
Abstract
Here we report whole genome sequencing of four individuals (H3, H4, H5, and H6) from a family of Pakistani descent. Whole genome sequencing yielded 1084.92, 894.73, 1068.62, and 1005.77 million mapped reads corresponding to 162.73, 134.21, 160.29, and 150.86 Gb sequence data and 52.49x, 43.29x, 51.70x, and 48.66x average coverage for H3, H4, H5, and H6, respectively. We identified 3,529,659, 3,478,495, 3,407,895, and 3,426,862 variants in the genomes of H3, H4, H5, and H6, respectively, including 1,668,024 variants common in the four genomes. Further, we identified 42,422, 39,824, 28,599, and 35,206 novel variants in the genomes of H3, H4, H5, and H6, respectively. A major fraction of the variants identified in the four genomes reside within the intergenic regions of the genome. Single nucleotide polymorphism (SNP) genotype based comparative analysis with ethnic populations of 1000 Genomes database linked the ancestry of all four genomes with the South Asian populations, which was further supported by mitochondria based haplogroup analysis. In conclusion, we report whole genome sequencing of four individuals of Pakistani descent.
Measurement(s) | SNV • genome |
Technology Type(s) | whole genome sequencing • DNA sequencing |
Factor Type(s) | individual |
Sample Characteristic - Organism | Homo sapiens |
Sample Characteristic - Location | Pakistan |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.12642761
Similar content being viewed by others


Background & Summary
The completion of Human Genome Project ignited several large scale efforts to characterize variations in the human genome, which led to a comprehensive catalog of the common variants including single-nucleotide polymorphisms (SNPs) and insertions/deletions (indels), across the entire human genome1,2. Population-based genome reference datasets played an important role in elucidation of rare variants in specific populations3,4. So far, comprehensive genome reference datasets have been reported for African, Japanese, Korean, and Chinese populations5,6,7,8.
Advancements in next-generation sequencing technologies have impelled the development of a comprehensive catalog of genetic variants from different ethnic populations9,10,11,12,13,14,15. The 1000 Genomes Project reports human genetic variation profiles from 26 ethnic populations, including one Pakistani (Punjabi), two Indian (Gujarati and Telugu), one Bangladeshi (Bengali), and one Sri Lankan (Tamil) population—all descendants of the Indian subcontinent15.
Additionally, independent groups have recently published two Indian and two Pakistani genomes with an overall 25–30× sequencing coverage16,17,18,19. Recently, the GenomeAsia 100 K project reported genomes of 1,739 individuals, including 113 individuals of Pakistani origin (https://browser.genomeasia100k.org). We previously reported the whole genome sequencing of two Pakistani individuals20. Here, we report whole genome sequencing of four individuals of Pakistani descent.
Methods
Sample collection
The protocol for this study was approved by the Institutional Review Board of the Johns Hopkins University School of Medicine (Baltimore, MD), the National Centre of Excellence in Molecular Biology (Lahore, Pakistan), and the National Eye Institute (Bethesda, MD). The participating members provided informed written consent consistent with the tenets of the Declaration of Helsinki. A small aliquot (~10 ml) of a blood sample was collected from each individual and genomic DNA was extracted as previously described20.
Library preparation and next-generation sequencing
Whole genome sequencing was performed using the Illumina HiSeq X10 (Illumina, San Diego, CA, USA). Briefly, 1.0–2.0 µg of fragmented gDNA was used to prepare paired-end libraries with the TruSeq DNA PCR-Free Library Preparation Kit for four samples (H3, H4, H5, and H6) according to the manufacturer’s instructions (Illumina Inc., San Diego, CA). All four libraries were sequenced using Illumina HiSeq X10 in paired-end fashion (2 × 150 bp; Illumina Inc.). The base calls were assigned through Illumina Real-Time Analysis software (Ver. 1.17.20) and binary base call (BCL) files were converted to flat-file format (qseq.txt) using Illumina BCL Converter software (Ver. 1.9.4).
Bioinformatics analysis
Paired-end raw reads were aligned to the human reference genome (GRCh38.p13) using Burrows-Wheeler Aligner-MEM (BWA-MEM; Ver. 0.7.17-r1188) without ALT-aware mode21. The quality of the read alignments was examined using CollectAlignmentSummaryMetrics from Picard Tools (Ver. 2.19.0; http://broadinstitute.github.io/picard). The duplicate reads were removed from the mapped reads using MarkDuplicates from Picard Tools. The variants including SNPs and indels were called using the Genome Analysis Tool Kit (GATK; Ver. 4.0) best-practices12,22. Briefly, the recalibration of base qualities of input reads was performed using GATK tools (BaseRecalibrator and ApplyBQSR). Subsequently, the SNPs, indels, and genotype of variants were identified using multiple tools i.e. HaplotypeCaller (in GVCF mode), GenotypeGVCFs, and VCFtools (Ver. 0.1.15)23. Alignment metrics were generated using CollectAlignmentSummaryMetrics and CollectInsertSizeMetrics from Picard Tools. Genome-wide read coverage was generated using Bedtools (Ver. 2.26.0)24.
Variant filtering and annotation
The variants identified through the GATK tool kit were further screened using the high-confidence regions characterized by Genome in a Bottle (GIAB) database25. The variants aligned within the large segmental duplication regions of the human genome were discarded while variants mapped to the high-confidence regions of GIAB were used in downstream analyses including Venn diagram generation using VennPainter26. Note: An allele (variant) with a minimum of 40% of the total reads mapped to reference allele is considered authentic. The filtered variants were annotated using clinEff (Ver. 1.0 h; http://www.dnaminer.com/clineff.html), a professional version of SNPEff27, designed for the prediction of functional effects of variants.
Variant calling
The CNVnator (Ver. 0.4.1) algorithm was used for the identification of copy number variations (CNVs) with a bin size of 1,000 and 10,000028. The GIAB filtered variants (SNPs) were imported into the CNV analysis pipeline for plotting the B-allele frequency (BAF) along the read depths for all deletion and duplication events.
Ancestry prediction
The ancestral roots of H3, H4, H5, and H6 were examined using the algorithms of Peddy (Ver. 0.3.5)29. The study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD. PCA plots were created using SNPs genotype information obtained from VCF (variant call format) files (from whole genome sequencing data of H3, H4, H5, and H6) and comparing it with combined ethnic populations from the 1000 Genomes dataset.
In parallel, ancestral roots of H3, H4, H5, and H6 were examined through a comparative analysis with genomes of five different ethnic populations within the 1000 Genomes database. We randomly selected 96 samples from African, Ad Mixed American, East Asian, European, and South Asian populations for comparative analysis by the bcftools-isec algorithm. These variants from 1000 Genomes database and four genomes in VCF format were converted to BCF using bcftools (Ver. 1.8). The BCF files were converted to PLINK format using PLINK (Ver. 1.90b6.18) and PLINK algorithms were used to filter the variants to generate a list of markers in approximate linkage equilibrium for PCA analysis.
Haplogroup analysis
The mitochondrial sequencing reads were mapped to the revised Cambridge reference sequence (rCRS) of the human mitochondrial genome30. Mitochondrial variants were identified using GATK (Ver. 4.0) best practices and are classified into phylogenetic clusters in the haplogroup analysis using HaploGrep 2 (Ver. 2.1.25)31, with Kulczynski measure and Phylotree (build 17). The Y chromosomal haplogroup analysis for all four genomes was performed using Yleaf32.
Data Records
Technical Validation
The next-generaton whole genome sequencing generated 1344.74, 1110.55, 1200.77, and 1142.35 million total reads for H3, H4, H5, and H6, respectively (Table 1)33. Quality control (QC) examination of the sequencing reads revealed that >99% of the sequencing data yielded a PHRED score of 30 or above (PHRED score of 30 represents the probability of 0.001 that the base call is wrong). Subsequent to QC examination and the removal of PCR duplicates (~10–18% of reads were marked duplicates and subsequently removed in downstream analysis), the majority of the reads (>99% of reads with a PHRED score ≥ 30) mapped to reference human genome (GRCh38.p13; Table 1). Map** of the paired-end reads identified an estimated mean insert size of 390 bp in all four genomes. Majority of the total mapped reads showed paired-end alignment, resulting in 1084.92, 894.73, 1068.62, and 1005.77 million mapped reads corresponding to 162.73, 134.21, 160.29, and 150.86 Gb sequence data and 52.49x, 43.29x, 51.70x, and 48.66x average coverage for the genomes of H3, H4, H5, and H6, respectively (Table 1).
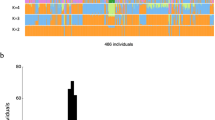
The evaluation of sequencing reads revealed that a significant fraction of the genomes of H3, H4, H5, and H6 exhibited 30–60x read coverage (Fig. 1 and Table 2). Importantly, 5–6% of the genomes of H3, H4, H5, and H6 were not captured, representing 0x read coverage while approximately, 1%, 2%, and 5% of four genomes exhibited 1–10x, 10–20x, and 20–30x read coverage, respectively (Fig. 1 and Table 2). A minor fraction i.e. <1% of the genomes of H3, H4, H5, and H6 exhibited 80–100x read coverage (Fig. 1 and Table 2).

Histogram depicting the sequence coverage of the genomes of H3, H4, H5, and H6. The x- and y-axes represent the genome coverage (sequencing depth) and genome fraction (%), respectively. The blue, yellow, green, and red colors represent the genomes of H3, H4, H5, and H6, respectively.
Sequence analysis of the genome of H3 revealed a total of 3,529,659 variants including 3,035,369 SNPs and 494,290 indels. The SNPs were annotated against dbSNP (Ver. 150) that identified 7,553 novel variants (0.21% of the total variants) in the genome of H334. A total of 494,290 indels including 34,869 novel indels (7.05% of the total indels) were identified in the H3 genome34.
Sequence analysis of the genome of H4 identifed 3,478,495 total variants including 2,996,403 SNPs and 482,092 indels while annotation of the SNPs identified 6,631 novel SNPs (0.19% of the total variants) in the genome of H434. A total of 482,092 indels including 33,193 novel indels (6.88% of the total indels) were identified in the genome of H434. Sequence analysis of the genome of H5 identifed 3,407,895 total variants including 2,983,279 SNPs and 424,616 indels while annotation of the SNPs identified 5,560 novel SNPs (0.16% of the total variants) in the genome of H534. A total of 424,616 indels including 23,039 novel indels (5.42% of the total indels) were identified in the genome of H534. Finally, sequence analysis of the genome of H6 identifed 3,426,862 total variants including 2,972,863 SNPs and 453,999 indels while annotation of the SNPs identified 6,703 novel SNPs (0.19% of the total variants) in the genome H634. A total of 453,999 indels including 28,503 novel indels (6.28% of the total indels) were identified in the genome of H634.
Importantly, we identified a total of 1,668,024 variants including 1,666,232 variants reported previously and 1,792 novel SNPs common in the four genomes (Fig. 2a–c). Altogether, the variants common in the four genomes constitute nearly half of the total variants identified in each genome.

Venn diagram illustration of the overlap** variants characterized by the Genome in a Bottle (GIAB) database in the genomes H3, H4, H5, and H6. (a) Venn diagram illustrating all variants, (b) Venn diagram illustrating all known variants, and (c) Venn diagram illustrating all novel variants identified in the high-confidence regions characterized by the GIAB database in H3, H4, H5, and H6 genomes. Note: Yellow, green, blue, and pink represent variants in the genomes of H3, H4, H5, and H6, respectively, whereas darker shades represent common variants in these genomes.
We examined the putative effect of the variants based on their location in the genome (exon, intron, etc.), functional impact (high, moderate, and low), and classification (synonymous vs. non-synonymous), etc. The analysis revealed that intergenic regions harbor the majority of SNPs consistent with the GIAB high-confidence variants. Furthermore, in contrast to intergenic variants, fewer variants were identified in the exons, splice site, and untranslated regions (UTRs) of the genome. Furthermore, >3 K, >82 K, and >28 K variants present in all four genomes were predicted to exhibit a putative high, moderate, and low impact, respectively.
We used CNVnator, an algorithm to characterize copy number variations (CNVs), to examine structural variants in the genomes of H3, H4, H5, and H6. The analysis identified a total of 4,269 copy number variation regions (CNVRs) common in four genomes, covering 305.95 Mb (9.53%) of the reference human genome (GRCh38.p13).
Although H3, H4, H5, and H6 belong to the Punjabi ethnic group of Pakistani population suggesting a close ancestral relationship with South Asian populations, we sought of genomic evidence to confirm the ancestral roots of the four genomes. We compared the SNP genotypes of H3, H4, H5, and H6 to the combined population of the 1000 Genomes project by the Peddy algorithm. The analysis localized the all four genomes within South Asian populations in principal component 1 and 3 (PC1 and PC3) (Fig. 3a–d; arrows pointing to samples shown as red circles in PCA plots) and on the edge of the South Asian populations in principal component 2 (PC2) towards the European populations (Fig. 3a–d). The localization of H3, H4, H5, and H6 in PC2 suggests some ancestral link with European populations.

Examining the ancestral roots of H3, H4, H5, and H6 through SNP-genotype comparative analysis with the combined ethnic populations of the 1000 Genomes dataset. The arrows point to (a) H3, (b) H4, (c) H5 and (d) H6 shown as red circles in the principal component analysis (PCA) plots with South Asian populations in principal components 1 and 3 (PC1 and PC3) and between South Asian, and European populations in principal component 2 (PC2). The x-axis represents PC1 while the y- and the z-axis represent PC2 and PC3, respectively. Note: South Asian: SAS; African: AFR; Ad Mixed American: AMR; and East Asian: EAS.
In parallel, we performed an additional comparative analysis of the four genomes with the genomes of five different ethnic populations in the 1000 Genomes database. The analysis identified >94% overlap of variants in the genomes of H3, H4, H5, and H6 with South Asian populations (Table 3) with a small number of variants in the genomes of H3 (157,110), H4 (166,633), H5 (159,515), and H6 (163,635) genomes not present in South Asian populations (Table 3). We identified > 92% overlap of variants in the genomes of H3, H4, H5, and H6 with both European and Ad Mixed American populations (Table 3). Likewise, we identified > 88% and > 90% overlap of variants in the genomes of H3, H4, H5, and H6 with East Asian and African populations, respectively (Table 3). These data support the notion that H3, H4, H5, and H6 have a close ancestral relationship with South Asian populations (Fig. 4a–c).

(a–c) Principal component analysis (PCA) plots illustrate three different angles of principal component # 1 (PC1), principal component # 2 (PC2), and principal component # 3 (PC3), respectively. The x-, y-, and z-axes depict the three largest components of the total variation in the percent of PC1, PC2, and PC3, respectively. The analysis illustrates an overlap** of four genomes with the South Asian population. Note: South Asian: SAS; African: AFR; Ad Mixed American: AMR; East Asian: EAS; and European: EUR.
To further confirm the results of SNP genotype based analysis, we performed mitochondria and Y chromosome based haplogroup analysis. The mitochondria genome analysis revealed M35b haplogroup in the H3 genome and M6 haplogroup in H4, H5, and H6 genomes. Both mitochondrial haplogroups (M35b and M6) have been mainly identified in South Asian populations35,36. The Y chromosome analysis identified G1a1b2a haplogroup in H3 and H5 genomes, suggesting a Middle Eastern origin. Taken together, the mitochondria haplogroup based analyses support the results of the SNP genotype based analysis and strengthen the notion that H3, H4, H5, and H6 have a close ancestral relationship with South Asian populations.
In conclusion, we have completed next-generation based whole genome sequencing of four individuals from a family of Pakistani descent. Importantly, nearly 1% of the total variants identified in each of the four genomes are novel and have not been reported previously. To the best of our knowledge, this is the first report of whole genome sequencing of four individuals from a family.
References
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Sachidanandam, R. et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409, 928–933 (2001).
Southam, L. et al. Whole genome sequencing and imputation in isolated populations identify genetic associations with medically-relevant complex traits. Nat. Commun. 8, 15606 (2017).
Ebenesersdottir, S. S. et al. Ancient genomes from Iceland reveal the making of a human population. Science 360, 1028–1032 (2018).
Gurdasani, D. et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 517, 327–332 (2015).
Nagai, A. et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Kim, J. et al. KoVariome: Korean National Standard Reference Variome database of whole genomes with comprehensive SNV, indel, CNV, and SV analyses. Sci. Rep. 8, 5677 (2018).
Gao, Y. et al. PGG.Han: the Han Chinese genome database and analysis platform. Nucleic Acids Res. 48, D971–D976 (2020).
Li, B. & Leal, S. M. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 83, 311–321 (2008).
Tucker, T., Marra, M. & Friedman, J. M. Massively parallel sequencing: the next big thing in genetic medicine. Am. J. Hum. Genet. 85, 142–154 (2009).
Frazer, K. A., Murray, S. S., Schork, N. J. & Topol, E. J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10, 241–251 (2009).
DePristo, M. A. et al. A framework for variation discovery and genoty** using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Sudmant, P. H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015).
Patowary, A. et al. Systematic analysis and functional annotation of variations in the genome of an Indian individual. Hum. Mutat. 33, 1133–1140 (2012).
Gupta, R. et al. Sequencing and analysis of a South Asian-Indian personal genome. BMC. Genomics 13, 440 (2012).
Azim, M. K. et al. Complete genome sequencing and variant analysis of a Pakistani individual. J. Hum. Genet. 58, 622–626 (2013).
Ilyas, M. et al. Whole genome sequencing of an ethnic Pathan (Pakhtun) from the north-west of Pakistan. BMC. Genomics 16, 172 (2015).
Khan, S. Y. et al. Whole genome sequencing data for two individuals of Pakistani descent. Sci. Data 5, 180174 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 43, 11 (2013).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics. 27, 2156–2158 (2011).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841–842 (2010).
Zook, J. M. et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol. 32, 246–251 (2014).
Lin, G. et al. VennPainter: A Tool for the Comparison and Identification of Candidate Genes Based on Venn Diagrams. PLoS. One. 11, e0154315 (2016).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin.) 6, 80–92 (2012).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011).
Pedersen, B. S. & Quinlan, A. R. Who’s Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy. Am. J. Hum. Genet. 100, 406–413 (2017).
Andrews, R. M. et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147 (1999).
Weissensteiner, H. et al. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 44, W58–W63 (2016).
Ralf, A., Montiel, G. D., Zhong, K. & Kayser, M. Yleaf: Software for Human Y-Chromosomal Haplogroup Inference from Next-Generation Sequencing Data. Mol. Biol. Evol. 35, 1291–1294 (2018).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP237990 (2020).
Khan, S. Y. et al. Whole-genome sequencing of multiple individuals of Pakistani descent. figshare https://doi.org/10.6084/m9.figshare.c.4787124 (2020).
Metspalu, M. et al. Most of the extant mtDNA boundaries in south and southwest Asia were likely shaped during the initial settlement of Eurasia by anatomically modern humans. BMC. Genet. 5, 26 (2004).
Malyarchuk, B. A. et al. Mitochondrial DNA variability in Slovaks, with application to the Roma origin. Ann. Hum. Genet. 72, 228–240 (2008).
Acknowledgements
We are thankful to individuals H3, H4, H5, and H6 for their participation in this study. This study was supported by the National Eye Institute Grant R01EY022714 (SAR).
Author information
Authors and Affiliations
Contributions
S.Y.K. and S.A.R.: conceived and designed the experiments; M.W.L., S. R., S.R., R.A., J.F.H. and S.A.R.: contributed reagents, materials, and analytical tools; S.Y.K., M.A., M.W.L., Z.M., P.B. and M.A.N.: performed experiments; S.Y.K., M.A., M.W.L., Z.M., P.B., A.A.K., M.A.N., S. R., S.R., R.A., J.F.H. and S.A.R.: analyzed the data; S.Y.K., M.A., M.W.L., S. R., S.R., R.A., J.F.H. and S.A.R.: contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Khan, S.Y., Ali, M., Lee, MC.W. et al. Whole genome sequencing data of multiple individuals of Pakistani descent. Sci Data 7, 350 (2020). https://doi.org/10.1038/s41597-020-00664-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-00664-2
- Springer Nature Limited
This article is cited by
-
A review of the pangenome: how it affects our understanding of genomic variation, selection and breeding in domestic animals?
Journal of Animal Science and Biotechnology (2023)