roles in modulating synaptic function by organizing the PSD, anchoring postsynaptic receptors, facilitating the trafficking of synaptic cargoes, and localizing the translation machinery in the synapses56,57. Therefore, impairments in signaling pathways that regulate synaptic markers such as PSD-95 and synaptophysin, and actin dynamics, such as profilin-1, could lead to the synaptic and cognitive deficits observed early in AD.
Dendritic spines are specialized anatomical structures in neuronal cells that serve as the postsynaptic element/component for the vast majority of CNS synapses. Structural changes at dendritic spines underlie learning and memory processes in the brain, and changes in spine structure and function might lead to cognitive impairments. Spines can be classified morphologically into four types: stubby, mushroom, thin and filopodia-like, which corresponds to their maturation and function as immature, stable and mature, transient or lacking synapses, respectively58,59. The structure and function of dendritic spines are dynamically regulated by cellular pathways acting on the actin cytoskeleton. In the data reported here, we observed that an increase in Aβ42 both intra- and extracellularly was coupled with a reduction in important synaptic proteins and changes in the density and morphology of dendritic spines that may constitute the primary cause for the synaptic inhibition and memory impairments.
Synaptic deficits and synapse loss occur early in AD and MCI, before the onset of plaques, being some of the first signs of the neurodegenerative process60,61. As Aβ aggregates it can adopt different shapes such as fibrils and non-fibrillar aggregates62,63, and there is a consensus that Aβ alone is not the main mediator responsible for AD. However, its precise pathogenic roles, subcellular location and state are still being discussed. Here we have provided evidence that low intraneuronal accumulation of Aβ peptides provoked by hippocampal infusion of UBI-Aβ42, as well as combined intraneuronal and extracellular accumulation of Aβ provoked by BRI-Aβ42, can disrupt cognitive behavior, synaptic plasticity and spine morphology.
Some insights on the relationship of different amyloid structures have already been discussed64,65,66. Further studies are required to determine the roles of these AAV vectors in promoting specific Aβ isoforms. It is of great importance to the advancement of AD research the acknowledgment of how the available tools work, so they can be appropriately employed in order to provide relevant results.
Methods
Infusion of vectors
Stereotaxic injection of AAV-EGFP, AAV-BRI-Aβ42 and AAV-UBI-Aβ42 into the hippocampus was performed according to previously described surgical protocols67,68. Viral preps were generated as described previously69. Briefly, AAV vectors expressing the Aβ peptides under the control of the cytomegalovirus enhancer/chicken beta actin (CBA) promoter, a WPRE, and the bovine growth hormone polyA were generated by plasmid transfection with helper plasmids in HEK293T cells. 48 hours after transfection cells were harvested and lysed in the presence of 0.5% Sodium Deoxycholate and 50U/ml Benzonase (Sigma) by freeze thawing, and the virus isolated using a discontinuous Iodixanol gradient, and affinity purified on a HiTrap HQ column (Amersham). The genomic titer of each virus was determined by quantitative PCR.
4–5-month-old male C57/BL6 mice (purchased from the Jackson Laboratory, Maine, USA) were anesthetized and placed in stereotaxic frame under continuous isoflurane anesthesia. Using a 10-μl Hamilton syringe and 30-gauge needle, mice received 2-μl injections of AAV-EGFP, AAV-BRI-Aβ42 and AAV-UBI-Aβ42 (1 × 1010 genome particles/ul) in the right and left hemispheres at the following stereotaxic coordinates: anterior-posterior (AP) −2.06 mm; dorsoventral (DV) −1.95 mm; mediolateral (ML) ± 1.75 mm. Animals were allowed to recover on a heating pad before being placed back in their home cages. All animal procedures are in accordance with National Institutes of Health and University of California guidelines and were approved by the Use Committee at the University of California, Irvine.
Cell culture
Primary hippocampal neurons were collected from postnatal day 0 C57BL/6 J mice. Cells were grown and fed twice a week with Neurobasal media with antibiotics and supplemented with GlutaMAX and B-27 (ThermoFisher Scientific). For experiments presented in Fig. 1a, cells were incubated for 24 h with AAV-EGFP, AAV-BRI-Aβ42 ([1] 4.7 × 1010 genome particles/ml, [2] 9.3 × 1010 genome particles/ml, [3] 2.8 × 1011 genome particles/ml) and AAV-UBI-Aβ42 ([1] 1.5 × 1010 genome particles/ml, [2] 7.7 × 1010 genome particles/ml, [3] 4.6 × 1011 genome particles/ml) and the media was collected and analyzed for Aβ42 levels with a sandwich ELISA system as described previously70. For experiments presented in Fig. 1b,c, cells were transduced with AAV-EGFP 7.6 × 1010 genome particles/ml, AAV-BRI-Aβ42 2.2 × 1011 genome particles/ml, and AAV-UBI-Aβ 1.6 × 1012 genome particles/ml. 72 h later, media was collected for analysis and cells were washed with ice-cold PBS. Then, M-PER complemented with proteases and phosphatases inhibitors (ThermoFisher Scientific) was added and cells were scrapped. Media and lysates were centrifuged at 12,000xg for 10 min at 4 °C. Protein concentration in the lysates was determined using a commercial Bradford assay (Biorad). Media and cell lysates were analyzed for high molecular Aβ oligomers using an ELISA kit (Wako, cat# 298–80101) following the manufacturer’s protocol.
Aβ derived diffusible ligand (ADDL) preparation
ADDLs were prepared according to previous publications71. Briefly, Aβ1–42 was dissolved in hexafluoro-2-propanol (HFIP) and aliquoted to microcentifuge tubes. HFIP was removed by evaporation under vacuum and an aliquot of Aβ42 was dissolved in anhydrous dimethyl sulfoxide (DMSO), which was then added to ice-cold F12 medium without phenol red. This solution was incubated at 4 °C for 24 h and then centrifuged at 14 000 g for 10 min. Centrifugation produced a small pellet and the supernatant is defined as the ADDL preparation, which comprises fibril-free solutions of oligomers as well as monomers. Cells were incubated with 10 mM ADDLs for 24 h.
Morris Water Maze
Three months after infusion of vectors, behavioral analyses were performed. Mice were trained to swim to a circular clear Plexiglas platform submerged 1.5 cm beneath the water’s surface. Four trials were performed per day, for 60 seconds each with 5 minutes between trials. Mice were trained for as many days as needed for the group to reach the training criterion of 25 seconds. The probe test was assessed 24 hours after the last trial, with the platform removed. Performance was monitored with the EthoVision XT video-tracking system (Noldus Information Technology, Leesburg, VA, USA).
Contextual Fear Conditioning
During training, mice were placed in the fear conditioning chamber and allowed to explore for 2 minutes before receiving three electric foot shocks (duration: 1 s, intensity: 0.2 mA, intershock interval: 2 minutes). Animals were returned to the home cage 30 seconds after the last foot shock. Twenty-four hours later, behavior in the conditioning chamber was video recorded for 5 minutes and subsequently analyzed for freezing behavior.
Fluorescence analysis of single-synapse long-term potentiation (FASS-LTP)
Activity-dependent responses in hippocampal synaptosomes were analyzed by FASS-LTP, as previously described26,27. FASS-LTP consists of chemical LTP (cLTP) stimulation directly in crude P2 synaptosomal fractions, immunofluorescence labeling for surface GluA1 and neurexin-1β, and flow cytometry analysis. Briefly, fresh crude synaptosome P2 fractions were obtained from the hippocampi of mice injected with AAVs and stimulated with glycine (500 μM) and KCl (50 mM). For surface immunolabeling, primary antibodies were rabbit anti-GluA1 (Cell Signaling #13185; 1:400) and mouse anti-Nrx1β (UC Davis/NIH NeuroMab Facility, 75–216; 2.5 μg/ml). Secondary antibodies were anti-rabbit-Alexa-405, anti-rabbit-Alexa-488 and anti-mouse-Alexa-647 antibodies (Life Sciences), at 2.5 μl/ml. Samples were protected from light, maintained at 4 °C and immediately run on a flow cytometer (Novocyte, ACEA Biosciences, Inc); 20,000 events were collected and analyzed for each sample with an event rate of approximately 500/sec. Analysis was performed using the FlowJo v10.3 software (LLC).
Golgi staining
Following transcardial perfusion with 0.1 M phosphate-buffered saline (PBS, pH 7.4), mice brains were removed and processed using a superGolgi Kit (Bioenno Tech LLC, Santa Ana, CA), as described previously24,72
Dendritic and spine analysis
Stereological quantifications were performed using Neurolucida software from Microbrightfield Bioscience (MBF Bioscience, Williston, VT, USA) to determine the number of spines in the stratum radiatum (SR) and the molecular layer (ML) of the hippocampal CA1 region, respectively. Briefly, every second section was used through the entire antero-posterior extent of the hippocampus (between −1.46 mm anterior and −3.40 mm posterior to Bregma according to Franklin and Paxinos, Third Edition, 2007). The SR and ML in the CA1 region were defined using a 5x objective and spines were counted using a 100x/1.4 objective. The coefficient of error (CE) value for each animal ranged between 0.03 and 0.08. Dendritic spine length was traced using a 100x/1.4 objective and data were analysed via Neurolucida Explorer software. For dendritic morphological analysis, 5 neurons per animal (n = 6) in the CA1 hippocampal area were traced using Neurolucida software. Dendritic width was measured using Image J software in electronic microscopic images (10 images per animal for a total of 6 mice per group).
Immunoblotting
Equal amounts of protein (30 μg) were separated on 10% Bis-Tris gel (Invitrogen, Carlsbad, CA), and transferred to nitrocellulose membranes that were blocked in a 5% (w/v) suspension of Bovine Serum Albumin (BSA; Gemini Bio-Products, West Sacramento, CA, USA) in 0.2% Tween 20 Tris-buffered saline (TBS-T, pH 7.5) for 1 h. Next, membranes were incubated overnight at 4 °C with the following primary antibodies: synaptophysin 1:1000 (Abcam, Cambridge, UK), PSD-95 1:1000 (Abcam, Cambridge, UK), profilin-1 1:1000 (Abcam, Cambridge, UK) and GAPDH 1:1000 (Santa Cruz Biotechnology, CA, USA). Membranes were then washed in Tween-TBS for 20 min and incubated with specific secondary antibodies at a dilution of 1:10,000 (Pierce Biotechnology) for 60 min. Immunocomplexes were visualized using Super Signal (ThermoFisher Scientific, Rockford, IL, USA) and band density measurements were made using ImageJ imaging software version 1.36b (NIH).
Immunohistochemistry
For immunohistochemistry, sections (40μm thick) were pretreated with 3% H2O2/3% methanol in Tris-buffered saline (TBS) for 30 min, followed by a TBS wash. Sections were then incubated in TBS with 0.1% Triton X-100 (TBST) for 15 min, followed by TBST with 2% BSA (Sigma-Aldrich) for 30 min. Sections were incubated with anti-6E10 (1:1000; Biolegend, San Diego, CA, USA) in TBS + 5% normal horse serum overnight at 40 C. Sections were then incubated with the appropriate secondary biotinylated antibody (1:500) in TBS containing 2% BSA plus 5% normal serum for 1 hour at room temperature, followed by Vector ABC Kit and DAB reagents (Vector Laboratories, Burlingame, CA, USA) to visualize staining.
Electrochemiluminescence-linked immunoassay
Quantitative biochemical analyses of human Aβ and inflammatory cytokines in mouse tissue were performed using a commercially available electrochemiluminescence-linked immunoassay from Meso Scale Discovery (MSD, Gaithersburg, MD, USA). The V-PLEX Aβ Peptide Panel 1 (6E10) was used and plates were analyzed on the MS2400 imager (MSD). Assays were performed according to the manufacturer’s instructions, and all standards and samples were measured in duplicate.
Enzyme-linked immunosorbent assay for Aβ42
Aβ1–42 was measured in the primary neuronal hippocampus cell culture medium using a sensitive sandwich enzyme-linked immunosorbent assay system as previously described70.
Statistical analysis
All data between two groups were analyzed by Student’s t-test comparisons, and one- or two-way analysis of variance (ANOVA), followed by Bonferroni’s test for comparisons among more than 2 groups. Mann-Whitney U Test was used for the FASS-LTP data. Graphpad Prism software (Graphpad Prism Inc., San Diego, CA, USA) was used, and the significance was set at 95% of confidence. Values are presented as mean ± SEM.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Alzheimer’s, A. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement 12(4), 459–509 (2016).
Querfurth, H. W. & LaFerla, F. M. Alzheimer’s disease. N Engl J Med 362(4), 329–344 (2010).
Walsh, D. M. & Selkoe, D. J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44(1), 181–193 (2004).
Selkoe, D. J. Alzheimer’s disease is a synaptic failure. Science 298(5594), 789–791 (2002).
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W. & Strittmatter, S. M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457(7233), 1128–1132 (2009).
Yamin, G. NMDA receptor-dependent signaling pathways that underlie amyloid beta-protein disruption of LTP in the hippocampus. J Neurosci Res 87(8), 1729–1736 (2009).
Brito-Moreira, J. et al. Interaction of amyloid-beta (Abeta) oligomers with neurexin 2alpha and neuroligin 1 mediates synapse damage and memory loss in mice. J Biol Chem 292(18), 7327–7337 (2017).
Jurgensen, S. et al. Activation of D1/D5 dopamine receptors protects neurons from synapse dysfunction induced by amyloid-beta oligomers. J Biol Chem 286(5), 3270–3276 (2011).
Szekely, C. A. et al. No advantage of A beta 42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology 70(24), 2291–2298 (2008).
Rota, E. et al. Increased intrathecal TGF-beta1, but not IL-12, IFN-gamma and IL-10 levels in Alzheimer’s disease patients. Neurol Sci 27(1), 33–39 (2006).
Baker-Nigh, A. et al. Neuronal amyloid-beta accumulation within cholinergic basal forebrain in ageing and Alzheimer’s disease. Brain 138(Pt 6), 1722–1737 (2015).
Iulita, M. F. et al. Intracellular Abeta pathology and early cognitive impairments in a transgenic rat overexpressing human amyloid precursor protein: a multidimensional study. Acta Neuropathol Commun 2, 61 (2014).
Gouras, G. K. et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156(1), 15–20 (2000).
Takahashi, R. H. et al. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161(5), 1869–1879 (2002).
Olsson, T. T., Klementieva, O. & Gouras, G. K. Prion-like seeding and nucleation of intracellular amyloid-beta. Neurobiol Dis 113, 1–10 (2018).
Takeda, S. et al. Brain interstitial oligomeric amyloid beta increases with age and is resistant to clearance from brain in a mouse model of Alzheimer’s disease. FASEB J 27(8), 3239–3248 (2013).
Heras-Sandoval, D., Ferrera, P. & Arias, C. Amyloid-beta protein modulates insulin signaling in presynaptic terminals. Neurochem Res 37(9), 1879–1885 (2012).
LaFerla, F. M. & Green, K. N. Animal models of Alzheimer disease. Cold Spring Harb Perspect Med 2(11) (2012).
Lawlor, P. A. et al. Novel rat Alzheimer’s disease models based on AAV-mediated gene transfer to selectively increase hippocampal Abeta levels. Mol Neurodegener 2, 11 (2007).
Prasad, T. et al. Amyloid beta peptides overexpression in retinal pigment epithelial cells via AAV-mediated gene transfer mimics AMD-like pathology in mice. Sci Rep 7(1), 3222 (2017).
Lewis, P. A. et al. Expression of BRI-amyloid beta peptide fusion proteins: a novel method for specific high-level expression of amyloid beta peptides. Biochim Biophys Acta 1537(1), 58–62 (2001).
Sabin, E. A., Lee-Ng, C. T., Shuster, J. R. & Barr, P. J. High-Level Expression and In Vivo Processing of Chimeric Ubiquitin Fusion Proteins in Saccharomyces Cerevisiae. Bio/Technology 7, 705 (1989).
Moore, B. D. et al. Short Abeta peptides attenuate Abeta42 toxicity in vivo. J Exp Med 215(1), 283–301 (2018).
Baglietto-Vargas, D. et al. Impaired AMPA signaling and cytoskeletal alterations induce early synaptic dysfunction in a mouse model of Alzheimer’s disease. Aging Cell, e12791 (2018).
Nabavi, S. et al. Engineering a memory with LTD and LTP. Nature 511(7509), 348–352 (2014).
Prieto, G. A. et al. Pharmacological Rescue of Long-Term Potentiation in Alzheimer Diseased Synapses. J Neurosci 37(5), 1197–1212 (2017).
Prieto, G. A. et al. Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1beta in the aged hippocampus. Proc Natl Acad Sci USA 112(36), E5078–5087 (2015).
Prieto, G. A., Tong, L., Smith, E. D. & Cotman, C. W. TNFalpha and IL-1beta but not IL-18 Suppresses Hippocampal Long-Term Potentiation Directly at the Synapse. Neurochem Res (2018).
Lu, W. et al. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29(1), 243–254 (2001).
Fu, Y. & Huang, Z. J. Differential dynamics and activity-dependent regulation of alpha- and beta-neurexins at develo** GABAergic synapses. Proc Natl Acad Sci USA 107(52), 22699–22704 (2010).
Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18(6), 794–799 (2015).
Hardy, J. A. & Higgins, G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256(5054), 184–185 (1992).
Armstrong, R. A. A critical analysis of the ‘amyloid cascade hypothesis’. Folia Neuropathol 52(3), 211–225 (2014).
Tu, S., Okamoto, S., Lipton, S. A. & Xu, H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener 9, 48 (2014).
Forner, S., Baglietto-Vargas, D., Martini, A. C., Trujillo-Estrada, L. & LaFerla, F. M. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci 40(6), 347–357 (2017).
Shankar, G. M. et al. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27(11), 2866–2875 (2007).
Shankar, G. M. et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14(8), 837–842 (2008).
Knobloch, M., Konietzko, U., Krebs, D. C. & Nitsch, R. M. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging 28(9), 1297–1306 (2007).
Leon, W. C. et al. A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J Alzheimers Dis 20(1), 113–126 (2010).
Oddo, S. et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39(3), 409–421 (2003).
Oakley, H. et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26(40), 10129–10140 (2006).
Gouras, G. K., Tampellini, D., Takahashi, R. H. & Capetillo-Zarate, E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol 119(5), 523–541 (2010).
Counts, S. E., Alldred, M. J., Che, S., Ginsberg, S. D. & Mufson, E. J. Synaptic gene dysregulation within hippocampal CA1 pyramidal neurons in mild cognitive impairment. Neuropharmacology 79, 172–179 (2014).
Mueller, S. G. et al. Hippocampal atrophy patterns in mild cognitive impairment and Alzheimer’s disease. Hum Brain Mapp 31(9), 1339–1347 (2010).
Sze, C. I. et al. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 56(8), 933–944 (1997).
Nomura, I., Takechi, H. & Kato, N. Intraneuronally injected amyloid beta inhibits long-term potentiation in rat hippocampal slices. J Neurophysiol 107(9), 2526–2531 (2012).
Puzzo, D. et al. LTP and memory impairment caused by extracellular Abeta and Tau oligomers is APP-dependent. Elife 6 (2017).
Ripoli, C. et al. Intracellular accumulation of amyloid-beta (Abeta) protein plays a major role in Abeta-induced alterations of glutamatergic synaptic transmission and plasticity. J Neurosci 34(38), 12893–12903 (2014).
Malenka, R. C. & Bear, M. F. LTP and LTD: an embarrassment of riches. Neuron 44(1), 5–21 (2004).
Sheng, M. & Hoogenraad, C. C. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 76, 823–847 (2007).
Cheng, D. et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics 5(6), 1158–1170 (2006).
Kim, E. & Sheng, M. PDZ domain proteins of synapses. Nat Rev Neurosci 5(10), 771–781 (2004).
Terry, R. D. et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30(4), 572–580 (1991).
Masliah, E. et al. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett 174(1), 67–72 (1994).
Masliah, E., Terry, R. D., Mallory, M., Alford, M. & Hansen, L. A. Diffuse plaques do not accentuate synapse loss in Alzheimer’s disease. Am J Pathol 137(6), 1293–1297 (1990).
Ackermann, M. & Matus, A. Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci 6(11), 1194–1200 (2003).
Hotulainen, P. & Hoogenraad, C. C. Actin in dendritic spines: connecting dynamics to function. J Cell Biol 189(4), 619–629 (2010).
Bourne, J. & Harris, K. M. Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol 17(3), 381–386 (2007).
Rochefort, N. L. & Konnerth, A. Dendritic spines: from structure to in vivo function. EMBO Rep 13(8), 699–708 (2012).
Mucke, L. & Selkoe, D. J. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2(7), a006338 (2012).
Scheff, S. W., Price, D. A., Schmitt, F. A., DeKosky, S. T. & Mufson, E. J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68(18), 1501–1508 (2007).
Haass, C. & Selkoe, D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8(2), 101–112 (2007).
Glabe, C. G. Structural classification of toxic amyloid oligomers. J Biol Chem 283(44), 29639–29643 (2008).
Ji, L., Zhao, X., Lu, W., Zhang, Q. & Hua, Z. Intracellular Abeta and its Pathological Role in Alzheimer’s Disease: Lessons from Cellular to Animal Models. Curr Alzheimer Res 13(6), 621–630 (2016).
Oddo, S., Caccamo, A., Smith, I. F., Green, K. N. & LaFerla, F. M. A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol 168(1), 184–194 (2006).
Takamura, A. et al. Sortilin is required for toxic action of Abeta oligomers (AbetaOs): extracellular AbetaOs trigger apoptosis, and intraneuronal AbetaOs impair degradation pathways. Life Sci 91(23-24), 1177–1186 (2012).
Sudduth, T. L., Greenstein, A. & Wilcock, D. M. Intracranial injection of Gammagard, a human IVIg, modulates the inflammatory response of the brain and lowers Abeta in APP/PS1 mice along a different time course than anti-Abeta antibodies. J Neurosci 33(23), 9684–9692 (2013).
Blurton-Jones, M. et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci USA 106(32), 13594–13599 (2009).
Chakrabarty, P. et al. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol 184(9), 5333–5343 (2010b).
Baglietto-Vargas, D. et al. Endogenous murine tau promotes neurofibrillary tangles in 3xTg-AD mice without affecting cognition. Neurobiol Dis 62, 407–415 (2014).
Lambert, M. P. et al. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J Neurochem 79(3), 595–605 (2001).
Baglietto-Vargas, D. et al. Short-term modern life-like stress exacerbates Abeta-pathology and synapse loss in 3xTg-AD mice. J Neurochem 134(5), 915–926 (2015).
Acknowledgements
The authors are supported by grants from the Alzheimer’s Association #AARF-16-440760 (S.F.), NIRG-15-363477 (D.B.V.) and MNIRGD-15-363229 (C.J.R.O.). The Larry L. Hillblom Foundation [2016-A-016-FEL (A.C.M.) and #2013-A-016-FEL (D.B.V.), the National Institute of Health (NIH) NIH/NIA AG027544, AG00538, and AG054884 (F.M.L.), R21-AG048506, P01-AG000538 and RO1-AG34667 (C.W.C.), U01AG046139 (T.G), the UC MEXUS-CONACYT Grant CN-16-170 (G.A.P. and C.W.C.), the Australian National Health and Medical Research Council GNT112919, GNT1128436 and GNT1139469 (R.M.), and the BrightFocus Foundation A2015535S (F.M.L.). The authors would like to thank Professor Giles Alexander Rae and Ms. Rowan Tweedale for their diligent proofreading of this manuscript.
Author information
Authors and Affiliations
Contributions
S.F., A.C.M., D.B.V. and G.A.P. designed the studies; S.F., A.C.M., D.B.V., A.G.P., C.J.R.O., J.M.R.R., R.A., C.T.D., L.T.E., C.D.C., E.J.A., J.P., J.V.H. and A.Z.V.D.C. performed the experimental work; S.F., A.C.M., D.B.V., A.G.P., C.J.R.O., J.M.R.R., L.T.E., Y.L., P.E.C., R.M. and F.M.L. performed the data analyses and participated in manuscript writing; M.K., C.G.G., C.W.C., T.G., D.B.V. and F.M.L. supported and supervised the study and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Forner, S., Martini, A.C., Prieto, G.A. et al. Intra- and extracellular β-amyloid overexpression via adeno-associated virus-mediated gene transfer impairs memory and synaptic plasticity in the hippocampus. Sci Rep 9, 15936 (2019). https://doi.org/10.1038/s41598-019-52324-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52324-0
- Springer Nature Limited