
Abstract
Growth differentiation factor 15 (GDF15) has recently been shown to have an important role in the regulation of mitochondrial function and in the pathogenesis of complex human diseases. Nevertheless, the role of GDF15 in alcohol-induced or fibrotic liver diseases has yet to be determined. In this study, we demonstrate that alcohol- or carbon tetrachloride (CCl4)-mediated hepatic GDF15 production ameliorates liver inflammation and fibrosis. Alcohol directly enhanced GDF15 expression in primary hepatocytes, which led to increased oxygen consumption. Moreover, GDF15 reduced the expression of pro-inflammatory cytokines in liver-resident macrophages, leading to an improvement in inflammation and fibrosis in the liver. GDF15 knockout (KO) mice had more TNF-α-producing T cells and more activated CD4+ and CD8+ T cells in the liver than wild-type mice. Liver-infiltrating monocytes and neutrophils were also increased in the GDF15 KO mice during liver fibrogenesis. These changes in hepatic immune cells were associated with increased tissue inflammation and fibrosis. Finally, recombinant GDF15 decreased the expression of pro-inflammatory cytokines and fibrotic mediators and prevented the activation of T cells in the livers of mice with CCl4-induced liver fibrosis. These results suggest that GDF15 could be a potential therapeutic target for the treatment of alcohol-induced and fibrotic liver diseases.
Similar content being viewed by others


Introduction
The liver can be considered an immunologic organ, in which antigen-rich blood from the gastrointestinal tract interacts with diverse innate and adaptive immune cells1,2. These immune cells play important roles in the development of hepatic inflammation, steatohepatitis, and fibrotic liver diseases3,4. Hepatic macrophages have been implicated in the inflammation induced by hyperglycemia in mice5. Natural killer T cells and gamma delta T cells are involved in alcoholic liver injury and hepatic fibrosis, respectively6,7. Additionally, migration and activation of neutrophils increase alcohol-induced liver injury8. Therefore, many studies have focused on alleviating the secretion of pro-inflammatory cytokines produced by immune cells to treat chronic liver diseases9,10.
Growth differentiation factor 15 (GDF15), a member of the transforming growth factor beta superfamily, has anti-inflammatory activities through a currently unknown receptor11. In previous reports, serum levels of GDF15 were enhanced in patients with viral hepatitis, cancer, or metabolic disease compared to healthy controls12,13,14,15. Mitochondrial dysfunction was also associated with elevated serum GDF15 levels in obese mice, which may be a physiologic response to restore metabolic homeostasis16. Therefore, GDF15 induction in various inflammatory diseases is thought to be an adaptation to stress response signaling pathways activated by mitochondrial stress.
Hepatocytes display an eosinophilic cytoplasm upon hematoxylin-eosin staining, reflecting abundant mitochondria17. Thus, it is not surprising that mitochondrial dysfunction promotes cellular damage and is linked to liver diseases. Chronic alcohol consumption alters mitochondrial oxidative phosphorylation in the liver by suppressing the synthesis of respiratory complex proteins18. Alcohol-mediated damage of mitochondrial DNA (mtDNA) also impairs cellular energy metabolism via enhanced formation of reactive oxygen species (ROS)19. Additionally, carbon tetrachloride (CCl4) reduces mitochondrial respiratory chain complex IV activity and depletes mtDNA in the liver20. Moreover, the radicals produced by cytochrome P450 2E1-mediated CCl4 metabolism bind to mtDNA directly and also promote lipid peroxidation, which results in degradation of mtDNA20,21. Although hepatotoxic molecules such as alcohol and CCl4 promote mitochondrial dysfunction in the liver, the role of GDF15 as a mitohormetic factor in alcohol- and CCl4-induced liver injury remains to be elucidated.
In this study, we aimed to establish a direct link between mitochondrial function and GDF15 induction. We also explored the anti-inflammatory role of GDF15 in the development of alcohol- and CCl4-induced liver injury and examined whether deficiency of GDF15 exacerbates liver injury and fibrosis in mice. Therefore, this study provides a range of pathophysiological insights into alcoholic and fibrotic liver diseases.
Research Design and Methods
Mice and ethical considerations
Wild-type (WT) mice on a C57BL/6 background were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). GDF15 KO mice derived from the inbred C57BL/6 strain were provided by Dr. S. Lee (Johns Hopkins University School of Medicine, Baltimore, MD, USA). All mice were maintained in a specific pathogen-free animal facility (Chungnam National University Hospital Preclinical Research Center) in a controlled environment (12 h light/12 h dark cycle; humidity, 50–60%; ambient temperature, 22 ± 2 °C). Mice were placed on a Lieber-DeCarli low-fat liquid diet (Dyets, Dyets, Inc., Bethlehem, PA, USA) containing 1 kcal/ml, of which 18% was derived from protein, 12% from fat, and either 70% from carbohydrate (control diet) or 43% from carbohydrate and 27% from ethanol (alcohol diet). Alcohol was administered gradually by escalating the content by 1% (v/v) each day until the mice were consuming a diet containing 5% (v/v) ethanol. This was continued for six more weeks. At the end of this period, mice were sacrificed, and liver tissues and whole blood were collected for further analysis. All animal experiments were approved by the Institutional Review Board on Animal Experimentation of Chungnam National University School of Medicine (CNUH-A0001) and performed in accordance with the guidelines and regulations of Chungnam National University.
Carbon tetrachloride (CCl4)-induced liver fibrosis in mice
In age-matched (8-week-old) WT and GDF15 KO male mice, liver fibrosis was induced by intraperitoneal injection of CCl4 (Sigma-Aldrich, MO; 2 mL/kg in olive oil, 20% v/v) three times per week for 3 weeks. Mice were sacrificed 12 hours after the final CCl4 injection.
Serum biochemical measurements
Serum was collected from whole blood by centrifugation at 10,000 rpm for 5 minutes at room temperature and was assayed for aspartate aminotransferase (AST) and alanine aminotransferase (ALT) using a Fuji Dri-Chem 4000i analyzer (Fujifilm, Tokyo, Japan). Serum or hepatic levels of tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) were measured using an enzyme linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA).
Histological analyses
Sections of the left and medial lobes of the liver were fixed with 10% neutral buffered formalin (BBC Biochemical, Mt. Vernon, WA, USA) or frozen in Tissue-Tek OCT compound (Sakura, Tokyo, Japan). For formalin-fixed samples, after deparaffinization in xylene and rehydration with ethanol, 4 μm-thick sections were stained with hematoxylin and eosin (H&E) and Sirius Red (Sigma-Aldrich, MO, USA) for detection of collagen deposition in the liver. Frozen sections (10 μm thick) were stained with Oil Red O Kit (ab150678; Abcam, Cambridge, UK) for visualization of lipid accumulation in the liver tissue.
Measurement of hepatic triglyceride levels
Hepatic lipids were extracted from 100 mg liver tissue using a mixture of chloroform and methanol (2:1 ratio), as previously described22. Hepatic lipid extracts were lyophilized with nitrogen gas, and the triglyceride (TG) levels in resuspended lipids were analyzed on a Fuji Dri-Chem 4000i analyzer according to the manufacturer’s instructions (Fujifilm, Tokyo, Japan).
Isolation of hepatocytes in mice
As previously described23,24, mouse livers were perfused in situ with EGTA solution (5.4 mM KCl, 0.44 mM KH2PO4, 140 mM NaCl, 0.34 mM Na2HPO4, 0.5 mM EGTA, 25 mM Tricine, pH 7.2), followed by the perfusion with a digestion buffer (0.8 mg/ml collagenase type I in HBSS; Worthington, Freehold, NJ, USA) for 30 minutes. The liver cell suspensions were filtered through 70 μm nylon cell strainers (BD Falcon, Millville, NJ, USA), and then centrifuged at 1,000 × g for 5 minutes. The pellet was suspended in 40% Percoll (GE Healthcare, Buckingham, UK) and centrifuged at 1,200 × g for 10 minutes at 4 °C. Isolated cells were cultured in DMEM (Welgene, Daegu, South Korea) supplemented with 10% fetal bovine serum (ThermoFisher Scientific, Waltham, MA, USA) and 1% penicillin/streptomycin (Welgene, Daegu, South Korea) at a density of 2 × 105 cells/well on 6-well plates.
Isolation of liver mononuclear cells
Liver mononuclear cells (MNCs) were isolated as described previously5,23. Briefly, liver tissues were cut into small pieces and incubated with pre-warmed media containing dissociation enzymes (Miltenyi Biotec, Bergisch Gladbach, Germany) for 30 minutes at 37 °C. After enzymatic digestion, the liver cell suspensions were quickly homogenized in C-Tubes using the GentleMACS Dissociator (Miltenyi Biotec) on the m_liver_03 program. After removal of debris, the cells were resuspended in phosphate-buffered saline and centrifuged at 1,000 × g for 5 minutes for the elimination of hepatocytes. The supernatant was removed with mechanical suction and filtered through a 70 μm nylon cell strainer (BD Falcon, Millville, NJ). Hepatic MNCs were isolated by centrifugation at 1,200 × g for 10 minutes at 4 °C and resuspended in RPMI-1640 medium (Welgene, Daegu, South Korea).
Isolation of hepatic stellate cells and Kupffer cells
As described previously5,23, hepatic stellate cells (HSCs) were isolated by in situ collagenase perfusion of the liver. After anesthesia by intraperitoneal injection with 100–200 mg/kg ketamine/xylazine cocktails, the liver was perfused in situ with collagenase type I through the portal vein. Non-parenchymal cells were separated by centrifugation at 500 rpm and 4 °C for 5 minutes. Then, the HSCs and Kupffer cells were isolated by differential centrifugation on a density gradient of 11.5% and 20% OptiPrep (Sigma-Aldrich, MO, USA) at 3,000 rpm and 4 °C for 17 minutes. HSCs and Kupffer cells were located in the upper and lower layers, respectively. Kupffer cells were positively purified by labeling with anti-mouse F4/80 microbeads (Miltenyi Biotec) and subjected to real-time PCR analysis. HSCs and Kupffer cells were treated with alcohol (50–200 mM), lipopolysaccharide (10 ng/mL; Sigma-Aldrich, MO, USA), or recombinant GDF15 (100 ng/mL; R&D Systems, Minneapolis, MN, USA).
Co-culture hepatocyte with Kupffer cells or HSCs
Mouse Kupffer cells were cultured in the lower chambers of 12-well plates with lipopolysaccharide (10 ng/mL) overnight. Isolated HSCs were cultured in the lower chambers of 12-well plates with lipopolysaccharide (10 ng/mL) for 4 days. The cells were then rinsed two times with PBS, and WT or GDF15 KO hepatocytes were seeded into the Transwell inserts (Corning Inc., Corning, NY). The two cell lines were co-cultured for 3 hours and the co-cultured Kupffer cells and HSCs were subjected to real-time PCR analysis.
Fluorescence-activated cell sorting
Isolated liver MNCs were resuspended in Dulbecco’s phosphate-buffered saline (DPBS; Welgene, Daegu, South Korea) containing 0.5% BSA and 0.05% sodium azide, and then labeled with fluorescence-activated cell sorting (FACS) antibodies. For blocking of non-specific binding, cells were pre-incubated with mouse CD16/32 Fc block (eBioscience/ThermoFisher Scientific, Waltham, MA, USA) prior to labeling with FACS antibodies. The cells were also stained with eFluor 780-labeled Fixable Viability Dye (eBioscience/ThermoFisher Scientific, Waltham, MA, USA) to exclude dead cells from the analysis. Lymphocytes (CD4 T, CD8 T, and regulatory T cells), monocytes, and neutrophils were analyzed using anti-CD45, anti-CD4, anti-CD8, anti-CD11b, anti-Ly6C, anti-Ly6G, anti-CD25, and anti-Foxp3 antibodies (eBioscience/ThermoFisher Scientific, Waltham, MA, USA). For intracellular cytokine staining, the cells were re-stimulated with phorbol-myristate acetate/ionomycin for 1 hour in the presence of brefeldin A for 5 hours, and then fixed and permeabilized using the BD Cytofix/Cytoperm kit (BD Pharmingen, San Jose, CA). Stained liver MNCs were read on a FACS Canto II flow cytometer (BD Biosciences, San Jose, CA), and data were analyzed using FlowJo software (FlowJo, LLC, Ashland, OR).
Cell culture and treatment with alcohol or mitochondrial inhibitors
Mouse primary hepatocytes were prepared from 8–10-week-old male WT and GDF15 KO mice (all on a C57BL/6 background) using in situ collagenase perfusion25. Isolated hepatocytes were maintained in DMEM (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in a humidified incubator containing 5% CO2. Primary murine hepatocytes were treated with alcohol (50–100 mM), recombinant GDF15 (100 ng/mL; R&D Systems, Minneapolis, MN, USA) or the oxidative phosphorylation (OXPHOS) complex inhibitors oligomycin (2 µg/mL; Sigma-Aldrich, MO, USA) and rotenone (1 µM; Sigma-Aldrich, MO, USA) for 24 hours.
Western blot analysis
Western blot analysis was performed using standard methods with commercially available antibodies. Anti-alpha smooth muscle actin (α-SMA, #ab5694) and anti-cytochrome p450 2E1 (CYP2E1, #ab151544) antibodies were purchased from Abcam (Cambridge, UK). Anti-phospho-NF-kB p65 (#3303), anti-NF-kB p65 (#4764), anti-phospho-p38 (#9215), anti-p38 (#9213) and anti-JNK (#9252) antibodies were purchased from Cell Signaling Technology (Beverley, MA, USA). Anti-phospho-JNK (#700031) and anti-GAPDH (#MA5-15738) antibodies were purchased from ThermoFisher Scientific. Anti-GDF15 (#sc-377195) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Immunoreactive bands were visualized using alkaline phosphatase-linked anti-rabbit or anti-mouse secondary antibodies, and images were scanned on an ODYSSEY instrument and Image Studio Software (LI-COR Biosciences; Lincoln, NE, USA).
Real-time polymerase chain reaction
Total RNA was extracted from cells or tissues using TRIzol reagent (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Complementary DNA (cDNA) was prepared from the same quantity of total RNA using M-MLV reverse transcriptase and oligo-dT primers (Invitrogen/ThermoFisher Scientific, Waltham, MA, USA). The resulting cDNA was amplified on a 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA) using 2X SYBR Green mix (Applied Biosystems). All reactions were performed in triplicate with the primers listed in Supplementary Table 1. Relative gene expression was calculated using the ΔΔCT method, with normalization to 18 s expression. Values are expressed as the fold change versus the control group.
Measurement of the oxygen consumption rate
The mitochondrial oxygen consumption rate (OCR) was measured using a Seahorse XF-24 analyzer (Seahorse Bioscience Inc., North Billerica, MA, USA) in 24-well plates. Hepatocytes were seeded at 2 × 104 cells per well 24 h before the analysis. On the day before the OCR measurement, the sensor cartridge was placed into calibration buffer (Seahorse Bioscience) and incubated in a non-CO2 incubator at 37 °C. Hepatocytes were washed and incubated in DMEM without sodium bicarbonate. The medium and mitochondrial OXPHOS inhibitors were adjusted to pH 7.4 on the day of the OCR assay. The basal OCR was measured three times, and three readings were taken after the addition of each mitochondrial OXPHOS inhibitor [oligomycin (2 µg/mL) and rotenone (1 µM)]. The basal and post-oligomycin OCRs were calculated by averaging the last three measurements after maintaining a steady state. Coupled respiration was expressed as the percent decrease from basal respiration. Additionally, carbonyl cyanide m-chlorophenyl hydrazone (CCCP; 5 µM) was used to measure maximal mitochondrial respiration of the cells. OCR was automatically calculated and recorded by the sensor cartridge and the Seahorse XF-24 software.
Statistical analysis
Statistical analyses were performed using Stat Graph Prism 6 (GraphPad, San Diego, CA, USA). Data are reported as the mean ± SEM. All data from animal studies were analyzed by two-way repeated-measures ANOVA followed by Bonferroni’s multiple comparisons, one-way ANOVA followed by Tukey’s post-hoc test, or a two-tailed Student’s t-test. P-values < 0.05 were considered statistically significant.
Results
Alcohol and mitochondrial inhibitors increase hepatic expression of GDF15 in vitro and in vivo
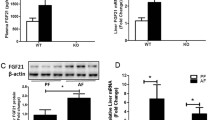
Alcohol decreases the activity of the oxidative phosphorylation (OXPHOS) complexes26 and increases ROS production27, leading to mitochondrial stress or dysfunction. Recently, mitochondrial stress has been shown to promote the induction of GDF15, a mitokine affecting systemic energy metabolism16. Based on previous observations that GDF15 expression is associated with the mitochondrial stress response, we examined the effect of alcohol on GDF15 expression in hepatocytes and non-parenchymal cells. As shown in Fig. 1a, Gdf15 was exclusively expressed in hepatocytes and not HSCs or liver-resident macrophages. Gdf15 expression was increased in hepatocytes treated with alcohol in a concentration-dependent manner (Fig. 1b). Treatment with alcohol also increased the levels of secreted GDF15 in supernatants of cultured hepatocytes (Fig. 1c). Additionally, we found HSC activation was not related to Gdf15 expression (Fig. 1d), but that treatment with alcohol increased Gdf15 expression in D1 HSCs (Fig. 1e). We next investigated the effect of mitochondrial dysfunction on GDF15 expression in hepatocytes. Inhibition of the mitochondrial OXPHOS complex using oligomycin or rotenone significantly increased Gdf15 expression and GDF15 secretion by cultured hepatocytes (Fig. 1f and g). To confirm this observation in vivo, WT mice were pair-fed an alcohol or control liquid diet for 6 weeks to investigate the effects of alcohol on GDF15 expression. No significant difference was detected in body weight gain or food intake between the two groups (Fig. 1h and i). Hepatic Gdf15 expression and serum levels of GDF15 were remarkably increased in alcohol-fed mice (Fig. 1j–l). Taken together, these data demonstrate that treatment with alcohol induces hepatic GDF15 expression in vitro and in vivo.

Treatment with alcohol induces GDF15 production in primary murine hepatocytes. (a) Gdf15 expression in liver tissue, hepatocytes, Kupffer cells, and hepatic stellate cells (HSCs). (b) Real-time PCR analysis of primary hepatocytes treated with vehicle or alcohol. (c) Levels of GDF15 in culture supernatants of primary hepatocytes treated with vehicle or alcohol. (d) Gdf15 expression in D1 HSCs and D7 HSCs (cultured for 1 day and 7 days, respectively). (e) Gdf15 expression in D1 HSCs treated with alcohol. (f) Real-time PCR analysis of primary hepatocytes treated with vehicle, oligomycin, or rotenone. (g) Levels of GDF15 in culture supernatants of primary hepatocytes treated with vehicle, oligomycin, or rotenone. (h,i) Body weight gain and dietary intake of mice fed a control or alcohol liquid diet for 6 weeks (n = 5/group). (j,k) Gdf15 mRNA and GDF15 protein expression in the livers of WT and GDF15 KO (GKO) mice fed a control or alcohol liquid diet for 6 weeks. Relative quantification of each protein was done by densitometry. (l) Serum levels of GDF15 in WT or GKO mice fed vehicle or an alcohol liquid diet for 6 weeks. All data are representative of three independent experiments and are expressed as the mean ± SEM. *P < 0.05 and **P < 0.01, versus the corresponding controls.
Recombinant GDF15 enhances oxygen consumption by hepatocytes and reduces inflammatory cytokine production by liver-resident macrophages
Although GDF15 is mainly expressed and secreted by hepatocytes, several types of liver-resident cells, including HSCs, macrophages, and liver sinusoidal endothelial cells as well as hepatocytes can be affected by GDF15. Therefore, hepatocytes were treated with recombinant GDF15 (rGDF15) and the OCR was measured to determine the effect of GDF15 on hepatocyte mitochondrial function. Treatment with rGDF15 increased the basal OCR and the maximal respiration in hepatocytes (Fig. 2a and b). Mitochondrial oxidative metabolism is linked to anti-inflammatory macrophage activation28. Therefore, Kupffer cells were treated with lipopolysaccharide (LPS) in the presence or absence of rGDF15 to determine the anti-inflammatory effect of GDF15 on hepatic resident macrophages. GDF15 reduced the expression of pro-inflammatory cytokines in Kupffer cells treated with LPS (Fig. 2c). The anti-inflammatory effect of rGDF15 on LPS-treated Kupffer cells was dose-dependent (Fig. 2c). To further evaluate the paracrine effect of GDF15 released from hepatocytes on Kupffer cells, WT or GDF15 KO hepatocytes were seeded into Transwell inserts, and lipopolysaccharide-treated Kupffer cells were placed in the lower chambers (Fig. 2d). The expression of pro-inflammatory cytokines by the LPS-treated Kupffer cells was significantly increased by co-culture with GDF15 KO hepatocytes as compared to co-culture with WT hepatocytes (Fig. 2e). However, co-culturing intermediate activated D4 HSCs with WT or GDF15 KO hepatocytes in the presence of LPS (10 ng/mL) did not result in any differences in the expression of fibrotic mediators in co-cultured D4 HSCs (Fig. 2f). These data suggest that GDF15 may regulate the hepatic immune microenvironment by inhibiting pro-inflammatory cytokine production from liver-resident macrophages.

Recombinant GDF15 enhances oxygen consumption in hepatocytes and inhibits the expression of pro-inflammatory cytokines in Kupffer cells. (a,b) Oxygen consumption rates were measured in primary murine hepatocytes treated with vehicle or recombinant GDF15 (rGDF15, 100 or 200 ng/mL). Oligomycin (2 µg/mL) and rotenone (1 µM) were used as mitochondrial OXPHOS inhibitors. CCCP (5 µM), a mitochondrial uncoupler, was used to measure maximal mitochondrial respiration. (c) Real-time PCR analysis of Kupffer cells treated with lipopolysaccharide (LPS; 10 ng/mL) and/or rGDF15 (50 or 100 ng/mL). (d) Schematic description of co-culturing Kupffer cells with hepatocytes. (e) Real-time PCR analysis of Kupffer cells co-cultured with WT or GDF15 KO hepatocytes in medium containing lipopolysaccharide (10 ng/mL). (f) Real-time PCR analysis of HSCs co-cultured with WT or GDF15 KO hepatocytes in medium containing lipopolysaccharide (10 ng/mL) for 3 hours. All data are representative of three independent experiments and are expressed as the mean ± SEM. *P < 0.05 and **P < 0.01, versus the corresponding controls.
GDF15 deficiency exacerbates alcoholic liver injury in mice
To investigate the effect of GDF15 depletion on alcoholic liver injury, WT and GDF15 KO mice were fed an alcohol liquid for 6 weeks. Blood chemistry analysis revealed that serum levels of AST and ALT were increased in alcohol-fed GDF15 KO mice compared to controls (Fig. 3a). Hepatic triglyceride content was also enhanced in the alcohol-fed GDF15 KO mice (Fig. 3b). As shown in Fig. 3c, H&E staining and Oil Red O staining showed that hepatic fat accumulation and immune cell infiltration were increased in alcohol-fed GDF15 KO mice compared to controls (Fig. 3c). Serum levels of TNF-α and IL-6 (Fig. 3d) and hepatic expression of pro-inflammatory cytokines were also increased in alcohol-fed GDF15 KO mice (Fig. 3e). Taken together, these data demonstrate that GDF15 is essential for the regulation of alcohol-mediated liver injury and steatosis.

GDF15 protects chronic alcohol-fed mice from liver injury and fat deposition. After 6 weeks of feeding with a control or alcohol liquid diet, WT and GDF15 KO (GKO) mice (n = 6/group) were sacrificed. (a) Serum levels of aspartate transaminase (AST) and alanine transaminase (ALT) were measured. (b) Triglyceride content in the liver of the mice was measured. (c) Hematoxylin-eosin (H&E) and Oil Red O staining of liver sections (original magnification, × 200; bars, 100 μm). (d) Serum levels of TNF-α and IL-6. (e) Real-time PCR analysis of hepatic Tnf-α, Il1b, Il-6, and Ccl2. All data are representative of three independent experiments and are expressed as the mean ± SEM. *P < 0.05 and **P < 0.01, versus the corresponding controls.
GDF15 is required for tissue homeostasis in CCl4-induced liver injury and fibrosis
Although several animal models for alcoholic liver disease have been developed, they mainly lead to steatosis and low-grade inflammation but not significant fibrosis, even after prolonged administration of alcohol29,30,31,32. Therefore, to determine the effect of GDF15 on advanced liver disease, we used the CCl4-induced model of liver injury. Interestingly, serum levels of AST and ALT were significantly increased in GDF15 KO mice compared to WT mice (Fig. 4a). In addition, serum levels of TNF-α were enhanced in GDF15 KO mice treated with CCl4 compared to controls (Fig. 4b). Although there was no significant difference between WT and GDF15 KO mice treated with vehicle, GDF15 KO mice showed more severe liver injury than WT mice treated with CCl4 (Fig. 4a and c). In addition, collagen deposition was significantly increased in the livers of GDF15 KO mice treated with CCl4 (Fig. 4c). Furthermore, Tnf-α and fibrotic mediators such as type 1 collagen 1 alpha 1 (Col1a1) and alpha smooth muscle actin (Acta2) were upregulated in the livers of GDF15 KO mice treated with CCl4 (Fig. 4d). These data demonstrate that GDF15 deficiency induces hepatic inflammation and fibrosis in the CCl4-induced model of liver fibrosis.

GDF15 deficiency exacerbates CCl4-induced liver fibrosis. (a) Serum levels of AST and ALT. (b) Serum levels of TNF-α. (c) H&E and Sirius Red staining of liver sections (original magnification, ×200; bars, 100 μm). (d) Real-time PCR analysis of hepatic Tnf-α, Acta2, and Col1a1 of the liver from the mice. All data are representative of three independent experiments and are expressed as the mean ± SEM. *P < 0.05, versus the corresponding controls.
GDF15 suppresses activation of hepatic immune cells in CCL4-induced liver injury
To further evaluate the role of GDF15 on hepatic inflammation induced by CCl4, the phenotype and function of hepatic immune cells in WT and GDF15 KO mice treated with CCl4 were analyzed by FACS. There were no significant differences in the numbers of liver-infiltrating CD4+ or CD8+ T cells between the two groups (Fig. 5a and b). However, the percentages of activated (CD44+) and CD4+ and CD8+ T cells were significantly increased in the GDF15 KO mice compared to WT mice (Fig. 5c–e). Moreover, the neutrophil (CD11b+Ly6G+) population was increased in GDF15 KO mice. However, the percentage of monocytes (CD11b+Ly6Chigh) in CCl4-injured livers was not changed by GDF15 deletion (Fig. 5f–h). Furthermore, the percentage of TNF-α+CD8 + T cells in the livers of GDF15 KO mice was higher than in WT mice, but the percentages of TNF-α+CD4+ T cells and Th17 cells were not significantly different between the two groups (Fig. 5i–l). Collectively, these data indicate that GDF15 is required for the regulation of the hepatic inflammatory response during CCl4-mediated liver fibrogenesis.

GDF15 suppresses activation of hepatic immune cells during CCl4-induced liver fibrogenesis. (a,b) Percentages of hepatic CD4+ and CD8+ T cells in WT and GDF15 KO (GKO) mice with liver fibrosis. (c–e) Percentages of hepatic CD4+CD44+ and CD8+CD44+ T cells in WT and GDF15 KO mice with liver fibrosis. (f–h) Percentages of hepatic monocytes (CD11b+Ly6Chigh) and neutrophils (CD11b+Ly6G+) in WT and GDF15 KO mice with liver fibrosis. (i–l) Intracellular staining for TNF-α and IL-17A in hepatic CD4+ and CD8+ T cells. All data are expressed as the mean ± SEM. *P < 0.05 and **P < 0.01, versus the corresponding controls.
Recombinant GDF15 attenuates CCl4-induced liver fibrosis by suppressing the hepatic inflammatory response
Based on the data described above, GDF15 deficiency exacerbates liver inflammation and fibrosis. Thus, to investigate the beneficial effect of GDF15 in liver fibrosis, mice were co-injected with CCl4 and rGDF15 for 3 weeks. rGDF15 administration reduced collagen accumulation in GDF15 KO mice during liver fibrogenesis (Fig. 6a). As shown in Fig. 6b, serum levels of AST and ALT were lower in GDF15 KO mice after treatment with rGDF15 than GDF15 KO mice treated with CCl4 alone. Treatment with rGDF15 also reduced the serum levels of pro-inflammatory cytokines such as TNF-α and IL-6 in the GDF15 KO mice (Fig. 6c). Consistent with these findings, hepatic expression of Tnf-α, Acta2, and Col1a1 was significantly downregulated in GDF15 KO by treatment with rGDF15 (Fig. 6d). To decipher the molecular basis by which rGDF15 reduces CCl4-induced liver injury and fibrosis, phosphorylation of NF-kB, JNK, and p38 was evaluated in the livers of WT and GDF15 KO mice treated with CCl4 and rGDF15. NF-kB phosphorylation was significantly increased by injection of CCl4 in GDF15 KO mice, while treatment with rGDF15 reduced CCl4-induced phosphorylation of NF-kB and JNK in the livers of GDF15 KO mice (Fig. 6e). CCl4-induced p38 phosphorylation was also significantly decreased in the livers of GDF15 KO mice treated with rGDF15 (Fig. 6e). Additionally, CYP2E1 expression was downregulated in the livers of GDF15 KO mice treated with rGDF15 (Fig. 6e). This pharmacologic inhibition of NF-kB, JNK and CYP2E1 expression and p38 phosphorylation may contribute to the suppression of α-SMA expression in the liver (Fig. 6e), leading to attenuation of CCl4-induced liver fibrosis. To analyze the effect of rGDF15 on hepatic resident immune cells, immune infiltrates in the livers of mice with CCl4-induced liver injury treated with rGDF15 were analyzed by FACS. Treatment with rGDF15 reduced the percentage of CD4+ and CD8+ CD44+ T cells and increased the percentage of CD4+ and CD8+ CD62L+ T cells in the liver (Fig. 6f). The numbers of monocytes (CD11b+Ly6Chigh) and neutrophils in the liver were also significantly decreased in the GDF15 KO mice treated with rGDF15 compared to GDF15 KO mice treated with vehicle (Fig. 6g). Moreover, treatment with rGDF15 reduced the numbers of CD8+TNF-α+ T cells in the livers of GDF15 KO mice (Fig. 6h). These data demonstrate that rGDF15 regulates hepatic immune homeostasis, thereby ameliorating CCl4-induced liver injury and fibrosis.

Recombinant GDF15 ameliorates CCl4-induced liver inflammation and fibrosis. WT and GDF15 KO (GKO) mice (n = 5/group) were treated with CCl4 (2 mL/kg in olive oil, 20% v/v) and recombinant GDF15 (rGDF15, 0.5 mg/kg). (a) H&E and Sirius Red staining of liver sections (original magnification, × 200; bars, 100 μm). (b) Serum levels of AST and ALT. (c) Serum levels of TNF-α and IL-6. (d) Real-time PCR analysis of hepatic Gdf15, Tnf-α, Acta2, and Col1a1. (e) Western blots of liver tissue. (f) Percentages of hepatic CD4+CD44+ and CD8+CD44+ T cells in GDF15 KO mice treated with vehicle or rGDF15. (g) Percentages of hepatic monocytes (CD11b+Ly6Chigh) and neutrophils (CD11b+Ly6G+) in GDF15 KO mice treated with vehicle or rGDF15. (h) Intracellular staining for TNF-α in CD8+ T cells in the livers of GDF15 KO mice treated with vehicle or rGDF15. All data are expressed as the mean ± SEM. **P < 0.01, versus the corresponding controls.
Discussion
The link between serum levels of GDF15 and liver disease is well described in mice and in humans, but it remains to be determined whether GDF15 has direct effects on hepatic inflammation and fibrosis. In the present study, we clearly show that alcohol and CCl4 promote hepatic GDF15 induction, which is required for protection from alcohol- and CCl4-induced liver injury in mice. The findings of this study are summarized in Fig. 7.

Graphical summary. Alcohol and CCl4 increase GDF15 production in hepatocytes. During alcohol-induced and fibrotic liver injury, GDF15 decreases hepatic inflammation and fibrosis via regulation of liver immune cells.
Although the GDF15 receptor and the signaling pathways that drive its biological action have not been unequivocally elucidated, GDF15 is currently considered a reliable biomarker for liver disease, cardiovascular disease, and cancer12,13,14,33. Moreover, GDF15 is induced by the cellular adaptive response to tissue injury or exposure to carcinogens in a number of solid organs34,35. However, most data on the relationship between GDF15 and disease come from observational studies, and there is no conclusive evidence on causality. In the current study, we show that alcohol and CCl4-induced liver inflammation and fibrosis are more severe in GDF15 KO mice, which suggests a causal relationship between GDF15 and liver disease. These data demonstrate that GDF15 is a negative regulator of hepatic inflammation and fibrosis.
Recent emerging evidence has suggested that cellular GDF15 is involved in mitochondrial dysfunction in humans and rodents. Specifically, serum levels of GDF15 are increased in patients with mitochondrial disease36. Moreover, mitochondrial OXPHOS dysfunction in skeletal muscle promotes GDF15 expression in mice16. Meanwhile, mitochondria are abundant in liver cells and are essential in hepatic metabolism, cell survival, and ROS homeostasis37. Specifically, mitochondria are more involved in glucose, lipid, and protein metabolism in the liver than in other organs37. Therefore, hepatic mitochondrial dysfunction is implicated in the pathogenesis of liver diseases such as alcoholic and non-alcoholic steatohepatitis, insulin resistance, and fibrosis. Chronic alcohol feeding alters the expression of respiratory chain complexes in the liver mitochondria38, and patients with non-alcoholic steatohepatitis show morphological changes in the liver mitochondria39. Heterozygosity for the mitochondrial trifunctional protein promotes hepatic fat accumulation and insulin resistance in mice40. Moreover, mitochondrial dysfunction in hepatocytes has a critical role in the progression of CCl4-induced liver fibrosis in mice41. However, recently, mild mitochondrial perturbation in the muscle increased the lifespan and delayed age-dependent locomotory impairment in Drosophila 42. The protective effects of muscle mitohormesis may be associated with the mitochondrial unfolded protein response, which is caused by dysfunction of the mitochondrial respiratory chain complex I42. Additionally, reduced mitochondrial OXPHOS function in muscle improved systemic energy homeostasis and glucose intolerance via GDF15 secretion from muscle in mice16. In the present study, we also showed that treatment with mitochondrial inhibitors such as oligomycin and rotenone induced GDF15 production in primary murine hepatocytes (Fig. 1), which may drive the protective effect against alcohol- or CCl4-induced liver injury. This evidence suggests that mitochondrial dysfunction may be a double-edged sword, as it may affect disease progression as well as protection.
The liver can be regarded as an immunologic organ, with diverse resident and infiltrating immune cells2. These immune cells contribute to the maintenance of hepatic immune homeostasis, but dysregulation of immune cells is involved in the development of inflammation and fibrosis in the liver. We have investigated the effect of GDF15 on the number and function of hepatic immune cells in WT and GDF15 KO mice with CCl4-induced liver injury. As shown in Fig. 5, the numbers of CD4+CD44+ T cells, CD8+CD44+ T cells, and TNF-α-producing CD8+ T cells were increased in GDF15 KO mice compared to WT mice during CCl4-induced liver fibrogenesis. Additionally, treatment with rGDF15 reduced the numbers of CD8+TNF-α+ T cells and the numbers of activated CD4+ and CD8+ T cells, but increased the numbers of naïve CD4+ and CD8+ T cells in the livers of GDF15 KO mice (Fig. 6). These findings suggest that GDF15 has an anti-inflammatory role in CCl4-induced liver injury, resulting in reduced collagen deposition in the liver. In agreement with our results, a recent study also showed that GDF15 inhibited the inflammatory response and septic heart and renal injury induced by lipopolysaccharide43. Furthermore, NF-kB, JNK, and p38 signaling pathways, which are closely associated with hepatic inflammation and fibrosis44, were inhibited by treatment with rGDF15 in a mouse model of liver fibrosis (Fig. 6). This result is also consistent with the anti-inflammatory effect of GDF15 in hepatic immune cells. However, further evaluation is required to clarify whether GDF15 directly reduces the numbers and functional activity of immune cells in the liver, and to identify the molecular mechanism responsible for the GDF15 effect on signaling pathways.
In a previous investigation, GDF15 did not affect the regeneration capacity of the liver after acute liver injury with a single injection of CCl4 45. Liver regeneration, as assessed by BrdU labeling, was also not different between WT and GDF15 KO mice after partial hepatectomy and a single CCl4 injection45. By contrast, our study showed that GDF15 has anti-inflammatory and anti-fibrotic roles in CCl4-induced chronic (3 week) liver fibrosis, which is more generally used to induce liver fibrosis than a single CCl4 injection. Additionally, we suggested that liver fibrosis can be attenuated by the anti-inflammatory action of GDF15, based on the observation that the numbers of neutrophils and activated T cells were lower in the inflamed liver of mice treated with rGDF15. However, the strain (C57BL/6) of GDF15 KO mice used in our study was different from the mouse strain (129/SvJ) used in their study, indicating that the differences between these two studies could be due to differences in experimental methodology and the genetic background of the mice.
Many previous investigations have revealed a remarkable induction of GDF15 in a number of diseases, such as inflammation, cancer, and various metabolic diseases, suggesting that it is induced as a compensatory measure. Alcohol- and CCl4-induced liver injury also induces a remarkable increase in GDF15 levels in WT mice. Because the existing high levels of GDF15 in various injury models make it difficult to pinpoint the role of rGDF15 in WT mice, we administered rGDF15 to GDF15 KO mice to obtain a clearer picture of the role played by GDF15 after hepatic cell injury. However, to determine the therapeutic role of GDF15, rGDF15 should be administered to WT mice rather than to GDF15 KO mice. Therefore, further study is required to establish a therapeutic role for alcoholic steatosis and fibrotic liver diseases.
In summary, we demonstrate that GDF15 has an anti-inflammatory role in alcohol- and CCl4-induced liver injury in mice. Mitochondrial dysfunction caused by alcohol and CCl4 promotes GDF15 production in hepatocytes, which inhibits alcohol-mediated fat accumulation and CCl4-induced fibrosis in the liver. Although future studies should be performed to elucidate the mechanism of cellular GDF15 induction and secretion in damaged hepatocytes, GDF15 could be a novel therapeutic target for the treatment of liver diseases.
References
Racanelli, V. & Rehermann, B. The liver as an immunological organ. Hepatology 43, S54–62, https://doi.org/10.1002/hep.21060 (2006).
Yi, H. S. & Jeong, W. I. Interaction of hepatic stellate cells with diverse types of immune cells: foe or friend. Journal of gastroenterology and hepatology 28(Suppl 1), 99–104, https://doi.org/10.1111/jgh.12017 (2013).
Tacke, F., Luedde, T. & Trautwein, C. Inflammatory pathways in liver homeostasis and liver injury. Clinical reviews in allergy & immunology 36, 4–12, https://doi.org/10.1007/s12016-008-8091-0 (2009).
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nature reviews. Gastroenterology & hepatology, doi:https://doi.org/10.1038/nrgastro.2017.38 (2017).
Lee, Y. S. et al. Hepatic immunophenoty** for streptozotocin-induced hyperglycemia in mice. Scientific reports 6, 30656, https://doi.org/10.1038/srep30656 (2016).
Cui, K. et al. Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1beta in mice. Journal of hepatology 62, 1311–1318, https://doi.org/10.1016/j.jhep.2014.12.027 (2015).
Seo, W. et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology 64, 616–631, https://doi.org/10.1002/hep.28644 (2016).
Kono, H. et al. ICAM-1 is involved in the mechanism of alcohol-induced liver injury: studies with knockout mice. American journal of physiology. Gastrointestinal and liver physiology 280, G1289–1295 (2001).
Hill, D. B. et al. A role for interleukin-10 in alcohol-induced liver sensitization to bacterial lipopolysaccharide. Alcoholism, clinical and experimental research 26, 74–82 (2002).
Louis, H. et al. Interleukin-10 controls neutrophilic infiltration, hepatocyte proliferation, and liver fibrosis induced by carbon tetrachloride in mice. Hepatology 28, 1607–1615, https://doi.org/10.1002/hep.510280621 (1998).
Breit, S. N. et al. The TGF-beta superfamily cytokine, MIC-1/GDF15: a pleotrophic cytokine with roles in inflammation, cancer and metabolism. Growth factors 29, 187–195, https://doi.org/10.3109/08977194.2011.607137 (2011).
Hong, J. H. et al. GDF15 Is a Novel Biomarker for Impaired Fasting Glucose. Diabetes & metabolism journal 38, 472–479, https://doi.org/10.4093/dmj.2014.38.6.472 (2014).
Liu, X. et al. Association of serum level of growth differentiation factor 15 with liver cirrhosis and hepatocellular carcinoma. PloS one 10, e0127518, https://doi.org/10.1371/journal.pone.0127518 (2015).
Mehta, R. S. et al. A prospective study of macrophage inhibitory cytokine-1 (MIC-1/GDF15) and risk of colorectal cancer. Journal of the National Cancer Institute 106, dju016, https://doi.org/10.1093/jnci/dju016 (2014).
Wang, X. et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) as a novel diagnostic serum biomarker in pancreatic ductal adenocarcinoma. BMC cancer 14, 578, https://doi.org/10.1186/1471-2407-14-578 (2014).
Chung, H. K. et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. The Journal of cell biology 216, 149–165, https://doi.org/10.1083/jcb.201607110 (2017).
Nassir, F. & Ibdah, J. A. Role of mitochondria in alcoholic liver disease. World journal of gastroenterology 20, 2136–2142, https://doi.org/10.3748/wjg.v20.i9.2136 (2014).
Wei, Y. H., Chen, Y. S., Lee, J. F., Huang, J. Y. & Lee, C. H. Effect of ethanol intake on rat liver mitochondrial respiration and oxidative phosphorylation. Proceedings of the National Science Council, Republic of China. Part B, Life sciences 14, 61–68 (1990).
Mansouri, A. et al. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology 117, 181–190 (1999).
Knockaert, L. et al. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Laboratory investigation; a journal of technical methods and pathology 92, 396–410, https://doi.org/10.1038/labinvest.2011.193 (2012).
Levy, G. N. & Brabec, M. J. Binding of carbon tetrachloride metabolites to rat hepatic mitochondrial DNA. Toxicology letters 22, 229–234 (1984).
Folch, J., Lees, M. & Sloane Stanley, G. H. A simple method for the isolation and purification of total lipides from animal tissues. The Journal of biological chemistry 226, 497–509 (1957).
Yi, H. S. et al. Alcohol dehydrogenase III exacerbates liver fibrosis by enhancing stellate cell activation and suppressing natural killer cells in mice. Hepatology 60, 1044–1053, https://doi.org/10.1002/hep.27137 (2014).
Kang, S. G. et al. ANGPTL6 expression is coupled with mitochondrial OXPHOS function to regulate adipose FGF21. The Journal of endocrinology 233, 105–118, https://doi.org/10.1530/JOE-16-0549 (2017).
Chung, H. K. et al. The indole derivative NecroX-7 improves nonalcoholic steatohepatitis in ob/ob mice through suppression of mitochondrial ROS/RNS and inflammation. Liver Int 35, 1341–1353, https://doi.org/10.1111/liv.12741 (2015).
Bailey, S. M. & Cunningham, C. C. Effect of dietary fat on chronic ethanol-induced oxidative stress in hepatocytes. Alcoholism, clinical and experimental research 23, 1210–1218 (1999).
Robin, M. A. et al. Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS letters 579, 6895–6902, https://doi.org/10.1016/j.febslet.2005.11.029 (2005).
Vats, D. et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell metabolism 4, 13–24, https://doi.org/10.1016/j.cmet.2006.05.011 (2006).
Bertola, A., Mathews, S., Ki, S. H., Wang, H. & Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nature protocols 8, 627–637, https://doi.org/10.1038/nprot.2013.032 (2013).
Brandon-Warner, E., Schrum, L. W., Schmidt, C. M. & McKillop, I. H. Rodent models of alcoholic liver disease: of mice and men. Alcohol 46, 715–725, https://doi.org/10.1016/j.alcohol.2012.08.004 (2012).
Leo, M. A. & Lieber, C. S. Hepatic fibrosis after long-term administration of ethanol and moderate vitamin A supplementation in the rat. Hepatology 3, 1–11 (1983).
Mathews, S., Xu, M., Wang, H., Bertola, A. & Gao, B. Animals models of gastrointestinal and liver diseases. Animal models of alcohol-induced liver disease: pathophysiology, translational relevance, and challenges. American journal of physiology. Gastrointestinal and liver physiology 306, G819–823, https://doi.org/10.1152/ajpgi.00041.2014 (2014).
Wallentin, L. et al. GDF-15 for prognostication of cardiovascular and cancer morbidity and mortality in men. PloS one 8, e78797, https://doi.org/10.1371/journal.pone.0078797 (2013).
Zimmers, T. A. et al. Growth differentiation factor-15/macrophage inhibitory cytokine-1 induction after kidney and lung injury. Shock 23, 543–548 (2005).
Zimmers, T. A. et al. Growth differentiation factor-15: induction in liver injury through p53 and tumor necrosis factor-independent mechanisms. The Journal of surgical research 130, 45–51, https://doi.org/10.1016/j.jss.2005.07.036 (2006).
Montero, R. et al. GDF-15 Is Elevated in Children with Mitochondrial Diseases and Is Induced by Mitochondrial Dysfunction. PloS one 11, e0148709, https://doi.org/10.1371/journal.pone.0148709 (2016).
Degli Esposti, D. et al. Mitochondrial roles and cytoprotection in chronic liver injury. Biochemistry research international 2012, 387626, https://doi.org/10.1155/2012/387626 (2012).
Han, D. et al. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: biogenesis, remodeling, and functional alterations. The Journal of biological chemistry 287, 42165–42179, https://doi.org/10.1074/jbc.M112.377374 (2012).
Caldwell, S. H. et al. Mitochondrial abnormalities in non-alcoholic steatohepatitis. Journal of hepatology 31, 430–434 (1999).
Ibdah, J. A. et al. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 128, 1381–1390 (2005).
Mitchell, C. et al. Protection against hepatocyte mitochondrial dysfunction delays fibrosis progression in mice. The American journal of pathology 175, 1929–1937, https://doi.org/10.2353/ajpath.2009.090332 (2009).
Owusu-Ansah, E., Song, W. & Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155, 699–712, https://doi.org/10.1016/j.cell.2013.09.021 (2013).
Abulizi, P. et al. Growth Differentiation Factor-15 Deficiency Augments Inflammatory Response and Exacerbates Septic Heart and Renal Injury Induced by Lipopolysaccharide. Scientific reports 7, 1037, https://doi.org/10.1038/s41598-017-00902-5 (2017).
Luedde, T. & Schwabe, R. F. NF-kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nature reviews. Gastroenterology & hepatology 8, 108–118, https://doi.org/10.1038/nrgastro.2010.213 (2011).
Hisao, E. C. et al. Characterization of growth-differentiation factor 15, a transforming growth factor beta superfamily member induced following liver injury. Molecular and Cellular Biology 20, 3742-3751, doi:https://doi.org/10.1128/MCB.20.10.3742-3751.2000.
Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2017R1D1A1B03028361, NRF-2015R1D1A1A01060529) and the Ministry of Science, Communications Technology (ICT), and Future Planning (NRF-2015R1C1A1A01052432, NRF-2015R1A2A1A13000951), and was also supported by the Korean Healthcare Technology R&D Project (HI16C0312).
Author information
Authors and Affiliations
Contributions
H.K.C. and H.Y. designed and performed most experiments and wrote the manuscript; J.T.K., H.K., S.Y.K., and M.K. performed some experiments; M.S. and K.S.K. analyzed the data and gave scientific advice. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chung, H.K., Kim, J.T., Kim, HW. et al. GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Sci Rep 7, 17238 (2017). https://doi.org/10.1038/s41598-017-17574-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17574-w
- Springer Nature Limited
This article is cited by
-
Mitochondrial stress: a key role of neuroinflammation in stroke
Journal of Neuroinflammation (2024)
-
Gomphrena globosa L. extract alleviates carbon tetrachoride-induced liver injury in mice by activating antioxidant signaling pathways and promoting autophagy
Molecular Biology Reports (2023)
-
Catecholamine induces Kupffer cell apoptosis via growth differentiation factor 15 in alcohol-associated liver disease
Experimental & Molecular Medicine (2023)
-
Biomarkers of mitochondrial dysfunction and inflammaging in older adults and blood pressure variability
GeroScience (2023)
-
Transplanted allogeneic cardiac progenitor cells secrete GDF-15 and stimulate an active immune remodeling process in the ischemic myocardium
Journal of Translational Medicine (2022)