
Abstract
Head and neck cancer is the sixth most common malignancy worldwide, predominantly develo** from squamous cell epithelia (HNSCC). The main HNSCC risk factors are tobacco, excessive alcohol use, and the presence of human papillomavirus (HPV). HPV positive (+) cancers are etiologically different from other HNSCC and often show better prognosis. The current knowledge regarding HNSCC miRNA profiles is still incomplete especially in the context of HPV+ cancer. Thus, we analyzed 61 freshly collected primary oral (OSCC) and oropharyngeal (OPSCC) SCC samples. HPV DNA and RNA was found in 21% cases. The Illumina whole-genome small-RNA profiling by next-generation sequencing was done on 22 samples and revealed 7 specific miRNAs to HPV+ OSCC, 77 to HPV+ OPSCC, and additional 3 shared with both; 51 miRNAs were specific to HPV− OPSCC, 62 to HPV− OSCC, and 31 shared with both. The results for 9 miRNAs (miR-9, -21, -29a, -100, -106b, -143 and -145) were assessed by reverse transcription-quantitative polymerase chain reaction on the whole study population. The data was additionally confirmed by reanalyzing publicly available miRNA sequencing Cancer Genome Atlas consortium (TCGA) HNSCC data. Cell signaling pathway analysis revealed differences between HPV+ and HPV− HNSCC. Our findings compared with literature data revealed extensive heterogeneity of miRNA deregulation with only several miRNAs consistently affected, and miR-9 being the most likely HPV related miRNA.
Similar content being viewed by others

Introduction
Head and neck cancer (HNC) is the sixth most common malignancy worldwide, predominantly arising within the mucosal linings of the upper aerodigestive tract1. Most HNC develop from squamous cell epithelia, which accounts for 95% of head and neck carcinoma (HNSCC)2,3. HNSCC often gets diagnosed in a late phase, when it is difficult to treat, with 5-year survival of only 40–50%4,5. HNSCC are further characterized according to their primary site of origin, with most common sites being oral cavity, oropharynx, pharynx, larynx, and sinonasal tract6. Globally, HNSCC accounts for approximately 550,000 cases annually7, while in Croatia, 896 new cases were estimated in 20158.
The main risk factors for HNSCC development are smoking and excessive alcohol use. Furthermore, the role of human papillomavirus (HPV) has emerged in recent years, particularly in oropharyngeal tumors9,10. In western countries, tobacco and alcohol induced HNSCC is declining, while HPV-driven HNSCC, especially oropharyngeal, is increasing in younger individuals9,10. HPV type 16 has been found in the majority of HPV associated HNSCC, and it is capable of transforming infected cells into cancerous by expressing oncoproteins E6 and E7, which bind, among others, to two important tumor suppressor proteins, p53 and pRB, respectively10.
Based on the HPV presence, HNSCC is broadly divided in two groups: HPV positive (+) with better prognosis and HPV negative (−) tumors with worse prognosis10. Even though these two groups are etiologically different, the treatment remains the same7. However, there are indications that the treatment could be optimized for each groups of patients. Therefore, it is crucial to find more sensitive and specific biomarkers, which could enable development of better diagnostic, prognostic and therapeutic approaches for HNSCC. The HPV positive oropharyngeal cancer in particular was found to be so different from other HNSCC subtypes that the new TNM classifications11 and the specific staging guidelines12 were made specifically for this subset of tumors. In 2015, The Cancer Genome Atlas (TCGA) consortium published a comprehensive molecular catalogue on HNSCC13. Frequent mutations of novel druggable oncogenes were not demonstrated, but the difference between the HPV associated and non-viral groups was confirmed. The TCGA study revealed that HNSCC lacked predominant gain-of-function mutations in oncogenes, whereas an essential role of epigenetics in oncogenesis has become apparent. The study of Masuda et al.14 emphasizes that HNSCC seems to be an epigenetic disease, rather than genetic. Studies on the epigenetic changes in HNSCC such as miRNA profiling are promising to find specific biomarkers for both groups of tumor patients15.
Small non-coding RNAs, such as miRNA (miR) are highly conserved and about 22 nucleotides long, with important role in a variety of processes, including development, cell proliferation, and differentiation16. Previous reviews24 and the amplicons (~260 and ~86 bp, respectively) were visualized on 3%-agarose gel electrophoresis. CaSki cell line cDNA was used as positive control, while the negative control contained all PCR reagents without cDNA. The suitability of cDNA for amplification was confirmed by beta-actin PCR25. In this study, samples positive for both HPV DNA, and E6 mRNA were considered HPV positive.
To try separating relevant HPV infections from those where other factors might confound HPV activity, patients were also classified into risk groups according to Ang et al.26. Briefly, low risk group is defined as HPV positive tumors from non-smoking patients or from smoking patients with lower nodal stage. High risk group consists of HPV negative smokers or tumors with high T classification in nonsmokers, while the intermediate risk group consisted of smoking patients with HPV positive N2b+ tumors or non-smoking HPV negative tumors with T classification less than 4.
miRNA next generation sequencing analysis
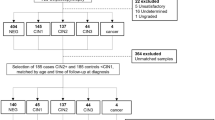
A subset of samples (19 cancer samples and 3 controls) was selected for high-throughput miRNA analysis by next generation sequencing (NGS). Samples with poor RIN (<7) scores were excluded. Thus, the following samples were selected for NGS library preparation: 6 HPV+ (DNA and RNA) and 4 HPV− oropharyngeal cancer samples (OP+ and OP−, respectively); 3 HPV+ (DNA and RNA) and 6 HPV− oral cancer samples (O+ and O−, respectively); and 3 healthy tonsil tissue samples (controls). Out of 6 selected OP+ samples, two were classified as intermediate risk group according to Ang et al.26 Twenty-two NGS libraries were constructed with TrueSeq Small RNA Library prep kit (Illumina) according to the manufacturer’s protocol. For multiplexing and library pooling, index pools A (1–12) and B (13–22) were used. Bioanalyser (Agilent) was used for quality control of indicated steps as recommended by the manufacturer. Library sequencing was done on NextSeq 500 sequencer (Illumina) using NextSeq 500 Mid output kit (Illumina).
Raw sequences were trimmed of adapter sequences using FastQ toolkit Basespace App (Illumina) by selecting TrueSeq Small RNA adapter sequences from the relevant app menu. Sequencing data was analyzed using Small RNA Basespace App v1.0.1 (Illumina) to determine significantly different miRNA expression between groups. The automated pipeline uses Bowtie to align reads against reference databases to determine counts, which are then assessed for differential expression using DESeq2. Within the pipeline, miRNA sequences with mean normalized counts across all samples ≤10 are filtered out before statistical analysis. Further analyses were performed by importing Small RNA Basespace App count data into R and independently analyzed by the DESeq2 package.
Technical validation
The NGS results were validated by real-time quantitative Reverse Transcription PCR (qRT-PCR) on the same samples tested by NGS. For technical validation of the NGS experiment, we have selected 9 miRNAs that were found to be differentially expressed by NGS (miR-9-5p, -21-3p, -27a-5p, -31-5p, -34a-5p, -100-5p, -143-3p, -145-5p, 218-5p). Assays were designed to cover both over and under expressed miRNA. Priority was given to miRs found in HPV positive samples but without being found as significant in HPV negative samples. In addition, miRNAs -21-3p, -31-5p, -100-5p were chosen, since they were often reported in many different cancer types and could represent positive control targets. The TaqMan Advanced miRNA synthesis kit (Applied Biosystems) was used to convert isolated RNA to cDNA following the manufacturers protocol. Following conversion, 5 μl of diluted cDNA was analyzed by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) using TaqMan Advanced miRNA single tube assays (Applied Biosystems). The three normal tonsillar samples were pooled in equal concentration before cDNA synthesis to be used as normal reference. Assays for miR-16-5p and -191-5p were evaluated as internal reference control (manufacturer’s recommendation) as well as miR-181a-5p that showed very low intra-sample variation in the NGS experiment. Calculations were performed using each of the 3 referent miRs individually (data not shown) and as average of all 3 values. As the results were similar, the final analysis was performed with the average value of all 3 reference miRs. The fold changes were calculated using the standard 2−ddCt method27. Briefly, dCt values were obtained by normalizing to the referent control sample, i.e. obtained by subtracting mean replicate Ct values of the combined referent sample from the mean replicate Ct value of each sample for each miR tested. Subsequently, dCt values for each miR were normalized to referent miRs in each sample to obtain ddCt value. The fold change was calculated by 2−ddCt formula. The statistical difference was tested by t-test on dCt values of each miR compared to dCt values of the referent miR within each subgroup of samples.
Clinical validation
For further validation of potentially relevant miRs, clinical samples not tested by NGS (independent set of 46 tumors and same controls used for technical validation) were tested with qRT-PCR individual assays in the same way as for technical validation. For clinically relevant validation, priority was given to miRNAs with at least 100 normalized mean count in OP+ subset since low count (expression) might lead to inconsistent results on routine samples. Even though NGS analysis indicated a very limited number of miRNAs exclusively associated with HPV, the following miRNAs were selected for analysis: miR-9-5p, -21-3p, -29a-3p, -100-5p, -106b-5p, -143-3p, and -145-5p. The miRNAs miR-9-5p, -106b-5p and -29a-3p were selected as they were deregulated in our OP+ subset and not found significant in HPV negative samples. As for technical validation, miRs -21-3p and -100-5p were chosen because of their relevance in different cancer types. Specifically miRs -143-3p and -145-5p were selected as they were most commonly found by other studies to be downregulated in HNSCC cases even though they were not found to be significant in our HPV positive samples. In both cases, the purpose was to assess the utility of selected miRs as potential biomarkers. Internal reference controls and combined sample pool of healthy tonsil samples was used as referent sample for fold change calculations; as done for the technical validation. Since the initial NGS set was selected with overrepresentation of HPV positive and oropharyngeal samples, the independent set was underrepresented in those samples and consisted of 35 O−, 6 OP−, 3 O+ and only 2 OP+ samples. To increase robustness, analysis was done on the independent set (n = 46) or the total set (n = 61) of clinical cancer samples.
Independent validation
To assess the validity of the results in a completely unrelated set of patients, we accessed publicly available miRNA sequencing data from TCGA data portal for oral and oropharyngeal cancer samples. Detailed clinical data and HPV status for cases with available miRNA sequencing data was obtained from the TCGA data portal as well as the TCGA consortium HNSCC focused publication13. We were able to match miRNA sequencing data and relevant information for 72 cancer samples (Supplementary Dataset SD1). There were 40 samples from oral cancer (12 HPV RNA positive) and 32 oropharyngeal cancer (21 HPV RNA positive) patients. We were also able to find miRNA sequencing results for matched normal solid tissue from two oropharyngeal cancer and two oral cancer patients. However, we chose to include only oropharyngeal tissue normal controls to make the control groups comparable to our sequencing experiment, where we have also used oropharyngeal normal samples as control. Briefly, raw counts of all miRNA sequences were tabulated (including isomiR sequences) and imported to R alongside annotation data (Supplementary Dataset SD1) for the analysis with DESeq2 package using identical R pipeline as for our samples. As before, miRNA sequences with 10 or less normalized reads across all samples were removed. Samples were also additionally classified according to Ang et al.26 risk factors from available clinical data, which included smoking and pack/year data for the majority of cases.
miRNA classifier
In attempt to create a miRNA classifier from the NGS data, we used multinomial sparse group lasso method as implemented within msgl R package28,29. Normalized counts of our and TCGA data were imported to R as “reads per million miRNA mapped”. Our sequencing data was used either as a training set for TCGA data classification, or as test set after training the classifier on TCGA data. Another set of classifier models were created where TCGA dataset was split in half with the first half used for training and the second for testing. Classification was performed for several variables: sample group (OP+, OP−, O+ and O−), HPV RNA presence (HPV+, HPV−) and risk group (high, intermediate, low) according to Ang et al.26.
Statistical analysis
Data management and basic analysis was done in Microsoft Excel, while statistical testing was done in Medcalc (v 11.4.2). R studio (v 1.1.383) was used to interface with R (v 3.4.2.) and perform miRNA differential expression using DEseq2 (1.18.1)30 or msgl classifier training and testing.
Literature review
So far, overlap of published results on miRNA deregulation in HNSCC, when only the validated or the most relevant miRs from each manuscript are considered, is relatively low35,36,37, thus the isomiRs cannot be simply disregarded.
Another important factor possibly confounding both previous and current findings is the fact that HPV itself does not need be a causal factor in tumor development even if found in the tumor as other factors like smoking might have a stronger impact. As emphasized in the study by Ang et al.26, three distinct survival profiles were observed. The greatest risk was for HPV negative OPSCC patients, however, HPV positive patients were in the lowest risk group only if they didn’t smoke or had tumors with lower nodal stage. It is possible that inclusion of samples classified as intermediate risk group might confound miRNA results and associations both herein and in other previous studies. Indeed, 2 of our 9 sequenced HPV positive samples could be classified as intermediate risk group, where in addition to HPV, other factors like smoking might further influence the miRNA profiles. However, classifying samples only according to Ang et al.26 risk factors still did not resolve the issue of suboptimal separation either in our samples or those in TCGA.
Another outcome of the miRNA NGS profiling was the apparent inability of this method to completely differentiate 4 specific subgroups of samples (Figs. 1 and 2); only control samples could be resolved clearly. Better separation of samples in OPSCC group (Fig. 2B) is possibly due to larger influence of HPV at oropharyngeal site noting that HPV is known to be less relevant for the development of oral cancer. Another interesting observation (but with very limited number of samples) is that samples positive for the unspliced form of HPV16-E6 mRNA clustered close to HPV negative samples (Fig. 2B), implying that HPV is also less etiologically relevant in that case. Previously transcriptionally negative HNSCC were also shown to have survival similar to HPV negative HNSCC38. While it is possible that the detected form of unspliced mRNA is due to DNA carryover, this is unlikely as RNAse-free DNAse step during RNA isolation was performed to minimize such possibility. It is also interesting to note that this sample was classified as an intermediate risk group sample (Supplementary Fig. S1), again implying that other factors could be confounding HPV activity.
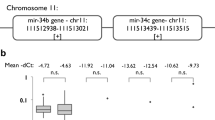
To verify the validity of NGS data, selected miRNA sequences were assessed on both the same samples (technical) and all clinical samples by qRT-PCR. The concordance of two methods on the same samples was very good (Supplementary Table S2); hence, validating the reliability of our results. The NGS results indicated the relevance of 16 mature miRNAs (miR-9-5p, 25-5p, -29a-3p, -29b-3p, -34a-5p, -93-5p, 106b-5p, -133a-5p, -133a-3p, -139-5p, -140-5p, -147b, -208b-3p, 210-5p, 328-3p, -1307-3p) for HPV positive oropharyngeal subset. Analyzing a further selection of potentially relevant miRs on the whole set of samples reinforced the relevance of miR-9 (p = 0.001) and miR-29a (p = 0.038) for HPV+ OPSCC. Results also reinforced the overall relevance of miR-21 (p < 0.0001 in OP+) and miR-100 (p < 0.0029 in OP+). However, in this study, as well as in the literature, those miRNAs are also associated with HPV negative tumors and thus are unlikely to be HPV associated.
Another validation was performed by reanalyzing miRNA sequencing data of a completely independent set of HNSCC cancer cases obtained from the publicly available TCGA portal. The analysis of this set of samples has also shown that miRNA sequencing cannot readily separate sample clusters, but somewhat better separation can be seen when oral and oropharyngeal subsets are analyzed separately (Supplementary Fig. S2). It is very important to highlight that miR-9 was found to be significantly upregulated in TCGA OP+ subset, its isomiRs in O+ subset, and was completely absent from HPV negative subsets. Another completely concordant result was for miR-21-3p, which was found significantly upregulated in all comparisons. Furthermore, other highlighted miRNAs selected for clinical set validation (-29a, -100, -106b, -143 and -145) were found to be significantly deregulated in the same direction at the isomiR level in the OP+ subset. It is important to note that our data and the data of TCGA was analyzed starting from raw count data, which was imported to R for DESeq2 analysis. However, there were some methodological differences up to that point, which might influence subsequent results. Namely, we used Illumina Basespace and SmallRNA app for alignment and counting, while TCGA data was aligned, counted and isomiRs presented differently. Despite that, results were highly comparable on clustering based on global miRNA profiles as well as deregulation of specific miRNA sequences. Reanalysis of TCGA data also indicates that miR-9 and -29a are relevant in OP+ subset and miR-9 is also relevant in O+ subset.
The expression of both miR-9 and miR-29a have previously been found deregulated in HNSCC (Supplementary Dataset SD2). Furthermore, both miRNAs were thoroughly reviewed very recently in the context of different cancer types39,40. Also, a similar systematic review of miRNA in cervical cancer indicated both miR-9 and miR-29 as consistently deregulated in literature and relevant in cervical cancer development41.
Briefly, miR-9 appears to be upregulated by HPV-E6 and when upregulated, it blocks keratinocyte differentiation and induces proliferation and migration39. However, its roles are very dependent on the context. Other studies have shown miR-9 to be upregulated in recurrent HNSCC42, but also as a potentially good salivary43 and even methylation biomarker44. It was one of the few overlap** miRs identified between HNSCC and cervical cancer22. More importantly, previous functional studies have already shown that miR-9 seems to be the miRNA most activated by HPV E6 protein in cervical cancer45.
MiR-29a was also shown to behave differently depending on the context40, but it is most often downregulated across cancers, and thus might be considered tumor suppressive. It was shown to influence proliferation, apoptosis, angiogenesis and metastasis depending on cancer type. Interestingly, miR-29a was also shown to induce drug resistance40, which might also explain some of the survival differences between HPV positive and HPV negative cancers if it is more often (but not exclusively) affected in HPV positive cancers.
Following examination of individual miRNAs, we also attempted to produce classifiers based on combinations of several miRNA sequences (Supplementary Table S3). A sparse group lasso method was employed with samples grouped according to different criteria (overall sample group, HPV status only and Ang et al.26 risk levels). We created different models where classifier was trained either on our data or TCGA data and then used to classify the other set. Since the maximal success rate was 64%, no classifier model was robust enough. Interestingly, models trained on one half of TCGA data and tested on the other half of TCGA data were also not successful even though their cross validation error rates were very low. It appears that large sample heterogeneity, even in the independent sample sets such as TCGA, might in part explain the discrepancies in previous literature. It probably is interesting to note that classifiers based on miRNA expression might need further specialized statistical modeling to decrease the impact of “normal tissue contamination” on the classifier29; however, this was beyond the scope of the current study.
The analysis of deregulated miRNAs in HPV+ and HPV− HNSCC using miRpath to assess KEGG pathway associations of functionally confirmed miR target genes revealed the main signaling pathways involved in the disease development (Table 2, Supplementary Dataset SD6). As expected, miRs found in both sets are associated with many other cancer related pathways, however, there were differences in strength of association with the particular pathways. The results indicated that HPV might be more involved with transcriptional dysregulation in cancer and adherens junction pathways, while miRNA profile of HPV− HNSCC implies stronger relevance of HIF-1 and TGF-beta pathways. The adherens junction pathway is of particular interest as high risk HPV types are known to interact and degrade many cell polarity proteins by E6-PDZ domain interactions46. It appears that this important viral process is also supported by consequent or parallel miRNA profile changes. In contrast, miRNA profile of HPV− HNSCC suggests stronger associations with more general pathways. These dissimilarities also support different etiologies of HPV positive and negative tumors.
In summary, miRNA landscape of HNSCC is very heterogeneous, primarily due to heterogeneity of sample material (and miRNA abundance therein), different methods (of miRNA detection, isomiR inclusion, HPV determination) and different grou** of samples during analysis (or lack of subgroup separation and even biological importance of HPV in cancer development). Despite this, some miRNAs show consistency, in particular miRs -21, -100 and -145 are overall relatively consistently detected in all HNSCC studies. However, of HPV specific miRs, only miR-9 seems to be consistently found in HPV positive and rarely in HPV negative subsets of HNSCC, including results from clinical samples in this study and specifically the analysis of TCGA data. Thus, miR-9 is the most likely miRNA specific for the HNSCC with the HPV etiology. While overall miRNA profiles also show lack of consistency and do not easily allow classification based on specific patterns, it appears that miRNAs identified in HPV positive and HPV negative cancers possibly affect cancer relevant pathways differently hence, reinforcing their etiological differences.
Data Availability
Raw sequences of the NGS experiment were uploaded to the ArrayExpress repository with the accession number E-MTAB-7030 (www.ebi.ac.uk/arrayexpress). All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
References
Argiris, A., Karamouzis, M. V., Raben, D. & Ferris, R. L. Head and neck cancer. Lancet Lond. Engl. 371, 1695–1709 (2008).
Leemans, C. R., Braakhuis, B. J. M. & Brakenhoff, R. H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 11, 9–22 (2011).
Vigneswaran, N. & Williams, M. D. Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral Maxillofac. Surg. Clin. N. Am. 26, 123–141 (2014).
Fakhry, C. et al. Human papillomavirus and overall survival after progression of oropharyngeal squamous cell carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 32, 3365–3373 (2014).
Leemans, C. R., Snijders, P. J. F. & Brakenhoff, R. H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 18, 269–282 (2018).
Yan, W., Wistuba, I. I., Emmert-Buck, M. R. & Erickson, H. S. Squamous cell carcinoma–similarities and differences among anatomical sites. Am. J. Cancer Res. 1, 275 (2011).
Keck, M. K. et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 21, 870–881 (2015).
Croatian National Cancer Registry. Cancer Incidence in Croatia. 1–39 (Croatian Institute of Public Health 2018).
Albers, A. E., Qian, X., Kaufmann, A. M. & Coordes, A. Meta analysis: HPV and p16 pattern determines survival in patients with HNSCC and identifies potential new biologic subtype. Sci. Rep. 7, 16715 (2017).
Taberna, M. et al. Human papillomavirus-related oropharyngeal cancer. Ann. Oncol. . Off. J. Eur. Soc. Med. Oncol. 28, 2386–2398 (2017).
O’Sullivan, B. et al. Development and validation of a staging system for HPV-related oropharyngeal cancer by the International Collaboration on Oropharyngeal cancer Network for Staging (ICON-S): a multicentre cohort study. Lancet Oncol. 17, 440–451 (2016).
Lydiatt, W., O’Sullivan, B. & Patel, S. Major Changes in Head and Neck Staging for 2018. Am. Soc. Clin. Oncol. Educ. Book 505–514, https://doi.org/10.1200/EDBK-199697 (2018).
TCGA. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015).
Masuda, M., Wakasaki, T. & Toh, S. Stress-triggered atavistic reprogramming (STAR) addiction: driving force behind head and neck cancer? Am. J. Cancer Res. 6, 1149–1166 (2016).
Jung, A. C. et al. A Poor Prognosis Subtype of HNSCC Is Consistently Observed across Methylome, Transcriptome, and miRNome Analysis. Clin. Cancer Res. 19, 4174–4184 (2013).
Wang, Y., Wang, J. & Huang, Y. MicroRNAs as new biomarkers for human papilloma virus related head and neck cancers. Cancer Biomark. Sect. Dis. Markers 15, 213–218 (2015).
Liu, X., Chen, Z., Yu, J., **a, J. & Zhou, X. MicroRNA Profiling and Head and Neck Cancer. Comp. Funct. Genomics 2009, 1–11 (2009).
Kaczkowski, B., Morevati, M., Rossing, M., Cilius, F. & Norrild, B. A Decade of Global mRNA and miRNA Profiling of HPV-Positive Cell Lines and Clinical Specimens. Open Virol. J. 6 (2012).
John, K., Wu, J., Lee, B.-W. & Farah, C. S. MicroRNAs in Head and Neck Cancer. Int. J. Dent. 2013, 1–12 (2013).
Koshizuka, K. et al. The microRNA signatures: aberrantly expressed microRNAs in head and neck squamous cell carcinoma. J. Hum. Genet. 62, 3–13 (2017).
Sannigrahi, M. K., Sharma, R., Panda, N. K. & Khullar, M. Role of Non-coding RNAs in Head and Neck Squamous Cell Carcinoma: a narrative review. Oral Dis., https://doi.org/10.1111/odi.12782 (2017).
Vojtechova, Z. et al. Comparison of the miRNA profiles in HPV-positive and HPV-negative tonsillar tumors and a model system of human keratinocyte clones. BMC Cancer 16, 382 (2016).
Milutin-Gašperov, N., Sabol, I., Halec, G., Matovina, M. & Grce, M. Retrospective study of the prevalence of high-risk human papillomaviruses among Croatian women. Coll. Antropol. 31, 89–96 (2007).
Smeets, S. J. et al. A novel algorithm for reliable detection of human papillomavirus in paraffin embedded head and neck cancer specimen. Int. J. Cancer J. Int. Cancer 121, 2465–2472 (2007).
Lesnikova, I., Lidang, M., Hamilton-Dutoit, S. & Koch, J. Rapid, sensitive, type specific PCR detection of the E7 region of human papillomavirus type 16 and 18 from paraffin embedded sections of cervical carcinoma. Infect. Agent. Cancer 5, 2 (2010).
Ang, K. K. et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 363, 24–35 (2010).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods San Diego Calif 25, 402–408 (2001).
Vincent, M. & Hansen, N. R. Sparse group lasso and high dimensional multinomial classification. Comput. Stat. Data Anal. 71, 771–786 (2014).
Perell, K. et al. Development and validation of a microRNA based diagnostic assay for primary tumor site classification of liver core biopsies. Mol. Oncol. 9, 68–77 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Oliveros, J. V. An interactive tool for comparing lists with Venn’s diagrams (2007).
Vlachos, I. S. et al. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res. 43, W460–W466 (2015).
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34 (1999).
Gillison, M. L., Chaturvedi, A. K., Anderson, W. F. & Fakhry, C. Epidemiology of Human Papillomavirus–Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 33, 3235–3242 (2015).
Cloonan, N. et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 12, R126 (2011).
Guo, L., Yu, J., Liang, T. & Zou, Q. miR-isomiRExp: a web-server for the analysis of expression of miRNA at the miRNA/isomiR levels. Sci. Rep. 6, 23700 (2016).
Telonis, A. G. et al. Knowledge about the presence or absence of miRNA isoforms (isomiRs) can successfully discriminate amongst 32 TCGA cancer types. Nucleic Acids Res. 45, 2973–2985 (2017).
Vojtechova, Z. et al. Analysis of the integration of human papillomaviruses in head and neck tumours in relation to patients’ prognosis. Int. J. Cancer 138, 386–395 (2016).
Nowek, K., Wiemer, E. A. C. & Jongen-Lavrencic, M. The versatile nature of miR-9/9* in human cancer. Oncotarget 9, 20838–20854 (2018).
Wang, J.-Y. et al. MiR-29a: a potential therapeutic target and promising biomarker in tumors. Biosci. Rep. 38 (2018).
Pardini, B. et al. MicroRNAs as markers of progression in cervical cancer: a systematic review. BMC Cancer 18, 696 (2018).
Citron, F. et al. An Integrated Approach Identifies Mediators of Local Recurrence in Head and Neck Squamous Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 23, 3769–3780 (2017).
Salazar, C. et al. A novel saliva-based microRNA biomarker panel to detect head and neck cancers. Cell. Oncol. Dordr. 37, 331–338 (2014).
Minor, J. et al. Methylation of microRNA-9 is a specific and sensitive biomarker for oral and oropharyngeal squamous cell carcinomas. Oral Oncol. 48, 73–78 (2012).
Liu, W. et al. Activation of miR-9 by human papillomavirus in cervical cancer. Oncotarget 5, 11620–11630 (2014).
Thomas, M. et al. Human papillomaviruses, cervical cancer and cell polarity. Oncogene 27, 7018–7030 (2008).
Acknowledgements
This study was funded by the Croatian Science Foundation grant HRZZ-IP-2013-11-4758, Epigenetic changes in head and neck squamous cell carcinoma - EpiC-HNSCC. The acquisition of additional patient material was supported by the grants from Ministry of Education, Youth and Sports of the Czech Republic within the National Sustainability Program II (BIOCEV-FAR project) LQ1604, Charles University, Prague, project GA UK no. 80318, and by the project BIOCEV (CZ.1.05/1.1.00/02.0109). The authors are obligated to all medical doctors, nurses and administrators who have contributed to the collection of samples analyzed in this study. The authors are especially grateful to Mrs. Jasminka Golubić Talić for her expert technical assistance and Nathaniel Edward Bennett Saidu, PhD, for assistance with language editing and constructive discussion.
Author information
Authors and Affiliations
Contributions
K.B. performed HPV, miRNA NGS and qRT experiments and assisted with data analysis, TCGA data procurement and manuscript preparation. I.S. conceptualized the study, designed and assisted with the experiments, performed literature review, analyzed data and wrote the manuscript. E.D. performed surgery and patient data collection, assisted with study design and data interpretation. N.M.G. assisted with study design and manuscript preparation. S.M. provided pathological and other patient data. R.T. and Z.V. provided additional samples, assisted with study design and data analysis. M.G. conceived the project, obtained funding, and assisted with manuscript preparation. All authors reviewed the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Božinović, K., Sabol, I., Dediol, E. et al. Genome-wide miRNA profiling reinforces the importance of miR-9 in human papillomavirus associated oral and oropharyngeal head and neck cancer. Sci Rep 9, 2306 (2019). https://doi.org/10.1038/s41598-019-38797-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-38797-z
- Springer Nature Limited