
Abstract
The eel gobies fascinate researchers with many important features, including its unique body structure, benthic lifestyle, and degenerated eyes. However, genome assembly and exploration of the unique genomic composition of the eel gobies are still in their infancy. This has severely limited research progress on gobies. In this study, multi-platform sequencing data were generated and used to assemble and annotate the genome of O. rebecca at the chromosome-level. The assembled genome size of O. rebecca is 918.57 Mbp, which is similar to the estimated genome size (903.03 Mbp) using 17-mer. The scaffold N50 is 41.67 Mbp, and 23 chromosomes were assembled using Hi-C technology with a mounting rate of 99.96%. Genome annotation indicates that 53.29% of the genome is repetitive sequences, and 22,999 protein-coding genes are predicted, of which 21,855 have functional annotations. The chromosome-level genome of O. rebecca will not only provide important genomic resources for comparative genomic studies of gobies, but also expand our knowledge of the genetic origin of their unique features fascinating researchers for decades.
Similar content being viewed by others


Background & Summary
Gobies have evolved many distinctive morphological features. For example, their pelvic fins have healed to form a suction cup for clinging to rocks in case they are swept away by rapids1. The eel goby (Odontamblyopus rebecca) (Fig. 1), which belongs to the genus Odontamblyopus (Gobiidae: Amblyopinae)2, is an eel-like benthic burrowing fish that lives mainly in warm waters such as the South China Sea and the Indo-West Pacific3. As a new component of gobies named in 2003, O. rebecca has evolved even more fascinating features that include a unique eel-like body plan, particularly degenerated eyes, and a benthic lifestyle. In recent years, studies of O. rebecca have mainly focused on geographic distribution patterns4,5 and phylogeny based on mitochondrial genome data6. However, the unique phenotypic characteristics of O. rebecca and its molecular mechanism cannot be fully understood by analyzing the mitochondrial genome data alone. Therefore, a high-quality genome assembly to obtain an accurate annotation of the protein-coding genes as a basis for a full understanding of the genetic mechanism of the unique phenotypes is particularly important for O. rebecca.

Eel goby, O. rebecca collected from the intertidal zone of Zhangzhou, Fujian Province, China.
In recent years, the rapid development of high-throughput sequencing technology and their gradual reduction in costs has made large-scale genome sequencing and assembly feasible in non-model taxa. Among them, next-generation sequencing (NGS) is highly accurate but is limited to short-read-length (typically 100 bp or 150 bp) sequencing and is thus not ideal for handling repetitive sequences. Meanwhile, third-generation sequencing (TGS) takes the advantage of long-read-length (typically 20–30 Kb) sequencing, but compromise in sequencing accuracy at single-base level7. Therefore, the prevailing genome-assembly strategy is to incorporate the merits of both sequencing technology by assembling the reference genome using TGS data while correcting assembly errors using NGS data. In combination with high-throughput chromosome conformation capture technology (Hi-C), the genome can be further assembled to the chromosome-level8. Such genome assembly strategy has been employed to address many important scientific problems in teleost fishes, to date9,10,11.
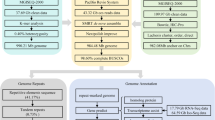
In this study, we used next-generation DNBSEQ short reads (MGI Tech Co., Ltd, Shenzhen, China), third-generation Nanopore long reads (Oxford Nanopore Technologies (ONT)), Hi-C and RNA-Seq sequencing data to assemble and annotate the O. rebecca genome. The results revealed an assembled genome size of 918.57 Mb with 23 pseudochromosomes anchored. The completeness of the genome assembly was assessed using a number of parameters, which include scaffold N50 score (41.67 Mbp), BUSCO score (97.75%), map** ratio of short reads (99.65%) and transcripts (99.82%), indicating the high contiguity and quality of the genome assembly. In addition, 22,999 protein-coding genes were successfully predicted, of which 21,855 gene were functionally annotated in the public database, indicating the reliability of our predictions. The assembled chromosome-level genome of O. rebecca would not only provide important genomic resources for phylogenetic and comparative genomic studies of eel gobies, but also expand our understanding on the possible genetic origin of their unique features such as eel-like body plan, particular degenerated eyes fascinating researchers for decades.
Methods
Sampling, library construction, and sequencing
The O. rebecca sample was collected from the intertidal zone of Zhangzhou, Fujian Province, China. Briefly, dissection was performed in a sterilized environment, and organs including muscle, liver, and intestine were sampled and snap-frozen in liquid nitrogen for nucleic acid extraction. All anatomical procedures comply with relevant ethical regulations provided by the Institutional Animal Care and Use Committee of Zhejiang Ocean University, Zhejiang, China (Protocol Number: 2023082). Genomic DNA was extracted from muscle using the QIAGEN kit (QIAGEN, Cat. No. 13343). The total RNA was extracted from muscle, liver, and intestine using TRIzol reagent (Invitrogen, Carlsbad, CA, USA)12. After extraction, the size and integrity of the extracted DNA and RNA were evaluated using 1% agarose gel electrophoresis, and the concentration and purity of DNA and RNA were further analyzed using a Nanodrop 2000c ultraviolet spectrophotometer. For genome assembly of O. rebecca, Nanopore sequencing libraries were first prepared with the SQK-LSK109 Ligation Sequencing Kit (Oxford Nanopore Technologies) following the manufacturer’s instruction. The prepared libraries were sequenced on R9.4.1 flow cells using a PromethION DNA sequencer (Oxford Nanopore Technologies) platform to generate the Nanopore long reads data. Secondly, short-insert (350–700 bp) paired-end libraries were constructed using the MGIEasy FS DNA Library Prep Kit (BGI, Cat. No.1000006988) and sequenced on the MGIDNB (MGIDNB T7) platform to generate the DNBSEQ short reads data to correct and evaluate the assembly from the extracted genomic DNA of O. rebecca. In addition, the Hi-C libraries were also constructed to generate Hi-C data to obtain chromosome-level genome assemblies using the isolated genomic DNA after fragmented and purified using magnetic beads. For genome annotation of O. rebecca, the complementary DNA libraries were constructed from RNA isolated from muscle, liver, and intestine using VAHTS Universal V6 RNA-seq Library Prep Kit for MGI (Vazyme, NRM604) according to the manufacturer’s instructions. For this purpose, the oligo dT magnetic beads were used to capture the mRNA, and then interrupted with the magnesium ions. The interrupted mRNA is reverse transcribed into a short cDNA using random primers, and end repair and A-tail addition were performed and sequenced also on the MGIDNB platform.
Quality control of raw sequencing data
All raw sequencing data generated in this study were filtered to remove adaptors, low-quality bases, and duplicate reads using different strategies depending on the platform used. For the DNBSEQ short reads, we used fastp software v0.23.213 to remove adaptor sequences, low-quality reads, and short sequences with parameters set as “-l = 50, -w = 6”. Then, we checked the quality of the cleaned data using FastQC software v0.11.914 and found very high base scores in these data, indicating the high-quality of the sequencing data we obtained (Fig. 2). For the Nanopore long reads, the reads were filtered using the NanoFilt software v2.8.015 with the parameter of “-q = 7”. The Hi-C data and RNA-seq data were filtered using the same method and parameter settings as for the DNBSEQ short reads. Finally, we obtained 48.21 Gbp of DNBSEQ short reads (Table 1), 84.64 Gbp of Nanopore long reads with an N50 length of 27.72 Kb (Table 2), and 146.02 Gbp of Hi-C sequencing data (Table 3). In addition, we obtained 41.10 Gbp of liver transcriptome data, 15.78 Gbp of muscle transcriptome data, and 6.62 Gbp of intestine transcriptome data (Table 4).

Base quality examined for DNBSEQ short reads using FastQC software. X-axis represents the position in reads, and the Y-axis represents the quality scores across the bases. The colored area illuminates the low (pink), middle (yellow) and high (green) quality scores of the bases.
Genome size estimation
DNBSEQ short reads were used to estimate the genome size based on k-mer analyses. To this end, all filtered high-quality DNBSEQ short reads data were calculated using kmerfreq v1.016 with the parameters of “-k = 17, -l”. Here, the 17-mer was selected because such k-mer size was demonstrated capable of generating adequate unique k-mer sequences for a sound genome size evaluation when the genome size falls into a scope of what is typical in Gobiidae17,18,19. The genome size was estimated using the formula: genome size = TKN17-mer/PKFD17-mer, where TKN17-mer is the total number of k-mers and PKFD17-mer is the peak frequency depth of the 17-mer. The estimated genome size was then used to evaluate the subsequent result of the genome assembly. The results revealed an estimated genome size of ~903.03 Mbp in O. rebecca. The kmer distribution of the genome consists of three peaks (Fig. 3), which may correspond to the heterozygous, homozygous, and repeated k-mers, respectively, as usually observed in many other teleost fishes20,21.

K-mer analysis for the genome size evaluation in O. rebecca. The distribution of 17-mer frequency in O. rebecca genome was shown. The X-axis represents the k-mer depth, and the Y-axis represents the frequency of the k-mer for a given depth. The first, second and third peaks in the figure corresponded to the heterozygous homozygous, and repeated Kmers, respectively.
Genome assembly
Nanopore long reads have a relatively higher error rate at the single-base level compared to DNBSEQ short reads. Therefore, we first performed error correction on the raw sequencing data, and the resulting Nanopore long clean reads were thereafter assembled into the genome using NextDenovo software v2.4.0 (https://github.com/Nextomics/Next Denovo) with parameters set as “read_type = ont, read_cutoff = 1k, and pa_correction = 3”. To this end, the filtered Nanopore clean data were split and compared with each other using Minimap2 software v2.922 to find overlap areas between reads and remove redundant overlap areas. The string graph algorithm was then applied to assemble high-quality genomes. NextPolish software v1.4.123 was further employed to correct the base errors (SNV/Indel) to improve the accuracy of the genome assembly using the DNBSEQ short reads with the parameters set as “sgs_options = -max_depth 100-bwa, lgs_options = -min_read_len 1k -max_depth 100, lgs_minimap2_options = -x map -ont”. The redundant heterozygous contigs were identified and removed based on sequence similarity and the proportion of redundant parts in total contig length calculated by the Purge_haplotigs software v1.0.424. The preliminary assembly yielded a genome size of 918.80 Mbp with 191 contigs and a contig N50 of 24.75 Mbp (Table 5). Hi-C sequencing data were further used for chromosome assembly by using 3D denovo assembly software v17012325 with parameters set as “rounds = 0, stage = polish”. Juicer software26 and JuiceBox software v1.11.0827 were then used for interaction map generation and error correction (Fig. 4). Finally, 23 chromosomes were obtained with a scaffold N50 of 41.67 Mb (Table 5; Figs. 4, 5), and the assembly rate of contigs into chromosomes was up to 99.96% (Table 6). Such a chromosomes number was consistent with what was observed in other closely related species of Boleophthalmus pectinirostris, Periophthalmus modestus (Gobiidae: Oxudercinae) and Taenioides sp (Gobiidae: Amblyopina). In addition, all the 23 pseudochromosomes could be distinguished easily based on the heatmap (Fig. 4), and the interaction signal around the diagonal was considerably strong, indicating the high-quality of this genome assembly.

A heatmap of chromosome interaction in O. rebecca. The blocks represent the 23 pseudochromosomes. The color bar illuminates the contact density from white (low) to red (high).

Circos plot of the distribution of the genomic elements in O. rebecca. From outside to inside are the distributions of protein-coding genes, tandem repeats (TRs), long tandem repeats (LTRs), long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), DNA elements, and GC content in the genome. The color bar illuminates the number/percent of each genomic element in the genome from light (low) to dark (high).
Genome evaluation
The completeness and accuracy of the genome assembly could have been reflected by the statistics of contig/scaffolds N50 analyses (contig: 24.75 Mbp; scaffolds: 41.67 Mbp) as indicated above. Here, the quality of the genome assembly was further assessed using three extra statistics resulting from BUSCO, short reads map** ratio, and transcripts map** ratio analyses. (1) For BUSCO analysis, the Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.3.128 software was used to search against a single-copy orthologous gene library of Actinopterygii (https://busco-data.ezlab.org/v5/data/lineages/ actinopterygii_odb10.2021-02-19.tar.gz) to assess the integrity of coding regions from the assembled genome. The results showed that a total of 3,640 core genes were identified, including 3,558 complete genes, 3,517 single-copy genes, 41 multi-copy genes, 30 fragmented genes, and 52 deletion genes, which account for 97.75%, 96.62%, 1.13%, 0.82% and 1.43% of the total genes, respectively (Table 7). (2) For short reads map** ratio analysis, the genome index was first built by the BWA-MEM software v0.7.17-r118829 using the parameters of “-a bwtsw”. The DNBSEQ short reads were then mapped to the genome to assess the completeness of the assembly. The map** ratio was calculated by the flagstat function of SAMtools software. The results showed that the total map** rate of DNBSEQ short reads to the genome was 99.65%, the paired map** rate was 99.64%, and the properly paired map** rate was 94.74% (Table 8). (3) For transcripts map** ratio analysis, all the RNA-Seq reads (99.16 Mb) were first assembled into transcripts using StringTie software 1.3.5. Linux_x86_6430, and then it was mapped to the genome using BLAT software v37x131. The results showed that a total of 41,624 reads were mapped to the genome, with a map** rate of 99.82% (Table 9). Taken together, all the results indicated that we had obtained a high-quality chromosome-level assembly of the O. rebecca genome.
Annotation of repetitive sequences
To annotate the repetitive elements in the O. rebecca genome, including tandem repeats and transposable elements (TEs), we integrated a homology prediction using the Repbase library32 (http://www.girinst.org/repbase) and a de novo prediction based on self-sequence alignment and repetitive sequence features. The tandem repeat was annotated using Tandem Repeat Finder software v4.0933 with parameters were set as “Match = 2, Mismatch = 7, Delta = 7, PM = 80, PI = 10, Minscore = 50, MaxPeriod = 2000 -d -h”. TEs were de novo predicted on both DNA and protein levels. On the DNA level, RepeatModeler software v1.0.1134 (-database mydb -pa 10) and LTR-FINDER v1.0.735 (-w 2 -o 3 -t 1 -e 1 -m 2 -u -2 -D 20000 -d 1000 -L 3500 -l 100 -p 20 -g 50 -G 2 -T 4 -S 6.00 -M 0.00 -B 0.400 -b 0.400 -O 40 -F 0) were used to build de novo repeat library. RepeatMasker software open-4.0.936 (http://repeatmasker.org) (-nolow -no_is -norna -parallel 2) was then run against the de novo library and repbase (RepBase v.16.02) separately to identify homologous repeats. On the protein level, RepeatProteinMask v4.0.9 was used to search TEs in its protein database. Finally, the annotation results of all repetitive sequences were merged as a final result. The results showed that a total of 489.68 Mb of sequences were identified as repetitive sequences (including TEs, satellite, simple repeat, others, and unknowns) in the O. rebecca genome, accounting for 53.29% of the genome size (Table 10). Among them, 297.90 Mb of transposable elements (TEs) were annotated, accounting for 32.43% of the genome (Table 11). There are four major types of TEs, of which, DNA elements, long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), and long erminal repeats (LTRs) (Fig. 5) account for 16.12% (148.10 Mbp), 8.49% (78.00 Mbp), 1.07% (9.82 Mbp), and 6.75% (61.98 Mbp) of the genome, respectively.
Prediction of protein-coding genes
To obtain a high-confidence gene set, a combination of three strategies of de novo prediction, homology-based prediction, and transcripts-based prediction were used to annotate the protein-coding genes. (1) For De novo prediction, GlimmerHMM software v3.0.437, Genscan software v1.038, and Augustus software v3.3.239 (-species = zebrafish, -uniqueGeneId = true, -noInFrameStop = true, -gff3 = on, -strand = both) were performed. (2) For homology-based prediction, the already predicted protein-coding gene sequences of close-related species, including Oryzias latipes (GCF_002234675.1), Boleophthalmus pectinirostris (GCF_026225935.1), Periophthalmus magnuspinnatus (GCF_009829125.3), Takifugu rubripes (GCF_901000725.2) and Danio rerio (GCF_000002035.6), were first downloaded from public databases. Then, all sequences were aligned to the O. rebecca genome using TBLASTN software v2.11.0+40 with an e-value of 0.01. The TBLASTN results were further processed to obtain the final homology-based prediction results for each species with parameters set as “model = protein2genome, showtargetgff = 1” using exonerate software v2.2.041. (3) For transcripts-based prediction, StringTie software v1.3.5 was first used to assemble transcripts with parameters set as “-f 0.1 -m 200 -a 10 -c 2.5 -g 50 -M 1.0”. HISAT2 software v2.1.042 was thereafter used to map the RNA-seq data to the genome with parameters set as “-dta -summary-file -S -x -1 -2”. TransDecoder software v5.5.0 (https://github.com/TransDecoder/TransDecoder) was used to predict the coding region of each transcript with parameters set as “-retain_long_orfs_mode dynamic -retain_long_orfs_length 150 -T 500”. Finally, Maker2 software v2.31.1043 was used to integrate the gene annotation results generated by the three methods to obtain the final gene set with parameters set as “-r local -o tmp -p 4”. The results revealed a total of 22,999 protein-coding genes that were successfully predicted in the O. rebecca genome (Table 12). We checked the quality of the annotated genes by comparing them with several species that share evolutionary affinity, and the results indicated a similarity in the distributions of mRNA length, CDS length, exon length, and intron length between genomes of O. rebecca and those closely related species (Fig. 6), possibly incating they share similar patterns of gene structure distribution as the published genomes.

The length of protein-coding genes in O. rebecca and their closely related species. The gene length, CDS length, exon length, and intron length were compared among O. rebecca and the other five teleost species to verify the quality of gene annotation.
Functional annotation of protein-coding genes
To evaluate the annotation quality and obtain the biological function information of the predicted protein-coding gene set, we compared the protein sequences output in this study with all the existing public protein databases, including InterPro44 (2021) (https://www.ebi.ac.uk/interpro/), GO45 (5.61–93.0) (http://geneontology.org/docs/go-annotations/), Kyoto Encyclopedia of Genes and Genomes (KEGG)46 (3.0) (http://www.genome.jp/kegg/), SwissProt47 (2021) (http://www.uniprot.org/), TrEMBL (2021) (http://www.uniprot.org/), TF (AnimalTFDB3.0), Pfam48 (01.34.0) (http://pfam.xfam.org), NCBI Non-Redundant Protein Sequence Database (NR) (2021) (https://www.ncbi.nlm.nih.gov/refseq/about/non-redundantproteins/), and Eukaryotic Orthologous Groups of Proteins (KOG) (2003) (ftp://ftp.ncbi.nih.gov/pub/COG/KOG/kyva). Functional information was analyzed using BLAST software v2.31.1049. The results showed that a total of 21,855 genes could be annotated, accounting for 95.03% protein-coding genes, and only 1,144 genes could not be annotated, accounting for 4.97% protein-coding genes (Table 12), further suggesting we got a reliable assembly and annotation of O. rebecca genome.
Data Records
The genomic DNBSEQ short-insert sequencing data were deposited in the Sequence Read Archive at NCBI SRR2506424450. The genomic Nanopore sequencing data were deposited in the Sequence Read Archive at NCBI SRR2506424251. The transcriptome sequencing data were deposited in the Sequence Read Archive at NCBI SRR2506423852, SRR2506423953, SRR2506424054, and SRR2506424355. The Hi-C sequencing data were deposited in the Sequence Read Archive at NCBI SRR2506424156. The final genome assembly was deposited in GenBank at NCBI with the accession number ASM3068695v157, the Submitted GenBank assembly number is GCA_030686955.1, the BioProject number is PRJNA977196, and the BioSample ID is SAMN35453534. The annotation results of repetitive sequences, gene structure, and functional prediction were deposited in the Figshare database under DOI code: https://doi.org/10.6084/m9.figshare.2368939858.
Technical Validation
Genome evaluation
The quality of O. rebecca genome assembly was evaluated using N50, BUSCO, short reads map** ratio, and transcripts map** ratio analyses. Results showed that the assembly contained good contiguity, a high percentage of complete and single-copy genes, had a high map** rate of short reads and transcripts, indicating a high-quality assembly.
Code availability
The software used in this study is in the public domain, with parameters clearly described in Methods. Where detailed parameters were not provided for the software, default parameters were used instead, as suggested by the developers. No custom script or code was used.
References
Forker, G. K., Schoenfuss, H. L., Blob, R. W. & Diamond, K. M. Bendy to the bone: Links between vertebral morphology and waterfall climbing in amphidromous gobioid fishes. J. Anat. 239, 747–754, https://doi.org/10.1111/joa.13449 (2021).
Murdy, E. O. & Shibukawa, K. A revision of the gobiid fish genus Odontamblyopus (Gobiidae: Amblyopinae). Ichthyol. Res. 48, 31–43, https://doi.org/10.1007/s10228-001-8114-9 (2001).
Murdy, E. O. & Shibukawa, K. Odontamblyopus rebecca, a new species of amblyopine goby from Vietnam with a key to known species of the genus (Gobiidae: Amblyopinae). Zootaxa 138, 1–6, https://doi.org/10.11646/zootaxa.138.1.1 (2003).
Lü, Z. M. Climate adaptation and drift shape the genomes of two eel-goby sister species endemic to contrasting latitude. Animals 13, 3240, https://doi.org/10.3390/ani13203240 (2023).
Tang, W. X. et al. Cryptic species and historical biogeography of eel gobies (Gobioidei: Odontamblyopus) along the Northwestern Pacific Coast. Zool. Sci. 27, 8–13, https://doi.org/10.2108/zsj.27.8 (2010).
Liu, Z. S. et al. Complete mitochondrial genome of three fish species (Perciformes: Amblyopinae): genome description and phylogenetic relationships. Pak. J. Zool. 49, 107–115, https://doi.org/10.17582/journal.pjz/2017.49.1.107 (2017).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351, https://doi.org/10.1038/nrg.2016.49 (2016).
Belton, J. M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276, https://doi.org/10.1016/j.ymeth.2012.05.001 (2012).
Bi, X. P. et al. Tracing the genetic footprints of vertebrate landing in non-teleost ray-finned fishes. Cell 184, 1377–1391, https://doi.org/10.1016/j.cell.2021.01.046 (2021).
Lü, Z. M. et al. Large-scale sequencing of flatfish genomes provides insights into the polyphyletic origin of their specialized body plan. Nature Genet. 53, 742–751, https://doi.org/10.1038/s41588-021-00836-9 (2021).
Wang, K. et al. African lungfish genome sheds light on the vertebrate water-to-land transition. Cell 184, 1362–1376, https://doi.org/10.1016/j.cell.2021.01.047 (2021).
Rio, D. C., Ares, M. Jr., Hannon, G. J. & Nilsen, T. W. Purification of RNA using trIzol (TRI reagent). Cold Spring Harb Protoc 6, pdb.prot5439, https://doi.org/10.1101/pdb.prot5439 (2010).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Andrews, S. FastQC A Quality Control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/ doi:citeulike-article-id:11583827 (2010).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669, https://doi.org/10.1093/bioinformatics/bty149 (2018).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
You, X. X. et al. Mudskipper genomes provide insights into the terrestrial adaptation of amphibious fishes. Nat. Commun. 5, 5594, https://doi.org/10.1038/ncomms6594 (2014).
Bian, C. et al. Genomics comparisons of three chromosome-level mudskipper genome assemblies reveal molecular clues for water-to-land evolution and adaptation. J. Adv. Res. 21, S2090–1232, https://doi.org/10.1016/j.jare.2023.05.005 (2023).
Liu, Y. T. et al. Genome sequencing provides novel insights into mudflat burrowing adaptations in eel goby Taenioides sp. (Teleost: Amblyopinae). Int. J. Mol. Sci. 24, 12892, https://doi.org/10.3390/ijms241612892 (2023).
Cai, M. Y. et al. Chromosome assembly of Collichthys lucidus, a fish of Sciaenidae with a multiple sex chromosome system. Sci. Data 6, 132, https://doi.org/10.1038/s41597-019-0139-x (2019).
Zhang, K. et al. A chromosome-level reference genome assembly of the Reeve’s moray eel (Gymnothorax reevesii). Sci. Data 10, 501, https://doi.org/10.1038/s41597-023-02394-7 (2023).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Hu, J., Fan, J. P., Sun, Z. Y. & Liu, S. L. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2020).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460, https://doi.org/10.1186/s12859-018-2485-7 (2018).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Kent, W. J. BLAT - The BLAST-like alignment tool. Genome Res. 12, 656–664, https://doi.org/10.1101/gr.229202 (2002).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467, https://doi.org/10.1159/000084979 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268, https://doi.org/10.1093/nar/gkm286 (2007).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protocols. BioInf. 25, 4–10, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM:: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94, https://doi.org/10.1006/jmbi.1997.0951 (1997).
Stanke, M. et al. AUGUSTUS:: ab initio prediction of alternative transcripts. Nucleic. Acids. Res. 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Gertz, E. M., Yu, Y. K., Agarwala, R., Schäffer, A. A. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biol. 4, 41, https://doi.org/10.1186/1741-7007-4-41 (2006).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31, https://doi.org/10.1186/1471-2105-6-31 (2005).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genoty** with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491, https://doi.org/10.1186/1471-2105-12-491 (2011).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199, https://doi.org/10.1093/nar/gkw1107 (2017).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nature Genet. 25, 25–29, https://doi.org/10.1038/75556 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic. Acids. Res. 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic. Acids. Res. 28, 45–48, https://doi.org/10.1093/nar/28.1.45 (2000).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic. Acids. Res. 49, D412–D419, https://doi.org/10.1093/nar/gkaa913 (2021).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410, https://doi.org/10.1016/s0022-2836(05)80360-2 (1990).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR25064244 (2023).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR25064242 (2023).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR25064238 (2023).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR25064239 (2023).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR25064240 (2023).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR25064243 (2023).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR25064241 (2023).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_030686955.1/ (2023).
Lü, Z. M. Chromosome-level genome assembly and annotation of eel goby, Odontamblyopus rebecca. figshare. Dataset. https://doi.org/10.6084/m9.figshare.23689398.v1 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (41976121) and (42171069).
Author information
Authors and Affiliations
Contributions
Yongxin Li and Zhenming Lü conceived and designed the research. Tianwei Liu, Yuzheng Wang, and Yantang Liu, collected the samples and extracted the genomic DNA. Zhenming Lü, Ziwei Yu, Wenkai Luo, Bingjian Liu, Li Gong, and Liqin Liu conducted the experiments, and analyzed part of the data, Ziwei Yu, WenkaiLuo and Zhenming Lü wrote the manuscript. All authors read, revised, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lü, Z., Yu, Z., Luo, W. et al. Chromosome-level genome assembly and annotation of eel goby (Odontamblyopus rebecca). Sci Data 11, 160 (2024). https://doi.org/10.1038/s41597-024-02997-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-02997-8
- Springer Nature Limited