
Abstract
Sugarcane (Saccharum officinarum L.) is an important crop for sugar production and bioenergy worldwide. In this study, we performed transcriptome sequencing for six contrasting sugarcane genotypes involved in leaf abscission, tolerance to pokkah boeng disease and drought stress. More than 465 million high-quality reads were generated, which were de novo assembled into 93,115 unigenes. Based on a similarity search, 43,526 (46.74%) unigenes were annotated against at least one of the public databases. Functional classification analyses showed that these unigenes are involved in a wide range of metabolic pathways. Comparative transcriptome analysis revealed that many unigenes involved in response to abscisic acid and ethylene were up-regulated in the easy leaf abscission genotype, and unigenes associated with response to jasmonic acid and salicylic acid were up-regulated in response to the pokkah boeng disease in the tolerance genotype. Moreover, unigenes related to peroxidase, antioxidant activity and signal transduction were up-regulated in response to drought stress in the tolerant genotype. Finally, we identified a number of putative markers, including 8,630 simple sequence repeats (SSRs) and 442,152 single-nucleotide polymorphisms (SNPs). Our data will be important resources for future gene discovery, molecular marker development, and genome studies in sugarcane.
Similar content being viewed by others


Introduction
Sugarcane produces more than 70% of the sugar worldwide and is also one of the most important crops for biofuels1. As an alternative energy source, many countries have implemented plans to produce alcohol from sugarcane2,3,4. China produced 10.556 million tons of sugarcane during the 2014/2015 harvest, which was 2.762 million tons less than in 2013/14. Production reduced to 9.3 million tons in 2015/16, with particularly sharp decreases in Guangxi, where the major constraints in sugar production were the increased labor and production cost, decreased sugar price and plantation area, and the more serious drought stress and disease incidence.
The cost for sugarcane production has quickly risen in China over the past several years, especially with regard to labor costs for manual harvest as well as production costs for over fertilization, pesticides and herbicides. Currently, more than 95% of sugarcane is manually harvested in China, in contrast to several other countries where mechanical harvesting dominates. Removal of dead leaves from cane stalks (defoliation) is a major task during harvesting, which contributes greatly to increased costs. One way to address these problems and facilitate green-cane harvesting is the development of easy-defoliating cultivars5. Biotic and abiotic stresses are becoming more serious problems due to the severe singleness of the sugarcane variety in the main sugarcane growing areas, where ROC22 has occupied almost 70% of the total sugarcane area for more than 10 years and more than 80% of sugarcane grown in the upland areas where irrigation is not available6. In recent years, the incidence of sugarcane pokkah boeng disease in China showed a trend of gradually increasing, and has become the main disease during the early growth of sugarcane. Therefore, the release and extension of new cultivars with strong resistance to disease and drought and higher ratooning ability is urgently needed for sugarcane production in China.
Modern sugarcane cultivars are derived from the interspecific hybridizations between S. officinarum, S. spontaneum, and other species in order to obtain disease resistance, high sucrose content, and high yield7. The chromosome number of these modern cultivars ranges from 100 to 130, indicating high levels of polyploidy and aneuploidy8. The genome size of sugarcane cultivar R570 was estimated to be approximately 10 Gb, and average monoploid genome sizes of S. officinarum and S. spontaneum were estimated to be 985 Mb and 843 Mb, respectively9,10. To date, no complete sugarcane genome sequence has been reported, which restricts the development of functional genomics and modern breeding.
Expressed sequence tags (ESTs) provide an important resource for discovering novel genes and assisting in the genome annotation of sugarcane. The first EST database for sugarcane was constructed from leaf roll tissue (meristematic region)11. The Brazilian SUCEST project, the largest EST database (about 238,000 ESTs), was carried out to construct a genetic linkage map and to identify cell wall-related genes in 26 cDNA libraries from many tissues and sugarcane cultivars12,13,14. All of the ESTs are deposited in the Sugarcane Gene Index (version 3.0), which contains 282,683 ESTs and 499 complete cDNA sequences, resulting in 121,342 unigenes. However, more than 10,000 sugarcane coding genes have yet to be identified, highlighting the necessity for further studies on the sugarcane transcriptome15.
RNA sequencing (RNA-seq) is an effective tool for deciphering the transcriptome and is particularly useful for species lacking a sequenced genome. The large quantity of reads obtained can be assembled for gene annotation, gene discovery, gene expression, and identification of regulation patterns in organisms. RNA-seq has also been used for discovery of putative molecular markers (SNPs, SSRs) to facilitate trait map** and marker-assisted selection without the requirement for a reference genome16. RNA-seq technology has been used for transcriptome analysis in many species, such as rice, maize, soybean, and sugarcane17,18,19,20.
In this study, the transcriptomic characterization of six contrasting sugarcane genotypes was compared in response to pokkah boeng, drought and leaf abscission. The sequenced data from the Illumina Hiseq. 2500 were de novo assembled and annotated against several public databases. The putative markers (SSRs, SNPs) were identified, which will be useful for screening variation among the contrasting sugarcane genotypes.
Results
Sequencing and assembly
Each RNA sample was extracted and sequenced using the Illumina paired-end sequencing technology. After quality assessment and data filtering, more than 465 million high-quality reads were used for de novo assembly (Supplementary Table S1). The raw reads were deposited in the Sequence Read Archive (SRA) at GenBank databases ID: SRP127762. A total of 471,654 transcripts were assembled with a mean length of 1,450 bp and an N50 length of 2,067 bp using Trinity software (Table 1). These transcripts represented a total of 93,115 unigenes, with a mean length of 910 bp and an N50 of 1,774 bp. The average length and N50 of the assembled unigenes was higher than those observed for S. spontaneum (801 bp and 1,337 bp) and sugarcane variety GT35 (460 bp and 640 bp) using similar sequencing technologies21,22, demonstrating the high quality of our sugarcane transcriptomic sequences. In total, more than 43,308 unigenes (46.51%) were over 500 bp in length, 25,486 (27.37%) over 1,000 bp, and 12,278 (13.19%) longer than 2,000 bp (Table 1).
Functional annotation
The lack of a reference sugarcane genome is a challenge for gene function prediction and utilization of the transcriptome dataset. Overall, 43,526 (46.74%) unigenes were annotated against at least one of the public databases (Table 2 and Supplementary Table S2). A total of 42,042 (45.15%) unigenes showed homologs in the NR database, while 22,660 (24.34%) unigenes had similarity to proteins in the Swiss-Prot database. However, a total of 49,589 (53.26%) unigenes could not be annotated, suggesting that these unannotated unigenes might be novel genes, although some of these unigenes may represent non-coding RNAs. Among the BLASTx top hits, 19,632 (46.71%) were matched to Sorghum bicolor proteins, followed by Zea mays (9,272; 22.06%), Setaria italica (3,812; 9.07%), and Oryza sativa Japonica Group (1,804; 4.29%) (Fig. 1). These results were consistent with previous reports due to the higher collinearity in the genic regions between sugarcane and sorghum genomes23,24. Interestingly, only 857 (2.04%) unigenes showed significant homology with those of the Saccharum hybrid cultivar R570, which was consistent with a previous report20. This result might be due to the lack of reference genome sequence and limited public data in sugarcane, and might also point to the high genetic variation among different sugarcane genotypes. A total of 20,738 (81.37%) of the unigenes with sizes over 1,000 bp showed homologous matches, whereas only 7,705 (27.67%) of the unigenes shorter than 300 bp were annotated.

Species distribution of the top BLAST hits against the NR database for the assembled unigene (E-value ≤ 1.0 × 10−5). Nearly 46.71% of unigenes showed significant homology with that of Sorghum bicolor and 22.06% showed significant similarity with that of Zea mays.
Cluster of Orthologous Group (COG)
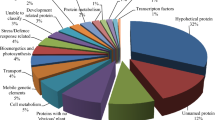
All assembled sugarcane unigenes were searched against the COG database for functional prediction and classification. A total of 10,575 (11.36%) unigenes were assigned and classified into 25 COG categories (Fig. 2). The cluster for ‘general function prediction only’ (2,833; 18.22%) represented the largest group, followed by ‘replication, recombination, and repair’ (2,047; 13.16%), ‘transcription’ (1,500; 9.64%), ‘translation, ribosomal structure and biogenesis’ (1,429; 9.19%) and ‘signal transduction mechanisms’ (1,333; 8.57%). Only a few unigenes were assigned to ‘cell motility’ and ‘nuclear structure’ (8 and 7 unigenes, respectively). In addition, 330 unigenes were assigned to a class representing unknown function. Unigenes in categories representing ‘energy production and conversion’ (745; 4.79%), ‘carbohydrate transport and metabolism’ (712; 4.58%), ‘signal transduction mechanisms’ (1,295; 8.53%), and ‘defense mechanisms’ (178; 1.14%) may be used to develop molecular markers of agronomic traits, such as biomass, sugar content, and abiotic and/or disease resistance.

COG classification of sugarcane assembled unigenes with an E-value threshold of 1.0 × 10−5 against COG databases. Total 10,575 unigenes were grouped into 25 COG classifications, and the cluster of general function prediction represented the largest group, accounting for 18.22%.
Gene Ontology (GO)
GO was used to classify the unigene function predictions according to three main categories: molecular function, biological process, and cellular component. A total of 30,677 (32.95%) unigenes were classified into 55 GO functional sub-categories (Fig. 3). Cellular components represented the majority of the functional terms (77,191; 40.19%), followed by biological processes (76,711; 39.94%) and molecular functions (38,159; 19.87%). Within the cellular components category, ‘cell’ and ‘cell part’ (19,953; 10.39%) was the most dominant group, followed by ‘organelle’ (17,728; 9.23%), ‘membrane’ (7,600; 3.96%), and ‘organelle part’ (3,559; 1.85%). Within the molecular function category, ‘binding’ (17,434; 9.08%) and ‘catalytic activity’ (15,430; 8.03%) were prominently represented, and within the biological process category, ‘metabolic process’ (19,385; 10.09%) and ‘cellular process’ (17,026; 8.86%) were the most enriched. The unigenes involved in the categories representing ‘signaling’ (1,235), ‘receptor activity’ (229), ‘response to stimulus’ (5,937), and ‘antioxidant activity’ (276), might be closely related to drought or/and disease response and provided valuable information for further studies.

Gene ontology (GO) classification of the assembled unigenes. A total of 30,677 unigenes with BLASTx matches were assigned to three main categories: biological processes (76,711; 39.94%), cellular components (77,191; 40.19%), and molecular functions (38,159; 19.87%).
KEGG pathway
To further understand the biology and intricate metabolic pathways of sugarcane, all assembled unigenes were annotated using the KEGG pathway database. In total, 12,367 (13.28%) unigenes were annotated to 126 pathways (Supplementary Table S3). The most highly represented pathways were ‘ribosome’ (1,009; 8.16%), followed by ‘carbon metabolism’ (452; 3.66%), ‘biosynthesis of amino acids’ (393; 3.18%) and ‘protein processing in endoplasmic reticulum’ (388; 3.14%). Unigenes in the ‘plant-pathogen interaction’ pathway (358; 2.90%) may be useful for studying the resistance mechanisms to pokkah boeng disease in sugarcane. Pathways associated with leaf abscission and drought stress, such as ‘plant hormone signal transduction’ (276; 2.23%), ‘glutathione metabolism’ (177; 1.43%) and ‘ubiquitin mediated proteolysis’ (189; 1.53%) will be the focus of future studies.
Comparative Transcriptomic Characterization of Contrasting Sugarcane Cultivars
Based on the RNA-seq data, the unique and shared unigenes were determined among the contrasting sugarcane cultivars (Fig. 4A). GXU-34140 and GXU-34176 shared a total of 55,461 (75.82%) unigenes, and GXU-34176 exhibited a higher number of unique unigenes (9,987) than those of GXU-34140 (7,703). A total of 72,562 (87.29%) unigenes were co-expressed in GN18 and FN95–1702 under drought stress, but the number of uniquely expressed unigenes in GN18 (6,855) was larger than that in FN95–1702 (3,709). GUC2 and GUC10 shared 36,854 (72.65%) unigenes, while the pokkah boeng disease susceptible GUC10 exhibited a higher number of unique unigenes (7,573) than the resistant cultivar GUC2 (6,301).

Venn diagram showing the number of unique and shared unigenes and SNPs among the contrasting sugarcane genotypes. (A) The number of unique and shared unigenes determined based on RSEM analysis. (B) The number of identified unique and shared putative SNPs based on GATK2 analysis. Only SNPs with distance >5 were retained.
A total of 7,838 differentially expressed genes (DEGs) were identified between GXU-34140 and GXU-34176, with 4,582 up-regulated and 3,062 down-regulated in GXU-34176 (both +1 and +3 leaf sheath samples) when compared with GXU-34140. The remaining 194 DEGs showed the opposite expression pattern. According to GO functional enrichment analysis, GO terms, including ‘abscisic acid-activated signaling pathway’ (22 unigenes) and ‘jasmonic acid mediated signaling pathway’ (24 unigenes), were enriched in the up-regulated DEGs, while ‘cellulose synthase (UDP-forming) activity’ (26 unigenes), ‘cellulose biosynthetic process’ (30 unigenes), ‘plant-type primary cell wall biogenesis’ (7 unigenes), and ‘cell wall’ (56 unigenes) were enriched in the down-regulated DEGs (Supplementary Table S4). These results suggested that those genes might play an important role in responsive to leaf abscission for the easy defoliation GXU-34176.
In the comparison of GN18 with FN95–1702, a total of 5,314 DEGs were identified, of which 3,428 DEGs were up-regulated and 1,839 DEGs were down-regulated in GN18 responses to drought stress (both in mild drought and severe drought). The remaining 47 DEGs showed the opposite pattern. GO functional enrichment analysis indicated that ‘photosynthesis’ (23 unigenes), ‘chloroplast thylakoid membrane’ (36 unigenes), and ‘photosystem I’ (7 unigenes) were enriched in the up-regulated DEGs (Supplementary Table S4). These results suggested that interference of photosynthesis was less affected by drought stress in GN18 when compared with FN95–1702.
A total of 3,645 DEGs were identified in the comparison between GUC2 and GUC10, of which 2,175 DEGs were up-regulated and 1,454 DEGs were down-regulated in pokkah boeng resistant GUC2 (both in healthy and infected samples). Only 16 DEGs showed the opposite pattern. GO functional enrichment analysis indicated that ‘protein phosphorylation’ (162 unigenes), ‘protein serine/threonine kinase activity’ (139 unigenes), ‘transmembrane receptor protein serine/threonine kinase signaling pathway’ (16 unigenes), and ‘response to salicylic acid’ (19 unigenes) were enriched in the up-regulated DEGs (Supplementary Table S4), suggesting that they might represent special mechanisms in GUC2 responses to pokkah boeng disease.
Quantitative Real-time PCR (qRT-PCR) validation of target genes
To validate the reliability and reproducibility of the Illumina RNA-Seq results, fourteen unigenes based on their characterizations of contrasting sugarcane genotypes were validated via qRT-PCR analysis. The primers used for qRT-PCR are listed in Supplementary Table S5. In GXU-34176, c71654.graph_c0 (encoding lipase-like PAD4) and c65832.graph_c0 (encoding probable protein phosphatase 2 C) related to ‘abscisic acid-activated signaling pathway’, c67492.graph_c0 (encoding putative serine/threonine-protein kinase-like protein CCR3) associated with ‘response to ethylene’ and c64240.graph_c0 (encoding zinc finger CCCH domain-containing protein) related to ‘leaf senescence’ were up-regulated in +3 leaf sheath compared to GXU-34140 (Fig. 5). Interestingly, the c65986.graph_c0 (encoding COBRA-like protein) related to ‘cell wall modification involved in abscission’ was down-regulated in +3 leaf sheath. In GN18, c54647.graph_c0 (encoding putative leucine-rich repeat receptor-like protein kinase family protein), c56804.graph_c0 (encoding protein TIFY 9), and c61335.graph_c0 (encoding heat shock protein 90) associated with ‘response to water deprivation’ as well as c57471.graph_c0 (encoding peroxidase) related to ‘response to oxidative stress’ were up-regulated under mild or severe drought stress compared to FN95–1702 (Fig. 5). In resistant cultivar GUC2, c69746.graph_c0 (encoding U-box domain-containing protein) associated with ‘ubiquitin-protein transferase activity’, c72075.graph_c0 (encoding respiratory burst oxidase homolog protein) related to ‘defense response to fungus’, and c65355.graph_c0 (encoding allene oxide synthase 2) related to ‘response to jasmonic acid’ were up-regulated after infected with F. verticillioides compared to sensitive cultivar GUC10 (Fig. 5). Pearson correlation coefficient between RNA-Seq and qRT-PCR was 0.89 and the Significance (two-tailed t test) was 1.11 × 10−4. These results showed that the expression trend of these genes was consistent with the transcriptome data.

Verification of RNA-seq results by qRT-PCR. The DEGs in GXU-34176 vs GXU-34140 related to the category of ‘abscisic acid-activated signaling pathway’ (c71654.graph_c0 and c65832.graph_c0), ‘leaf senescence’ (c64240.graph_c0), ‘response to ethylene’ (c67492.graph_c0) and ‘cell wall modification involved in abscission’ (c65986.graph_c0). The DEGs in GN18 vs FN95–1702 related to the category of ‘response to water deprivation’ (c54647.graph_c0, c56804.graph_c0 and c61335.graph_c0) and ‘response to oxidative stress’ (c57471.graph_c0). The DEGs in GUC2 vs GUC10 related to the category of ‘ubiquitin-protein transferase activity’ (c69746.graph_c0), ‘defense response to fungus’ (c72075.graph_c0), ‘response to jasmonic acid’ (c65355.graph_c0). Data of qRT-PCR are presented as mean ± SD (n = 9) and error bars represent SD.
Putative marker discovery for marker-selection of important traits
SSR markers are important tools for studying genetic diversity, constructing genetic maps, and performing comparative genomics25. Potential SSR markers were detected from unigenes with lengths over 1,000 bp using MISA software. A total of 8,630 SSRs were mined from 6,883 unigenes, of which 1,413 sequences contained more than one SSR and 286 SSRs were present in a compound formation (Supplementary Table S6). On average, the distribution density of SSRs was 1/6.62 kb. The most abundant repeat motifs were mononucleotide (4,162; 48.23%) and trinucleotide (2,924; 33.88%), followed by dinucleotide (1,392; 16.13%) and tetranucleotide (120; 1.39%) (Fig. 6A). Pentanucleotide and hexanucleotide repeat motifs represented only 0.22% and 0.15% of the total SSRs, respectively. The identified proportion of trinucleotide repeats were similar to the survey in the sugarcane EST (SUCEST) database (30.5%), but the percentage of tetranucleotides was lower than that obtained in the previous report26. Taken together, 188 types of nucleotide motif repeats were detected among 8,630 SSR loci. The most abundant repeat type was A/T (4,084; 47.32%), followed by CCG/CGG (1,237; 14.33%), AG/CT (712; 8.25%) and AGC/CTG (460; 5.33%) (Fig. 6B). These results were similar to those of the SSR motif previously reported20. Based on the 8,630 SSRs, primer pairs were designed using Primer 3.0 and are listed in Supplementary Table S6. These data are valuable resources for further studies on marker-assisted selection in sugarcane breeding.

Characterization of potential SSR markers using MISA software. A total of 8,630 SSRs were mined from 6,883 unigenes. (A) Distribution of the different nucleotide repeat types. The most abundant repeat motifs were mononucleotide (4,162; 48.23%), followed by trinucleotide (2,924; 33.88%). (B) Frequency of the different classified repeat motifs. The repeat types of A/T and CCG/CGG accounted for 47.32% and 14.33%, respectively.
SNPs are also widely used as molecular markers for the identification of quantitative trait locus, evolutionary analysis, and development marker-assisted selection to accelerate plant breeding. A total of 442,152 putative SNP positions were identified in 55,659 different unigenes (Supplementary Table S7). The unique and shared SNPs between the contrasting sugarcane genotypes were evaluated in Fig. 4B. The number of shared SNPs were 313,666 (80.25%) for GXU-34140 and GXU-34176, 172,118 (68.59%) for GUC2 and GUC10, and 386,996 (91.42%) for GN18 and FN95–1702, respectively. More shared SNPs resulted from the same parent of GXU-34140 and GXU-34176 or GN18 derived from FN95–1702 mediated with the Ea-DREB2B gene. A total of 77,184 SNPs (in 30,171 unigenes), 78,807 SNPs (in 25,029 unigenes), and 36,332 SNPs (in 19,719 unigenes) were different between GXU-34140 and GXU-34176, GUC2 and GUC10, and GN18 and FN95–1702, respectively. According to the GO annotation of unigenes that contained the unique SNPs in each group (Supplementary Table S8), several important categories were found to be associated with the sugarcane genotypes. For GXU-34140 and GXU-34176, 138 unigenes with SNPs were confirmed in the ‘response to abscisic acid’ category, 34 unigenes with SNPs in the ‘response to ethylene’ category, 17 unigenes with SNPs in the ‘leaf senescence’ category, and 17 unigenes with SNPs in the ‘cell wall modification’ category. Only one unigene with SNP was identified in the ‘cell wall modification involved in abscission’. For GUC2 and GUC10, 58 unigenes with SNPs were observed in the ‘defense response to fungus’, 17 in the ‘defense response to fungus, incompatible interaction’, 25 in the ‘response to fungus’, one in the ‘regulation of defense response to fungus, incompatible interaction’, and four in the ‘jasmonic acid and ethylene-dependent systemic resistance’. For GN18 and FN95–1702, 164 unigenes with SNPs were observed in the ‘defense response’, 50 in the ‘response to stress’, five in the ‘response to desiccation’, 49 in the ‘response to oxidative stress’, 45 in the ‘response to water deprivation’, and seven in the ‘water transport’. The SNPs derived from transcriptome sequences (in the transcribed regions) may be directly linked to expressed genes. The SNPs unique to each genotype are useful for develo** markers associated with important agronomic traits of the contrasting genotypes.
Discussion
Biotic and abiotic stresses in nature seriously restrict the development of the sugarcane industry worldwide. Currently the major aims of sugarcane breeding programs are to develop cultivars with strong resistance to phytopathogens (particularly Ustilago scitaminea and Fusarium species complex) and abiotic stress (especially drought stress), high sucrose and yield, and suitable for mechanized harvest. In our experiments, six sugarcane genotypes with contrasting responses to stress (drought, pokkah boeng disease) and defoliation were sequenced using the Illumina sequencing platform and 93,115 unigenes were assembled with an N50 of 1,774 bp, of which 43,526 unigenes were annotated against at least one of the public databases. The more assembled and annotated sequences in our studies indicated a comprehensive reference transcriptome of sugarcane when compared with other reports20,21. Many unigenes were identified in the signal transduction (1,333) and response to stimulus (5,937) categories, which were conducive to understanding the resistance mechanism.
Leaf abscission, which is one of the important traits for the sugarcane breeding program, is beneficial for improving the efficiency of sugarcane harvest. Two contrasting sugarcane genotypes (GXU-34140 with difficult-defoliation and GXU-34176 with easy-defoliation) derived from the same cross Co1001 × ROC22 showed different defoliation ability from our previous studies. ABA and ethylene played a more important role in the abscission of organs27,28,29. Compared to the leaf packaging sugarcane varieties (Q2 and B), ten transcripts involved in ‘abscisic acid associated pathways’ were up-regulated in leaf abscission sugarcane varieties (Q1 and T)30. In this work, more unigenes involved in ‘abscisic acid-activated signaling pathway’ (22 unigenes) and ‘response to abscisic acid’ (38 unigenes) were up-regulated in easy-defoliation GXU-34176 when compared to difficult-defoliation GXU-34140. Interestingly, 15 unigenes involved in ‘response to ethylene’ were up-regulated in GXU-34176, which was not observed in leaf abscission sugarcane varieties (Q1 and T)30. The position of organ separation from the plant body is called abscission zones (AZs). In Phaseolus vulgaris petioles, ethylene may induced the formation of AZs31. A total of 10 unigenes related to ‘cell wall macromolecule catabolic processes’ were up-regulated and 30 unigenes involved in ‘cellulose biosynthetic processes’ were down-regulated in GXU-34176. It is well-known that the common feature of abscission processes is cell wall degradation32. Genes involved in cell-wall modification during the abscission period have also been identified in Citrus and Tomato33,34.
Sugarcane pokkah boeng disease, caused by Fusarium species complex, is one of the most serious and devastating diseases recorded in countries where sugarcane is grown35. It has reportedly caused a yield loss of 40.8–64.5% in infection-susceptible sugarcane cultivars36,37. Based on our field survey and inoculation test, GUC2 was a promising genotype resistant to pokkah boeng disease, whereas GUC10 was susceptible to pokkah boeng. Comparative transcriptomic analysis indicated that 12 and 19 unigenes involved in response to jasmonic acid and salicylic acid were up-regulated in resistant GUC2. Salicylic acid (SA)-dependent signaling pathways and jasmonic acid (JA)-dependent signaling pathways are thought to form the backbone of the plant defense system38. In general, SA-mediated defenses are activated to resist biotrophic pathogens, whereas JA-mediated defenses confer resistance against necrotrophic pathogens that kill host cells39,40. SA and JA has been reported as defense elicitors to enhance resistance to Dutch elm disease in the tolerant phenotype of American elm41. The ubiquitin-proteasome system plays important roles in the regulation of plant immunity, especially the E3 ubiquitin ligases42. The results presented here showed one and 14 unigenes encoding an ubiquitin conjugating enzyme and ubiquitin-protein transferase activity were up-regulated in GUC2, respectively. In addition, our data also revealed that more than 100 unigenes encoding protein serine/threonine kinases were up-regulated in response to Fusarium species infection in GUC2. Future studies should focus on how pathogens and elicitors are perceived by protein serine/threonine kinases and how protein serine/threonine kinases and their activated signaling pathways are regulated during the Fusarium infection process in GUC2.
Natural disasters, such as drought and low temperature, occur frequently and have caused serious loss to sugarcane production43. Therefore, the development of sugarcane cultivars tolerant to abiotic stresses is crucial to improve the profitability of sugarcane industries. Transgenic technology can improve the efficiency of genetic improvement in many crops44,59. Three biological and three technical replications were performed for each sample. The data were analyzed using LightCycler® 480 sofware version 1.5.1 provided by Roche. The relative fold change of the selected genes was calculated using 2−△△Ct algorithm60. The Pearson correlation test was performed by Origin 9.0 sofware.
References
Yearbook, C. Commodity Research Bureau. New York (1939).
Bizzo, W. A., Lenço, P. C., Carvalho, D. J. & Veiga, J. P. S. The generation of residual biomass during the production of bio-ethanol from sugarcane, its characterization and its use in energy production. Renewable and Sustainable Energy Reviews 29, 589–603 (2014).
Li, S. Z. & Chan-Halbrendt, C. Ethanol production in (the) People’s Republic of China: potential and technologies. Applied Energy 86, S162–S169 (2009).
Martines-Filho, J., Burnquist, H. L. & Vian, C. E. Bioenergy and the rise of sugarcane-based ethanol in Brazil. Choices 21, 91–96 (2006).
Cock, J., Amaya, A., Bohorquez, C. & Munchmeyer, B. Simulation of production potential of self-defoliating sugarcane cultivars. Field crops research 54, 1–8 (1997).
Zhang, M. Q., Wang, H. Z. & Bai, C. Genetic improvement and high efficient breeding of sugar crops. (Chinese Agricultural Publishing 2005).
Ming, R. et al. Detailed alignment of Saccharum and Sorghum chromosomes: comparative organization of closely related diploid and polyploid genomes. Genetics 150, 1663–1682 (1998).
Ming, R. et al. Sugarcane improvement through breeding and biotechnology. Plant breeding reviews 27, 15 (2006).
Grivet, L. & Arruda, P. Sugarcane genomics: depicting the complex genome of an important tropical crop. Current Opinion in Plant Biology 5, 122–127 (2002).
Zhang, J. et al. Genome size variation in three Saccharum species. Euphytica 185, 511–519 (2012).
Carson, D. L. & Botha, F. C. Preliminary analysis of expressed sequence tags for sugarcane. Crop Science 40, 1769–1779 (2000).
Vettore, A. L., Silva, F. R. D., Kemper, E. L. & Arruda, P. The libraries that made SUCEST. Genetics and Molecular Biology 24, 1–7 (2001).
Oliveira, K. M. et al. Functional integrated genetic linkage map based on EST-markers for a sugarcane (Saccharum spp.) commercial cross. Molecular Breeding 20, 189–208 (2007).
Lima, D., Santos, H., Tiné, M., Molle, F. & Buckeridge, M. Patterns of expression of cell wall related genes in sugarcane. Genetics and Molecular Biology 24, 191–198 (2001).
Vicentini, R., Del Bem, L., Van Sluys, M., Nogueira, F. & Vincentz, M. Gene content analysis of sugarcane public ESTs reveals thousands of missing coding-genes and an unexpected pool of grasses conserved ncRNAs. Tropical plant biology 5, 199–205 (2012).
D’Hont, A. Unraveling the genome structure of polyploids using FISH and GISH; examples of sugarcane and banana. Cytogenetic and genome research 109, 27–33 (2005).
Lu, T. et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome research 20, 1238–1249 (2010).
Hansey, C. N. et al. Maize (Zea mays L.) genome diversity as revealed by RNA-sequencing. PloS one 7, e33071 (2012).
Deschamps, S. et al. Rapid genome-wide single nucleotide polymorphism discovery in soybean and rice via deep resequencing of reduced representation libraries with the Illumina genome analyzer. The Plant Genome 3, 53–68 (2010).
Cardoso-Silva, C. B. et al. De novo assembly and transcriptome analysis of contrasting sugarcane varieties. PloS one 9, e88462 (2014).
Dharshini, S. et al. De novo sequencing and transcriptome analysis of a low temperature tolerant Saccharum spontaneum clone IND 00-1037. Journal of biotechnology 231, 280–294 (2016).
Huang, D. L. et al. Transcriptome of High-sucrose sugarcane variety GT35. Sugar Tech 18, 520–528 (2016).
Grivet, L. et al. Comparative genome map** of sugar cane with other species within the Andropogoneae tribe. Heredity 73, 500–508 (1994).
Wang, J. et al. Microcollinearity between autopolyploid sugarcane and diploid sorghum genomes. BMC genomics 11, 261 (2010).
Singh, R., Mishra, S. K., Singh, S. P., Mishra, N. & Sharma, M. Evaluation of microsatellite markers for genetic diversity analysis among sugarcane species and commercial hybrids. Australian Journal of Crop Science 4, 116 (2010).
Pinto, L. R., Oliveira, K. M., Ulian, E. C., Garcia, A. A. F. & De Souza, A. P. Survey in the sugarcane expressed sequence tag database (SUCEST) for simple sequence repeats. Genome 47, 795–804 (2004).
Taylor, J. E. & Whitelaw, C. A. Signals in abscission. New Phytologist 151, 323–340 (2001).
Davis, L. A. & Addicott, F. T. Abscisic acid: correlations with abscission and with development in the cotton fruit. Plant Physiology 49, 644–648 (1972).
Reid, M. S. Ethylene and abscission. HortScience 20, 45–50 (1985).
Li, M. et al. De novo analysis of transcriptome reveals genes associated with leaf abscission in sugarcane (Saccharum officinarum L.). BMC genomics 17, 195 (2016).
McManus, M. T., Thompson, D. S., Merriman, C., Lyne, L. & Osborne, D. J. Transdifferentiation of mature cortical cells to functional abscission cells in bean. Plant Physiology 116, 891–899 (1998).
Roberts, J. A., Elliott, K. A. & Gonzalez-Carranza, Z. H. Abscission, dehiscence, and other cell separation processes. Annual review of plant biology 53, 131–158 (2002).
Agustí, J., Merelo, P., Cercós, M., Tadeo, F. R. & Talón, M. Ethylene-induced differential gene expression during abscission of citrus leaves. Journal of Experimental Botany 59, 2717–2733 (2008).
Nakano, T. et al. MACROCALYX and JOINTLESS interact in the transcriptional regulation of tomato fruit abscission zone development. Plant physiology 158, 439–450 (2012).
Rott, P. A guide to sugarcane diseases. (Editions Quae, 2000).
Patil, A. & Hapase, D. Studies on Pokkah boeng disease in Maharashtra. Ind Phytopath 40, 290 (1987).
Dohare, S., Mishra, M. & Kumar, B. Effect of wilt on juice quality of sugarcane. Annals of Biology 19, 183–186 (2003).
Pieterse, C. M. et al. Hormonal modulation of plant immunity. Annual review of cell and developmental biology 28, 489–521 (2012).
Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 43, 205–227 (2005).
Pieterse, C. M., Leon-Reyes, A., Van der Ent, S. & Van Wees, S. C. Networking by small-molecule hormones in plant immunity. Nature chemical biology 5, 308–316 (2009).
Sherif, S., Shukla, M., Murch, S., Bernier, L. & Saxena, P. Simultaneous induction of jasmonic acid and disease-responsive genes signifies tolerance of American elm to Dutch elm disease. Scientific reports 6, 21934 (2016).
Craig, A., Ewan, R., Mesmar, J., Gudipati, V. & Sadanandom, A. E3 ubiquitin ligases and plant innate immunity. Journal of Experimental Botany 60, 1123–1132 (2009).
Li, Y. R. & Yang, L. T. Sugarcane agriculture and sugar industry in China. Sugar Tech 17, 1–8 (2015).
Dos Santos, T. B. et al. Expression of three galactinol synthase isoforms in Coffea arabica L. and accumulation of raffinose and stachyose in response to abiotic stresses. Plant Physiology and Biochemistry 49, 441–448 (2011).
Chen, J. Q., Meng, X. P., Zhang, Y., **a, M. & Wang, X. P. Over-expression of OsDREB genes lead to enhanced drought tolerance in rice. Biotechnology letters 30, 2191–2198 (2008).
Wu, Y., Zhou, H., Que, Y. X., Chen, R. K. & Zhang, M. Q. Cloning and identification of promoter Prd29A and its application in sugarcane drought resistance. Sugar Tech 10, 36–41 (2008).
Agarwal, P. K., Agarwal, P., Reddy, M. K. & Sopory, S. K. Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Reports 25, 1263–1274 (2006).
Augustine, S. M. et al. Overexpression of EaDREB2 and pyramiding of EaDREB2 with the pea DNA helicase gene (PDH45) enhance drought and salinity tolerance in sugarcane (Saccharum spp. hybrid). Plant Cell Reports 34, 247–263 (2015).
Mittler, R., Vanderauwera, S., Gollery, M. & Van Breusegem, F. Reactive oxygen gene network of plants. Trends in plant science 9, 490–498 (2004).
Lin, Z. et al. Species-specific detection and identification of fusarium species complex, the causal agent of sugarcane pokkah boeng in China. PloS one 9, e104195 (2014).
Hsiao, T. C., Acevedo, E., Fereres, E. & Henderson, D. Water stress, growth, and osmotic adjustment. Philosophical Transactions of the Royal Society B: Biological Sciences 273, 479–500 (1976).
Patel, R. K. & Jain, M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS one 7, e30619 (2012).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology 29, 644–652 (2011).
**e, C. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic acids research 39, W316–W322 (2011).
Finn, R. D. et al. Pfam: the protein families database. Nucleic acids research 42, D222–D230 (2013).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303 (2010).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 12, 323 (2011).
Young, M. D., Wakefield, M. J., Smyth, G. K. & Oshlack, A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome biology 11, R14 (2010).
Iskandar, H. M. et al. Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression in sugarcane. Plant Molecular Biology Reporter 22, 325–337 (2004).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Acknowledgements
We greatly appreciate Bioscience Editing Solutions for critically reading this paper and providing helpful suggestions. Financial support was provided by Natural Science Foundation of Guangxi (2014GXNSFFA118002) and National Natural Science Foundation of China (31460374 and 31660420).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Z.M.Q. and C.B.S. Performed the experiments: X.S.Q., W.J.H., S.H.Y., J.H.T. and H.Y.Z. Analyzed the data: X.S.Q., W.J.H., S.H.Y., Y.W. and Z.M.Q. Contributed reagents/materials/analysis tools: Z.M.Q. and C.B.S. Wrote the paper: X.S.Q. and Z.M.Q. All authors read and approved the final version of the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, S., Wang, J., Shang, H. et al. Transcriptomic characterization and potential marker development of contrasting sugarcane cultivars. Sci Rep 8, 1683 (2018). https://doi.org/10.1038/s41598-018-19832-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19832-x
- Springer Nature Limited
This article is cited by
-
Sugar Transporters, Sugar-Metabolizing Enzymes, and Their Interaction with Phytohormones in Sugarcane
Journal of Plant Growth Regulation (2023)
-
Sugarcane Transcriptomics in Response to Abiotic and Biotic Stresses: A Review
Sugar Tech (2022)
-
Transcriptome changes in the develo** sugarcane culm associated with high yield and early-season high sugar content
Theoretical and Applied Genetics (2022)
-
Leaf transcriptome profiling of contrasting sugarcane genotypes for drought tolerance under field conditions
Scientific Reports (2022)
-
Comparative analysis of drought-responsive transcriptomes of sugarcane genotypes with differential tolerance to drought
3 Biotech (2020)