

Abstract
In all organisms, regulation of gene expression must be adjusted to meet cellular requirements and frequently involves helix–turn–helix (HTH) domain proteins1. For instance, in the arms race between bacteria and bacteriophages, rapid expression of phage anti-CRISPR (acr) genes upon infection enables evasion from CRISPR–Cas defence; transcription is then repressed by an HTH-domain-containing anti-CRISPR-associated (Aca) protein, probably to reduce fitness costs from excessive expression2,3,4,5. However, how a single HTH regulator adjusts anti-CRISPR production to cope with increasing phage genome copies and accumulating acr mRNA is unknown. Here we show that the HTH domain of the regulator Aca2, in addition to repressing Acr synthesis transcriptionally through DNA binding, inhibits translation of mRNAs by binding conserved RNA stem-loops and blocking ribosome access. The cryo-electron microscopy structure of the approximately 40 kDa Aca2–RNA complex demonstrates how the versatile HTH domain specifically discriminates RNA from DNA binding sites. These combined regulatory modes are widespread in the Aca2 family and facilitate CRISPR–Cas inhibition in the face of rapid phage DNA replication without toxic acr overexpression. Given the ubiquity of HTH-domain-containing proteins, it is anticipated that many more of them elicit regulatory control by dual DNA and RNA binding.






Similar content being viewed by others

Data availability
The cryo-EM map of the Aca2–RNA complex has been deposited in the Electron Microscopy Data Bank under accession code EMD-43762. Raw cryo-EM micrographs have been deposited to EMPIAR under accession code EMPIAR-11918. The coordinates of the atomic model have been deposited in the Protein Data Bank under accession code 8W35. Source data are provided with this paper.
Code availability
Code for the mathematical modelling is available at https://github.com/JacksonLab/Modelling_Aca-Acr/ and code for bioinformatic analysis of Aca2 family proteins and their associated motifs is available at https://github.com/MaxFeussner/Aca2_Bioinformatics.
References
Aravind, L., Anantharaman, V., Balaji, S., Babu, M. M. & Iyer, L. M. The many faces of the helix–turn–helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 29, 231–262 (2005).
Birkholz, N., Fagerlund, R. D., Smith, L. M., Jackson, S. A. & Fineran, P. C. The autoregulator Aca2 mediates anti-CRISPR repression. Nucleic Acids Res. 47, 9658–9665 (2019).
Stanley, S. Y. et al. Anti-CRISPR-associated proteins are crucial repressors of anti-CRISPR transcription. Cell 178, 1452–1464 (2019).
Shehreen, S., Birkholz, N., Fineran, Peter, C. & Brown, C. M. Widespread repression of anti-CRISPR production by anti-CRISPR-associated proteins. Nucleic Acids Res. 50, 8615–8625 (2022).
Lee, S. Y., Birkholz, N., Fineran, P. C. & Park, H. H. Molecular basis of anti-CRISPR operon repression by Aca10. Nucleic Acids Res. 50, 8919–8928 (2022).
Jacob, F. & Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3, 318–356 (1961).
Laughon, A. & Scott, M. P. Sequence of a Drosophila segmentation gene: protein structure homology with DNA-binding proteins. Nature 310, 25–31 (1984).
McGinnis, W., Garber, R. L., Wirz, J., Kuroiwa, A. & Gehring, W. J. A homologous protein-coding sequence in Drosophila homeotic genes and its conservation in other metazoans. Cell 37, 403–408 (1984).
Bürglin, T. R. & Affolter, M. Homeodomain proteins: an update. Chromosoma 125, 497–521 (2016).
Biedenkapp, H., Borgmeyer, U., Sippel, A. E. & Klempnauer, K.-H. Viral myb oncogene encodes a sequence-specific DNA-binding activity. Nature 335, 835–837 (1988).
McKay, D. B. & Steitz, T. A. Structure of catabolite gene activator protein at 2.9 Å resolution suggests binding to left-handed B-DNA. Nature 290, 744–749 (1981).
Anderson, W. F., Ohlendorf, D. H., Takeda, Y. & Matthews, B. W. Structure of the cro repressor from bacteriophage λ and its interaction with DNA. Nature 290, 754–758 (1981).
Mayo-Muñoz, D., Pinilla-Redondo, R., Birkholz, N. & Fineran, P. C. A host of armor: prokaryotic immune strategies against mobile genetic elements. Cell Rep. 42, 112672 (2023).
Bondy-Denomy, J., Pawluk, A., Maxwell, K. L. & Davidson, A. R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432 (2013).
Pawluk, A. et al. Inactivation of CRISPR–Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat. Microbiol. 1, 16085 (2016).
Usher, B. et al. Crystal structure of the anti-CRISPR repressor Aca2. J. Struct. Biol. 213, 107752 (2021).
Lee, S. Y., Kim, G. E. & Park, H. H. Molecular basis of transcriptional repression of anti-CRISPR by anti-CRISPR-associated 2. Acta Crystallogr. D 78, 59–68 (2022).
Liu, Y. et al. Structural basis for anti-CRISPR repression mediated by bacterial operon proteins Aca1 and Aca2. J. Biol. Chem. 297, 101357 (2021).
Meaden, S. et al. Phage gene expression and host responses lead to infection-dependent costs of CRISPR immunity. ISME J. 15, 534–544 (2021).
Tovkach, F. I. Study of Erwinia carotovora phage resistance with the use of temperate bacteriophage ZF40. Microbiology 71, 72–77 (2002).
Zhang, K. et al. Inhibition mechanisms of AcrF9, AcrF8, and AcrF6 against type I-F CRISPR–Cas complex revealed by cryo-EM. Proc. Natl Acad. Sci. USA 117, 7176–7182 (2020).
Chen, Y.-J. et al. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 10, 659–664 (2013).
Osuna, B. A. et al. Critical Anti-CRISPR locus repression by a bi-functional Cas9 inhibitor. Cell Host Microbe 28, 23–30 (2020).
Kimanius, D. et al. Data-driven regularisation lowers the size barrier of cryo-EM structure determination. Nat. Methods https://doi.org/10.1038/s41592-024-02304-8 (2024).
Segall-Shapiro, T. H., Sontag, E. D. & Voigt, C. A. Engineered promoters enable constant gene expression at any copy number in bacteria. Nat. Biotechnol. 36, 352–358 (2018).
Bleris, L. et al. Synthetic incoherent feedforward circuits show adaptation to the amount of their genetic template. Mol. Syst. Biol. 7, 519 (2011).
Rivera-Pomar, R., Niessing, D., Schmidt-Ott, U., Gehring, W. J. & Jacklë, H. RNA binding and translational suppression by bicoid. Nature 379, 746–749 (1996).
Alfano, C. et al. Structural analysis of cooperative RNA binding by the La motif and central RRM domain of human La protein. Nat. Struct. Mol. Biol. 11, 323–329 (2004).
Dong, G., Chakshusmathi, G., Wolin, S. L. & Reinisch, K. M. Structure of the La motif: a winged helix domain mediates RNA binding via a conserved aromatic patch. EMBO J. 23, 1000–1007 (2004).
Tan, D., Zhou, M., Kiledjian, M. & Tong, L. The ROQ domain of Roquin recognizes mRNA constitutive-decay element and double-stranded RNA. Nat. Struct. Mol. Biol. 21, 679–685 (2014).
Schlundt, A. et al. Structural basis for RNA recognition in roquin-mediated post-transcriptional gene regulation. Nat. Struct. Mol. Biol. 21, 671–678 (2014).
Soler, N., Fourmy, D. & Yoshizawa, S. Structural insight into a molecular switch in tandem winged-helix motifs from elongation factor SelB. J. Mol. Biol. 370, 728–741 (2007).
Yoshizawa, S. et al. Structural basis for mRNA recognition by elongation factor SelB. Nat. Struct. Mol. Biol. 12, 198–203 (2005).
Morrison, J., Anderson, K., Beenken, K., Smeltzer, M. & Dunman, P. The staphylococcal accessory regulator, SarA, is an RNA-binding protein that modulates the mRNA turnover properties of late-exponential and stationary phase Staphylococcus aureus cells. Front. Cell. Infect. Microbiol. 2, 26 (2012).
Chu, L.-C. et al. The RNA-bound proteome of MRSA reveals post-transcriptional roles for helix–turn–helix DNA-binding and Rossmann-fold proteins. Nat. Commun. 13, 2883 (2022).
Conrad, T. et al. Serial interactome capture of the human cell nucleus. Nat. Commun. 7, 11212 (2016).
Oksuz, O. et al. Transcription factors interact with RNA to regulate genes. Mol. Cell 83, 2449–2463.e2413 (2023).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 (2019).
LaFleur, T. L., Hossain, A. & Salis, H. M. Automated model-predictive design of synthetic promoters to control transcriptional profiles in bacteria. Nat. Commun. 13, 5159 (2022).
Solovyev, V. & Salamov, A. in Metagenomics and its applications in agriculture, biomedicine and environmental studies (ed. Li, R. W.) 61–78 (Nova Science Publishers, 2011).
Proctor, J. R. & Meyer, I. M. CoFold: An RNA secondary structure prediction method that takes co-transcriptional folding into account. Nucleic Acids Res. 41, e102 (2013).
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neuböck, R. & Hofacker, I. L. The Vienna RNA Websuite. Nucleic Acids Res. 36, W70–W74 (2008).
Cai, Y. et al. A nucleotidyltransferase toxin inhibits growth of Mycobacterium tuberculosis through inactivation of tRNA acceptor stems. Sci. Adv. 6, eabb6651 (2020).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J 478, 4169–4185 (2021).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Casañal, A., Lohkamp, B. & Emsley, P. Current developments in Coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 29, 1055–1064 (2020).
Croll, T. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D 74, 519–530 (2018).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Kubitschek, H. E. & Friske, J. A. Determination of bacterial cell volume with the Coulter counter. J. Bacteriol. 168, 1466–1467 (1986).
Gillespie, D. T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81, 2340–2361 (1977).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Will, S., Reiche, K., Hofacker, I. L., Stadler, P. F. & Backofen, R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 3, e65 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Yao, Z., Weinberg, Z. & Ruzzo, W. L. CMfinder—a covariance model based RNA motif finding algorithm. Bioinformatics 22, 445–452 (2005).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Nordberg, H. et al. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 42, D26–D31 (2014).
Di Tommaso, P. et al. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17 (2011).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Acknowledgements
This work was supported by a University of Otago Research Grant, the former Bio-Protection Research Centre (Tertiary Education Commission, New Zealand) and Bioprotection Aotearoa (Tertiary Education Commission, New Zealand). N.B. and K.K. were supported by University of Otago Doctoral Scholarships. P.C.F. was supported by an Experienced Researcher Fellowship from the Alexander von Humboldt Foundation and a James Cook Research Fellowship from the Royal Society Te Apārangi of New Zealand. S.A.J. was supported by the Health Research Council of New Zealand (Sir Charles Hercus Fellowship). M.F. was supported by a fellowship of the German Academic Exchange Service (DAAD) and by German Research Foundation (DFG) grant 468749960 to Z.W. C.L.B. was supported by a European Research Council Consolidator Grant (865973) and the DFG (BE 6703/3-1). M.E.W. was supported by a Helen Hay Whitney Foundation/HHMI postdoctoral fellowship and thanks F. Zhang for funding support. S.C.W. was supported by an Engineering and Physical Sciences Research Council Molecular Sciences for Medicine Centre for Doctoral Training studentship (grant number EP/S022791/1). B.U. was supported by a Springboard Award from the Academy of Medical Sciences (grant number SBF002\1104). T.R.B. was funded by a Lister Institute Prize Fellowship. The authors thank S. Valabhji and T. Arrowsmith for assistance with size-exclusion chromatography; and E. Brignole and C. Borsa for the smooth running the MIT.nano cryo-EM facility, established in part with financial support from the Arnold and Mabel Beckman Foundation. We gratefully acknowledge use of the Otago Micro and Nanoscale Imaging (OMNI) Flow Cytometry unit.
Author information
Authors and Affiliations
Contributions
N.B., R.D.F., S.A.J. and P.C.F. conceived the study. N.B. and P.C.F. coordinated the study. All authors designed experiments. N.B., K.K., M.F., M.E.W., C.C.S., A.M., D.K., M.C., S.C.W., B.U., R.D.F. and S.A.J. performed experiments, modelling and bioinformatics, with additional analyses provided by T.R.B., C.M.B., C.L.B., Z.W. and P.C.F. N.B. generated figures, except for structure figures which were created by M.E.W. N.B. and P.C.F. wrote the manuscript with input from the other authors. N.B., T.R.B., C.M.B., C.L.B., Z.W., R.D.F., S.A.J. and P.C.F. provided supervision.
Corresponding author
Ethics declarations
Competing interests
C.L.B. is a co-founder of Leopard Biosciences, co-founder and member of the Scientific Advisory Board of Locus Biosciences and a member of the Scientific Advisory Board of Benson Hill. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Kevin Corbett and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Phage engineering and phage escape mutants.
a) Illustration of the two-step engineering approach for phage ZF40 using the endogenous type I-E CRISPR–Cas system of its host P. carotovorum RC5297. First, the locus to be altered (purple) is deleted using a “deletion plasmid” in which appropriate homologous regions (grey) flank a placeholder. The plasmid also contains a type I-E spacer (red) targeting the original locus, resulting in the selection for phages that have integrated the placeholder. Next, recombinant phages are used to infect cells containing an “addition plasmid” with any locus of choice (yellow) flanked by homology regions; the plasmid also contains a I-E spacer (pink) targeting the placeholder. This results in the selection for and release of phages containing the locus of choice. b) Sequences of wild-type and escape mutant phages in the region surrounding IR1, −10 and −35 motifs. Mutants 1 to 3, with their genotypes indicated, were isolated from plaques formed in the absence of the Aca2 helper plasmids. Point mutations in the sequence are indicated in red.
Extended Data Fig. 2 Investigation of the acrIF8–aca2 5′ UTR and its interaction with Aca2.
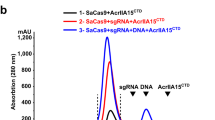
a) Sequencing trace of cDNA generated from the P. carotovorum ZM1 transcriptome using a template-switching reverse transcriptase. The 3′ end of the indicated template-switching oligo sequence borders the 5′ end of the transcript, thus allowing identification of the transcription start site. b) Size-exclusion chromatography traces of Aca2 alone or Aca2 incubated with the 37-nt RNA shown on the right.
Extended Data Fig. 3 Comparison of the acrIF8–eyfp reporter operon with the wild-type acrIF8–aca2 operon.
a) Overview of both operons. In the reporter construct, the red octagon indicates a premature stop codon and the downstream acrIF8 sequence is shown in a lighter shade. Areas in grey boxes are shown in panels b and c. b) Detailed view of the promoter and 5′ UTR region of both operons, with regulatory motifs indicated. c) Detailed view of the intergenic region between acrIF8 and aca2 (wild-type operon) or acrIF8 and eyfp (reporter construct). Note that in the reporter construct, the first three codons of aca2 are fused to eyfp.
Extended Data Fig. 4 Aca2 residues involved in DNA and RNA binding.
a) Cryo-EM density of the ~40-kDa Aca2–RNA complex at 2.6 Å resolution. b) Close-up view of residues Arg30 (R30), Tyr34 (Y34) and Arg39 (R39) in the Aca2–RNA complex (interacting with IR2 or IR-RBS) and the Aca2–DNA complex. c) Clustal Omega Alignment of Aca2 homologs from the indicated species (with accession numbers indicated on the right) to the first 60 amino acids of Aca2. Residues involved in DNA and RNA binding are highlighted in purple, the D45 residue only involved in RNA binding in turquoise. Asterisks indicate conserved residues whereas colons and periods indicate conservation between groups of strongly and weakly similar properties, respectively.
Extended Data Fig. 5 Impact of different regulation modes on AcrIF8 levels and production rate.
a) Modelled mean acrIF8–aca2 mRNA levels (solid green line) over a period of 60 min, with the standard deviation indicated by lighter shading. b-d) Acr production rates as in Fig. 5b but for different modes of regulation: only DNA-based transcriptional regulation (b), only RNA-based translational regulation (c), or no regulation (d). The green line in panels b-d represents the fully regulated state for comparison (same data as displayed in Fig. 5b). e) Time courses corresponding to the end point data in Fig. 5e for the indicated modes of regulation and final phage genome copy numbers. All modelling data shown is the mean (solid line) and standard deviation (shading) from 200 simulations. f) Experimental data for plasmids of different origins and unregulated eyfp–aca2 operons, with eYFP fluorescence detected by flow cytometry (mean and standard deviation of four biologically independent replicates). The determined copy numbers for each plasmid are indicated with a circle.
Extended Data Fig. 6 Aca2-encoding operons are associated with predicted 5′ UTR RNA motifs.
a) Phylogenetic tree of a set of 145 Aca2 homologs. Coloured boxes at the ends of the branches indicate the bacterial phylum of origin for the corresponding operon, and coloured dots indicate the presence or absence of proteo- and actino-motifs associated with the operons. b) Position plot displaying, relative to the transcription start site, the locations of the DNA binding motif (corresponding to IR1 in the ZF40 acrIF8–aca2 promoter), the actino-motif for RNA-based regulation and the start codon. Approximate locations of promoter motifs (−10, −35) are indicated on the x axis. c) Structure and sequence conservation within the actino-motif and its upstream DNA motif. Coloured boxes represent the presence of any nucleotide at this position, with the frequency indicated by the shade as described on the right; coloured letters indicate the likelihood of the nucleotide identity at this position. d) Co-variation analysis of a subset of aca2-containing operons for the Aca2 HTH domain (helix α3 shown) and the IR1 and IR2 sequences. Co-varying residues are indicated by vertical purple and yellow bars. Note that the nucleotide complementary to the co-varying nucleotide in IR2 (grey bar) does not significantly co-vary, likely because a T at this position can, as a U at the RNA level, base-pair with either A or G, thus requiring fewer evolutionary changes.
Supplementary information
Supplementary Information
Supplementary Tables 1–7 and references.
Supplementary Fig. 1
Uncropped gels. Boxed areas are shown in the indicated figures in the main text.
Supplementary Fig. 2
Modelling parameter sweeps and their impact on Acr levels and production rates. For each parameter, five different values were tested and are indicated in different colours; the intermediate values (black) were used in the base model described in the main text, unless otherwise indicated.
Supplementary Fig. 3
Flow cytometry gating. Cells were gated based on forward and side scatter and eYFP fluorescence of individual cells was measured within the gated cell population.
Supplementary Fig. 4
Cryo-EM data processing. a, Example cryo-EM micrograph of the Aca2–RNA complex. b, 2D class averages of the Aca2–RNA complex. c, Flowchart outlining the cryo-EM data processing scheme.
Supplementary Fig. 5
Cryo-EM map statistics and example densities. a, Gold-standard Fourier shell correlation curve for the final reconstruction. b, Orientation distribution plot for the final reconstruction. c, Map-to-model Fourier shell correlation (calculated in PHENIX), softly masking the map around the fitted model. d, Unsharpened cryo-EM map coloured by local resolution with RELION. e, Example cryo-EM densities.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Birkholz, N., Kamata, K., Feussner, M. et al. Phage anti-CRISPR control by an RNA- and DNA-binding helix–turn–helix protein. Nature (2024). https://doi.org/10.1038/s41586-024-07644-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41586-024-07644-1
- Springer Nature Limited