

Abstract
Cellular senescence is a ubiquitous process with roles in tissue remodelling, including wound repair and embryogenesis. However, prolonged senescence can be maladaptive, leading to cancer development and age-related diseases. Cellular senescence involves cell-cycle arrest and the release of inflammatory cytokines with autocrine, paracrine and endocrine activities. Senescent cells also exhibit morphological alterations, including flattened cell bodies, vacuolization and granularity in the cytoplasm and abnormal organelles. Several biomarkers of cellular senescence have been identified, including SA-βgal, p16 and p21; however, few markers have high sensitivity and specificity. In addition to driving ageing, senescence of immune and parenchymal cells contributes to the development of a variety of diseases and metabolic disorders. In the kidney, senescence might have beneficial roles during development and recovery from injury, but can also contribute to the progression of acute kidney injury and chronic kidney disease. Therapies that target senescence, including senolytic and senomorphic drugs, stem cell therapies and other interventions, have been shown to extend lifespan and reduce tissue injury in various animal models. Early clinical trials confirm that senotherapeutic approaches could be beneficial in human disease. However, larger clinical trials are needed to translate these approaches to patient care.
Key points
-
Cellular senescence regulates physiological and homeostatic processes, particularly during embryonic development and wound healing, but can also be a pathological process that contributes to ageing, various diseases and metabolic disorders.
-
Senescent cells are characterized by morphological alterations including large, flat bodies and organelle abnormalities, as well as loss of physiological functions, an inability to proliferate and the senescence-associated secretory phenotype.
-
SABG, p21 and p16 are the most commonly used senescence markers but have limitations; novel non-invasive approaches are needed to detect cellular senescence with high sensitivity and specificity in vitro.
-
Cellular senescence is involved in the pathogenesis of chronic kidney disease and acute kidney injury, but also seems to have a protective role in the early stages of acute kidney injury.
-
Senescence-targeting interventions, including senolytic drugs conjugated to antibodies against β2-microglobulin, chimeric antigen receptor T cells and anti-ageing vaccines, show promise for clinical application.
-
Clinical trials are needed to assess the safety and efficacy of senotherapeutic approaches, optimize treatment regimens and develop more individualized and standardized treatment strategies.
Similar content being viewed by others
Introduction
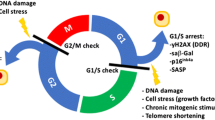
The phenomenon of cellular senescence was discovered in the 1960s in human diploid cell strains that had exhausted their replicative potential1. Senescence is characterized by cell-cycle arrest in the G1 or possibly G2 phase, which prevents the proliferation of damaged cells2,3. By contrast, cellular quiescence, a reversible growth arrest state secondary to scarce nutrition and growth factors, takes place in the G0 phase4. Cellular senescence occurs during embryonic development and can be induced by cellular impairment, including DNA damage, telomere shortening or dysfunction, oncogene activation or loss of tumour suppressor functions, epigenetic changes and organelle damage5.
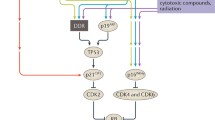
The principal cause of senescent stress is DNA damage6, which activates the DNA damage response (DDR) and the canonical p53–p21 pathway7 (Fig. 1). P21 (also known as cyclin-dependent kinase inhibitor 1) inhibits cyclin–cyclin-dependent kinase complexes that block the formation of the DREAM complex, which represses cell-cycle genes by binding their homology region8. Unlike DDR-induced senescence, epigenetic alterations cause senescence mainly via the p16–RB pathway9. p16 (also known as cyclin-dependent kinase inhibitor 2A) can inhibit the formation of cyclin D–CDK4/6 complexes and thereby prevent phosphorylation of RB and promote formation of the RB–E2F complex, which inhibits the transcription of cell-cycle genes8. Evidence suggests that p21 is mainly activated early during the evolution of senescence, whereas p16 maintains cellular senescence10.

DNA damage secondary to radiation, chemical agents or accumulation of reactive oxygen species (ROS) is the main cause of cellular senescence-inducing stress. Proliferation-induced telomere shortening can also activate the DNA damage response, which in turn leads to activation of the p53–p21 pathway, inhibition of cyclin-dependent kinase (CDK)–cyclin complexes and formation of the DREAM complex, which represses cell-cycle genes, leading to cell-cycle arrest and senescence. Activated p21 can also induce further ROS production, forming a vicious circle. In addition, impaired mitophagy can lead to mitochondrial dysfunction and excessive ROS production. ROS can induce senescence independently of the DNA damage response by activating the p16–RB pathway via activation of ERK, which inhibits BMI1 and thereby enables activation of p16, and by activating p38MAPK signalling, which upregulates ETS2 and in turn activates p16, and by inhibiting NAD+, which leads to reduced expression of sirtuin 1 and activation of FOXO3, which activates p53. P16 inhibits the formation of CDK4/6–cyclin D complexes and thereby promotes formation of the RB–2F complex, which inhibits the transcription of cell-cycle genes. Oncogene activation can activate the p53–p21 pathway not only via the DNA damage response, but also via the ARF–MDM2 signalling. In addition, oncogene activation can activate the p16–RB pathway via p38MAPK signalling. Loss of tumour suppressor genes induces senescence via Akt–mTOR signalling, which activates p53. Other factors that regulate the p53–p21 pathway include α-Klotho, MBP1 and p300. Epigenetic alterations such as methylation (Me) and acetylation (Ac) can induce senescence through the p16–RB pathway. NF-κB signalling regulates the senescence-associated secretory phenotype (SASP) and together with the transcription factor C/EBPβ, co-activates promoters of SASP genes, such as those that encode pro-inflammatory cytokines. The DNA damage response protein ATM together with NEMO activates the NF-κB–C/EBPβ signalling pathway. ROS can activate the SASP not only by promoting nuclear translocation of NEMO and activation of ATM, but also by inhibiting sirtuin 1 and activating p38MAPK and TGFβ, which in turn activate NF-κB. Heat shock, metabolic disorders, mechanical damage and endoplasmic reticulum (ER) stress can also activate the NF-κB–C/EBPβ pathway via p38MAPK signalling. Cytoplasmic DNA accumulation can trigger aberrant activation of cGAS-STING cytoplasmic DNA sensors and promote the SASP via activation of NF-κB. Impairment of autophagy hampers degradation of the transcription factor GATA4, which activates NF-κB and leads to initiation of the SASP. Notably, oncogenic Ras can activate C/EBPβ via ERK–p90 signalling and histone epigenetic changes can regulate the SASP independently of the NF-κB–C/EBPβ pathway. ARF, ADP-ribosylation factor; BMI1, B lymphoma Mo-MLV insertion region 1 homologue; cGAS, cyclic GMP–AMP synthase; CHK, checkpoint kinase; ERK, extracellular regulated protein kinases; ETS2, E26 transformation-specific proto-oncogene 2; E2F, early 2 factor; FOXO3, forkhead box protein O3; IKK, IκB kinase; MDM, mouse double minute 2; MSK, mitogen- and stress-activated protein kinase; NEMO, NF-κB essential modulator; STING, stimulator of interferon genes.
The senescence-associated secretory phenotype (SASP) is an important feature of senescent cells that comprises the release of numerous cytokines, chemokines, growth factors and proteases11, which are sometimes enclosed within microparticles, into the extracellular environment. Many cell types also release extracellular vesicles, which contain cellular contents, including proteins, lipids and nucleic acids. Extracellular vesicles have a role in inter-cellular communication, and altered extracellular vesicle cargoes are important components of the SASP12. Through the release of SASP factors, senescence can modulate pathways in neighbouring cells and tissues as well as at remote sites. Notably, senescent cells that are induced by different stress stimuli may manifest distinctive SASP components13.
Cellular senescence has beneficial biological functions in the regulation of embryonic development, wound healing, resolution of fibrosis and tumour suppression. However, prolonged senescence can result in deleterious sequelae, including tumour development, chronic inflammation, immune deficit and stem cell exhaustion. Interest in cellular senescence and in senescence-modulating interventions is increasing owing to observations that in addition to driving ageing, cellular senescence has important roles in the pathogenesis of chronic diseases, including osteoporosis, metabolic syndrome, type 2 diabetes mellitus, cancer, reproductive ageing, atherosclerosis, neurodegeneration, glaucoma and chronic kidney disease (CKD). In this Review, we describe the mechanisms, hallmarks and consequences of cellular senescence, as well as the therapeutic potential of senescence-targeting interventions.
Senescence in physiology and pathology
Diverse types of stimuli trigger senescence, reflecting its spectrum of roles under different conditions (Table 1). Developmental senescence and replicative senescence (which occurs secondary to telomere shortening) occur under physiological conditions during embryogenesis and ageing, respectively, whereas other types of senescence are often induced by pathological stressors, including tumorigenesis, diabetes mellitus, chemotherapy or radiation.
Embryogenesis and development
In 2006, a transcript of INK4b, which encodes a cyclin-dependent kinase (CDK) inhibitor that blocks progression of the cell cycle beyond the G1 phase, was detected in the roof plate of the develo** chicken hindbrain14, implicating senescence in the regulation of embryonic development. Subsequently, p66Shc, which regulates oxidative stress-induced senescence, was found to mediate early cleavage arrest in failed bovine embryonic development, suggesting that cellular senescence might fine-tune embryogenesis to prevent the continued development of poor-quality embryos15.
In the mammalian embryo, senescence occurs at multiple locations, including the limbs, nervous system and gut endoderm16. In the develo** kidney, accumulation of senescent cells signals immune cells to facilitate mesonephros regression through macrophage-mediated phagocytosis of these senescent cells17. Markers of senescence, including senescence-associated β-galactosidase (SABG), p21, p27 (encoded by CDKN1B) and p15 (encoded by CDKN2B), were detected in mouse mesonephros at embryonic day 12.5 to 14.5 (ref.17). Knocking down p21 in mouse embryos results in developmental abnormalities16. Senescence has also been shown to regulate the development of multiple tissues in zebrafish embryos18.
Importantly, senescent decidual cells in the mammalian endometrium can secrete multiple canonical implantation factors and form a suitable environment for embryonic implantation19. Cellular senescence may therefore have roles in pruning and remodelling develo** systems and modulating their microenvironment, and is thus required to ensure fetal integrity.
Wound healing
Wound healing is a physiological response for repairing tissue injury that involves inflammation, new tissue formation and tissue remodelling. Cellular senescence has an important role throughout the wound-healing process. In cutaneous wound healing, the matricellular protein CCN1 can induce fibroblast or myofibroblast senescence and thereby reduce fibrosis via activation of the DDR and ROS–p16 signalling20. Similarly, in corneal wound healing, fibroblast senescence manifests as an anti-fibrogenic phenotype, with reduced responses to fibroblast growth factor 2 (FGF2; also known as basic FGF) and platelet-derived growth factor-BB, and increased expression of MMP1, MMP3 and MMP13 (ref.21).
Conversely, cellular senescence can interfere with wound healing. For example, inflammation-mediated cellular senescence decreases fibroblast proliferation and migration, which is vital during new tissue formation22. Tenovin-1 treatment induced senescence of cultured astrocytes and thereby impaired their wound-healing activity23. Moreover, senescent lung fibroblasts induced G2/M cell-cycle arrest of alveolar epithelial cells, leading to aberrant wound repair and re-epithelialization24. Senescent mesenchymal stem cell (MSC)-derived extracellular vesicles also inhibit wound healing via a mechanism that involves downregulation of miR-146a25. In diabetes, oxidative stress activates caveolin 1–PTRF signalling, which leads to induction of cellular senescence via the p53–p21 pathway26. The resulting diabetes-induced senescence26, together with a CXCR2-enriched SASP27, impair wound healing. These findings indicate that although transient cellular senescence can promote tissue repair, the prolonged presence of senescent cells can hamper this process.
Cancer
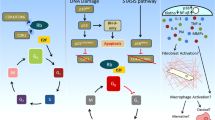
Cellular senescence can have beneficial and detrimental effects in cancer (Fig. 2). Oncogene activation, loss of anti-oncogenes and irreparable DNA damage not only induce apoptosis but also elicit cellular senescence to prevent tumour initiation. Transient insults may result in cell-cycle arrest, which can prevent carcinogenic mutations from being passed on to the next generation of cells and accelerate immune clearance.

Most carcinogenic mutations induce senescence through the p53–p21 and p16–RB pathways, although some mutations activate p21 directly. Reactive oxygen species (ROS)-induced senescence in cancer cells is mainly dependent on p16, p21 and/or p27. Senescence-induced cell-cycle arrest can prevent mutations from being passed on to the next generation of cells and accelerate immune clearance, resulting in suppression of tumurigenesis. However, the senescence-associated secretory phenotype (SASP) also contributes to a pro-inflammatory and growth-stimulatory microenvironment that can promote tumour development.
DDR signalling is the main mechanism of oncogene-induced senescence. Oncogene activation or irreparable DNA damage induce activation of ataxia telangiectasia mutated (ATM) and checkpoint kinase 2 (CHK2), leading to phosphorylation of histone H2AX and p53 and activation of p53–p21 signalling28. Additional pathways, including NLRP6–NF-κB–p14ARF–MDM2 and miR-203–ITPKA–MDM2, can also activate p53–p21 signalling. In breast carcinoma cells, overexpression of oncogenic ERBB2 elicits senescence by upregulating p21 independently of p53 (ref.29). Loss of anti-oncogenes such as PTEN can induce senescence through the Akt–mTOR–p53 and p19ARF–MDM2–p53 signalling pathways30.
Depletion of CSN5 induced senescence in p53-null cells, suggesting that alternative pathways of senescence exist31. Further studies demonstrated that the p16–RB and TAp63–p21–RB pathways are involved in oncogene-induced senescence and suppression of tumorigenesis32,33. In mice, inactivation of heat shock factor 4 (HSF4) enhanced senescence and suppressed tumorigenesis independently of p53 via a mechanism that is dependent on p21 and p27 (ref.34). Reactive oxygen species (ROS)-induced senescence in cancer cells is dependent on p16INK4A, p21Waf1/Cip1 and/or p27Kip1, but does not require p53 (ref.35).
The high-mobility group box-containing protein 1 (HBP1) transcription factor is a novel activator of p21 (and thereby senescence) that acts by attenuating degradation of p53 or by regulating Wnt–β-catenin–EZH2 signalling independently of p53 (ref.36). Wnt signalling also has a role in inducing the senescence of thyroid cancer cells146 and cell density might influence SABG staining regardless of cell proliferation status147. These findings suggest that SABG is not a sufficiently robust and specific marker of senescent cells.
Gene and protein components of senescence-related signalling pathways such as p16 and p21 are canonical markers of senescence148,149. Expression of Met is also considered to be a marker of cellular senescence150 and the accumulation of nuclear globular actin accumulation has been reported to be a more sensitive marker of cellular senescence than SABG activity151. Other markers of senescence include telomere shortening152, the DNA double-strand-break marker H2AX153, typical chromatin changes such as senescence-associated heterochromatin foci154, cytosolic double-stranded DNA and miR-146a155,156. Decreased cellular proliferation potential (which can be measured using BrdU or EdU incorporation assays) and increased apoptosis resistance (for example, as a result of upregulation of BCL proteins) also occur in some types of senescent cells157.
Potential blood or urine biomarkers of senescence include plasma angiopoietin-like 2, growth/differentiation factor 15, stanniocalcin 1 and serine protease inhibitors, serum T-kininogen and urinary 8-oxoguanosine11,158,159,160. Activin A is normally only expressed during embryonic development, but is copiously expressed in injured and senescent kidneys and can be detected in the plasma and urine of patients with kidney disease161. SASP signatures enable the detection and characterization of cellular senescence in vivo but need to be defined in specific biological contexts as they might overlap with inflammatory profiles that are not associated with senescence. Extracellular vesicles can be isolated from urine or peripheral blood and analysed as an index of parent cell senescence; for example, increased levels of urinary URAT1+p16+ extracellular vesicles reflect increased proximal tubular cell senescence162.
Several markers characterize specific senescent tissues or cells. For example, loss of EPC1 expression is a marker of senescence in human dermal fibroblasts163. In T cells, expression of NKG2D, KLRG1, CD57 and CD28 might reflect senescence64,164,165. NKG2D166 and activin A161 are potential markers of senescence in kidney tissue. Spectrin–phaemoglobin crosslinking is an important feature of senescence in red blood cells167, SRSF1 has been postulated to be a key marker of endothelial senescence168 and maspin is a marker of senescence in oral premalignant lesions169.
Endogenous autofluorescence of MSCs is considered to be a useful marker to rapidly determine their senescent status in vitro170. However, the small number of senescent cells relative to quiescent cells, robust tissue autofluorescence, and low penetration of fluorescence signals hamper the detection of senescent cells in vivo using fluorescence-based imaging. Near-infrared imaging methods are not clinically applicable because of the limited availability of high-performance second near-infrared region (NIR-II) fluorophores with high brightness and biocompatibility as well as the long-term health risks of using non-biodegradable quantum dots and lanthanide-doped nanoparticles. Positron emission tomography-based methods of detecting senescent cells are being investigated in animal studies but will require the development of sensitive clinical probes171. Advances in non-invasive imaging methods may enable the detection and spatial allocation of senescence-prone regions that warrant intervention.
Senescence in kidney diseases
Kidney cell senescence was first described in 1992 (ref.172). In addition to its role in physiological kidney ageing, senescence has important roles in the development of CKD and acute kidney injury (AKI) (Fig. 3). Interventions that clear senescent cells, including senolytic drugs, are therefore promising novel treatments for kidney diseases.

Cellular senescence is involved in the pathogenesis of acute kidney injury (AKI) and many types of chronic kidney disease (CKD). Data from animal models suggest that senescent interstitial and tubular epithelial cells contribute to ischaemia–reperfusion injury (IRI), septic shock-induced AKI and contrast-induced AKI, as well as to AKI-to-CKD progression. Tubular epithelial cell senescence has also been detected in many forms of CKD, including obesity-related nephropathy, membranous nephropathy, lupus nephritis, minimal change disease (MCD), unilateral ureteral obstruction (UUO), IgA nephropathy (IgAN) and diabetic kidney disease (DKD). Endothelial cell, podocyte and mesangial cell senescence might also contribute to DKD.
Acute kidney injury
Emerging evidence indicates that senescence contributes to the progression of AKI and that senescence inhibition can promote kidney recovery. For example, inhibition of tubular cell senescence using lipoxin A4 restored renal function in a rat model of septic shock-induced AKI173. Similarly, in a rat model of contrast-induced AKI, pre-treatment with paricalcitol before contrast medium administration reduced cellular senescence (that is, expression of SABG and p16INK4A) and tissue damage and prevented kidney dysfunction174. Notably, patients aged over 70 years have a 3.5-fold higher incidence of AKI than younger individuals175. This increased incidence has been linked to immunosenescence, amplification of the SASP176, and/or downregulation of the geroprotective protein α-Klotho177. Urine and plasma levels of p21 correlate with renal cortical expression of this protein and could be a useful non-invasive biomarker of AKI and kidney ageing178.
The complement system (C5a), DNA methylation, Wnt4–β-catenin signalling and ROS have been implicated in the development of cellular senescence in AKI179,180. Senescence has been shown to reduce the regenerative capacity of tubular, glomerular and interstitial cells181 and to delay recovery from AKI induced by ischaemia–reperfusion injury (IRI) in mice182. Following IRI, markers of kidney cell senescence (Bax and p16 mRNA) peaked at day 12, suggesting an increase in senescent cells in the chronic phase after AKI that might contribute to maladaptive repair and progression to CKD183. Treatment with nicotinamide mononucleotide reduced tubular cell DNA damage and senescence and attenuated renal fibrosis in mouse models of ischaemic AKI184. Increasing evidence suggests that cellular senescence might also have beneficial effects in AKI. A small-molecule inhibitor of CDK4 and CDK6 induced proximal tubule cell-cycle arrest and ameliorated kidney injury in a mouse IRI model185. Moreover, we found that inhibition of senescence within the first week after induction of renal ischaemia in a mouse model impeded functional recovery (measured using renal perfusion and plasma cystatin-c levels)186, suggesting a protective role early in the process of AKI. Delineating the time course of kidney injury and the role of cellular senescence during each phase might identify a therapeutic time window to target senescence and interrupt the development of AKI.
Chronic kidney disease
CKD is increasingly recognized to mimic age-related diseases and senescence and the SASP are important drivers of CKD progression187 (Fig. 4). CKD can accelerate the senescence of immune, endothelial and vascular smooth muscle cells via a process known as uraemia-associated ageing188,189, potentially constituting a feed-forward mechanism of cellular damage. Immunosenescence in CKD manifests as an increased proportion of terminally differentiated T cells, telomere shortening of mononuclear cells, low thymic output189 and reduced immune-mediated clearance of senescent kidney cells, which promotes CKD progression.

Several stimuli can trigger the senescence of various kidney cell types through different pathways. High glucose levels result in macrophage infiltration into the kidney, mitochondrial dysfunction and activation of NOX1–PKC signalling, which lead to an increase in reactive oxygen species (ROS) and senescence of tubular epithelial cells and endothelial cells. High glucose can also induce senescence of tubular epithelial cells by provoking endoplasmic reticulum (ER) stress, which activates ATF4–p16 signalling. In addition, high glucose induces mesangial cell senescence via AGE–STAT5 signalling, which leads to the inhibition of autophagy and therefore results in the accumulation of injured mitochondrial and ROS (not shown). Nephrotoxic drugs and ageing can induce podocyte and tubular epithelial cell senescence via inhibition of C/EBPα, which leads to a reduction in AMPK–mTOR signalling. Ageing and high glucose can also induce tubular epithelial cell senescence by inhibiting AMPK. Nephrotoxic drugs, ischaemia, radiation and unilateral ureteral obstruction (UUO) activate Wnt–β-catenin signalling, which inhibits autophagy and therefore induces the senescence of tubular epithelial cells. Notably, ischaemia and metabolic syndrome can induce the senescence of kidney scattered tubule-like cells (STCs) and mesenchymal stem cells (MSCs), respectively, resulting in impairment of kidney repair, which promotes progression to chronic kidney disease (CKD). Chronic kidney cell senescence promotes epithelial-to-mesenchymal transition (EMT) and results in the senescence-associated secretory phenotype (SASP), which increases inflammation, eventually leading to the development of fibrosis and CKD. Furthermore, CKD can lead to uraemia-induced senescence of immune cells, endothelial cells, vascular smooth muscle cells and MSCs. CKD is also associated with hyperphosphataemia, which induces senescence in myoblasts, endothelial cells and vascular smooth muscle cells and thereby contributes to sarcopenia and vascular calcification. ADAM, A-disintegrin and metalloproteinase; ATF4, activating transcription factor 4; BRG1, brahma-related gene 1; ILK, integrin-linked protein kinase; NOX1, NADPH oxidase 1; OPTN, optineurin.
The mechanisms that underlie CKD-induced senescence include hyperphosphataemia, a common complication of CKD that elicits senescence in myoblasts190, endothelial cells191 and aorta smooth muscle cells192 and contributes to sarcopenia and vascular calcification. Furthermore, uraemic toxins have been implicated in the senescence of proximal tubular cells193. Tubular epithelial cell senescence can be induced by inhibition of AMPK–mTOR signalling194, activation of the Wnt–β-catenin pathway195 or overexpression of Wnt9a196, and promotes epithelial to mesenchymal transition and consequent fibrosis. α-Klotho, an endogenous antagonist of Wnt–β-catenin signalling, was downregulated in unilateral ureteral obstruction, adriamycin nephropathy, and IRI models of fibrotic kidney disease197. Senolytic combination therapy with dasatinib and quercetin alleviated kidney fibrosis in mouse models of chronic renal ischaemia186 and abrogated the progression of AKI to CKD in mouse models of IRI and cisplatin-induced injury198.
Podocyte senescence induces glomerulosclerosis in the ageing kidney via AMPK–mTOR signalling199, whereas DKD or overfeeding can induce renal cellular senescence by decreasing sirtuin 1 expression200. In patients with DKD, the circulating senescence marker activin A is elevated161 and p16 is upregulated in tubular epithelial cells201. In mice with streptozotocin-induced diabetes, glomerular endothelial senescence is driven by M1 macrophages and largely dependent on intracellular ROS202. High blood glucose also induces mesangial cell senescence by activating RAGE–STAT5 signalling, which inhibits autophagy203 and therefore leads to accumulation of injured mitochondria and ROS, which are important inducers of senescence.
We detected cellular senescence (upregulation of p16, p19 and p21) in the kidneys of mice186, pigs204 and patients186 with renal artery stenosis and observed that ischaemic renovascular disease induces senescence of renal scattered tubular-like cells, which impairs their reparative capacity205. Senescence of renal tubular epithelial cells promotes progression of immunoglobulin A (IgA) nephropathy206 and the presence of p16INK4a-positive cells in kidney biopsy samples from patients with lupus nephritis is associated with renal injury and a worse prognosis207. Elevated gene expression of the senescence markers Tp16, Tp19 and Tp53 was also observed in mice with obesity-induced kidney injury208 and p16 expression was strikingly increased in biopsy samples from patients with glomerular disease209. Exposure of kidney cells to nephrotoxic factors including radiation125, TNF210, hypoxia or glucose oxidase211,212 can induce cell-cycle arrest in vitro. Replicative senescence is also involved in the development of chronic allograft nephropathy213. The complement factor H-related genes CFHR1 and CFHR3 have been implicated in tubular cell senescence in allografts in transplant recipients with IgA nephropathy214.
Extensive basic and clinical studies are required to elucidate the complex mechanisms of cellular senescence in CKD. Although these mechanisms are shared across many forms of kidney disease, the extent and unique features of cellular senescence likely vary with the severity and underlying aetiology of CKD.
Interventions targeting senescence
As cellular senescence has key roles in many age-related diseases, interventions targeting senescence, known as senotherapeutics, are potential therapies for these diseases (Table 2). The promise of such approaches is underscored by the observation that genetic animal models of senescent cell deficiency show improved recovery from kidney injury215 and extended lifespan128. Existing senotherapeutic approaches include drugs that selectively eliminate senescent cells, known as senolytics, drugs that inhibit the SASP, known as senomorphics, exogenous cell-based products and non-pharmacological therapies.
Senolytic interventions
Senolytic drugs were developed to overcome the characteristic resistance to apoptosis of senescent cells by inducing pre-programmed cell death216. Quercetin, a natural flavonoid found in some fruit and vegetables, has been shown to eliminate senescent vascular smooth muscle and endothelial cells in animal models by inducing apoptosis through activation of AMPK217,218, sirtuin 1–PINK1-mediated mitophagy219 and NRF2–NF-κB220 signalling. We found that quercetin blunted the expression of senescence markers in the kidneys of obese mice208 and had beneficial, senescence-independent effects on cardiac function in mice fed a high-fat diet owing to pro-angiogenic activity221, highlighting the multiple modes of action of senolytic drugs.
The plant flavonoid fisetin222,223 and procyanidin C1, which is a flavonoid found in grape seeds, are also senolytics224. Herbal extracts can also have senolytic activity. For example, one of the best-known anti-ageing drugs, ginsenoside, is an extract of ginseng that prevents senescence of bone marrow MSCs by activating NRF2 and PI3K–Akt signalling225. Ginsenoside can also modulate the SASP, reduce inflammation, balance redox status and attenuate organ ageing226,227. These observations suggest that selected dietary interventions with senolytic effects could potentially halt the progression of senescence-associated CKD.
AP20187, an FK1012 analogue, selectively induced apoptosis of p16Ink4a‐expressing senescent cells in various mouse models, resulting in improvements in age‐related brain inflammation, cognitive function and stenotic kidney function186,228. Navitoclax (also known as ABT-263) is an inhibitor of anti-apoptotic BCL-2 family proteins that induces apoptosis and exerts potent senolytic effects in ageing animal models and in some types of senescent cells in vitro229,230. Other BCL-2 family inhibitors, including A-1331852, A-1155463, EF24 and venetoclax, are also effective senolytics6,231,232. In addition, heat shock protein 90 inhibitors have senolytic activity233 and radio-electric asymmetric conveyer technology was shown to be effective in reducing the senescence of cultured stem cells234. Notably, these interventions do not selectively target senescent cells, and thus can be associated with off-target adverse effects, such as abnormal embryo development, dysregulated wound healing and tumorigenesis.
Organ-targeted or cell-targeted approaches using protein-based or peptide-based carriers, nanoparticles, extracellular vesicles or other vehicles could increase the specificity, decrease the off-target effects and facilitate the clinical translation of senolytic interventions. Current strategies to selectively target senescent cells include conjugating toxic drugs to antibodies against the senescent membrane marker β2-microglobulin235, which is upregulated via a p53-dependent mechanism, suggesting that it is a marker of stress-induced senescence. Another strategy involves activation of invariant NK T cells to improve immunosurveillance and removal of senescent cells236. Moreover, chimeric antigen receptor T cells that were engineered to specifically recognize the urokinase-type plasminogen activator receptor on the surface of senescent cells had senolytic effects in vitro and in vivo237. Finally, anti-ageing vaccines have been developed to target CD153+ senescent T cells or GPNMB+ senescent endothelial cells with promising results in obese mouse models238.
Senomorphic drugs
One of the best-studied senomorphic drugs is metformin, which reduces the incidence of age-related diseases and expands the lifespan of Caenorhabditis elegans, mice and patients with type 2 diabetes mellitus239. Metformin also enhances the anticancer efficacy of CDK4 and CDK6 inhibitors by modulating the SASP240, inhibits endothelial senescence caused by high glucose-induced metabolic memory by regulating the sirtuin 1–p300–p53–p21 pathway241, triggers immune‐mediated clearance of senescent cells and restores tumour immunosurveillance242.
Other senomorphic drugs include ruxolitinib, a JAK inhibitor that reduces inflammation and alleviates frailty in aged mice by suppressing inflammatory SASP factors243. mTOR inhibitors, such as rapamycin, inhibit senescence and suppress the SASP in endothelial cells244 and fibroblasts245 by inducing autophagy, thereby reducing the accumulation of damaged organelles. Activation of mTOR leads to peroxisome proliferator-activated receptor-γ coactivator 1β-dependent mitochondrial biogenesis, ROS production and persistent activation of the DDR246. Thus, inhibition of mTOR ameliorates cellular senescence.
Several herb extracts, such as resveratrol247, also show anti-SASP activity. Furthermore, the small molecule ML324 that inhibits KDM4, which is involved in the epigenetic regulation of SASP genes, was shown to decrease the SASP in senescent tumour stromal cells248. However, this approach needs to be used cautiously to avoid adverse effects given the sometimes incongruent behaviours of tumour and stromal microenvironments249 and possibly other cellular niches.
Some hormones also have senomorphic effects. For example, melatonin suppresses SASP gene expression by interrupting the recruitment of CREB-binding protein (CBP) by poly-(ADP-ribose) polymerase 1 (PARP1), which is a sensor of DNA damage250. Melatonin improves senescent T cell activity251, alleviated cardiac mitochondrial dysfunction in a mouse model of accelerated senescence252, and rescued MSCs from uraemic toxin-induced senescence in CKD253. Other hormones, including androgens254, oestrogens255, oestradiol256 and glucocorticoids257, can also modulate the release of inflammatory cytokines. However, glucocorticoids need to be used with caution as they can induce senescence in primary human tenocytes257.
Stem cells and extracellular vesicles
In addition to many other salutary effects, stem cells and their extracellular vesicles can exert senolytic activity. For example, bone-marrow-derived MSCs decreased senescence and improved cardiac function in aged mice258, pluripotent stem cells prevented stress-induced senescence in cardiomyocyte-derived cells259 and human umbilical cord-derived MSCs protected rat kidneys from AKI-induced senescence180. We observed relatively modest senolytic efficacy of adipose-derived MSCs in post-stenotic mouse and human kidneys260. In contrast to these senolytic effects, human umbilical cord-derived MSCs increased splenic CD4+ T cell senescence and alleviated symptoms of lupus in mice261. These differing findings suggest that the effects of MSCs are likely cell type and context dependent.
Stem cell extracellular vesicles have robust anti-senescence activity and might be less prone to rejection or tumour formation than their parent stem cells. MSC-derived extracellular vesicles inhibited oxidative stress-induced senescence in endothelial cells, promoted wound closure in ageing diabetic mice162, but methods for their harvesting and characterization require standardization. Clinical end-points and indices of the therapeutic success of senotherapeutics are required for clinical trials. In the future, novel biomarkers of senescence could potentially be used to direct the management of patients who might benefit from attenuation of cellular senescence.
References
Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37, 614–636 (1965).
Di Leonardo, A., Linke, S. P., Clarkin, K. & Wahl, G. M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 8, 2540–2551 (1994).
Gire, V. & Dulic, V. Senescence from G2 arrest, revisited. Cell Cycle 14, 297–304 (2015).
Sun, D. & Buttitta, L. States of G0 and the proliferation-quiescence decision in cells, tissues and during development. Int. J. Dev. Biol. 61, 357–366 (2017).
Herranz, N. & Gil, J. Mitochondria and senescence: new actors for an old play. EMBO J. 35, 701–702 (2016).
Di Micco, R., Krizhanovsky, V., Baker, D. & d’Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 22, 75–95 (2021).
Rossiello, F., Herbig, U., Longhese, M. P., Fumagalli, M. & d’Adda di Fagagna, F. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 26, 89–95 (2014).
Kumari, R. & Jat, P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 9, 645593 (2021).
Petrova, N. V., Velichko, A. K., Razin, S. V. & Kantidze, O. L. Small molecule compounds that induce cellular senescence. Aging Cell 15, 999–1017 (2016).
Dulić, V., Beney, G. E., Frebourg, G., Drullinger, L. F. & Stein, G. H. Uncoupling between phenotypic senescence and cell cycle arrest in aging p21-deficient fibroblasts. Mol. Cell Biol. 20, 6741–6754 (2000).
Basisty, N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020).
Terlecki-Zaniewicz, L. et al. Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype. Aging 10, 1103–1132 (2018).
Özcan, S. et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging 8, 1316–1329 (2016).
Kim, S. H. et al. Upregulation of chicken p15INK4b at senescence and in the develo** brain. J. Cell Sci. 119, 2435–2443 (2006).
Favetta, L. A. et al. The oxidative stress adaptor p66Shc is required for permanent embryo arrest in vitro. BMC Dev. Biol. 7, 132 (2007).
Storer, M. et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130 (2013).
Muñoz-Espín, D. et al. Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118 (2013).
Da Silva-Álvarez, S. et al. Developmentally-programmed cellular senescence is conserved and widespread in zebrafish. Aging 12, 17895–17901 (2020).
Rawlings, T. M. et al. Modelling the impact of decidual senescence on embryo implantation in human endometrial assembloids. Elife 10, e69603 (2021).
Jun, J. I. & Lau, L. F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685 (2010).
Wang, X. et al. Induction of fibroblast senescence during mouse corneal wound healing. Invest. Ophthalmol. Vis. Sci. 60, 3669–3679 (2019).
Younis, L. T., Abu Hassan, M. I., Taiyeb Ali, T. B. & Bustami, T. J. 3D TECA hydrogel reduces cellular senescence and enhances fibroblasts migration in wound healing. Asian J. Pharm. Sci. 13, 317–325 (2018).
Bang, M. et al. Tenovin-1 induces senescence and decreases wound-healing activity in cultured rat primary astrocytes. Biomol. Ther. 27, 283–289 (2019).
Blokland, K. E. C. et al. Senescence of IPF lung fibroblasts disrupt alveolar epithelial cell proliferation and promote migration in wound healing. Pharmaceutics 12, 389 (2020).
Xu, M. et al. Hydrogen peroxide-induced senescence reduces the wound healing-promoting effects of mesenchymal stem cell-derived exosomes partially via miR-146a. Aging Dis. 12, 102–115 (2021).
Bitar, M. S., Abdel-Halim, S. M. & Al-Mulla, F. Caveolin-1/PTRF upregulation constitutes a mechanism for mediating p53-induced cellular senescence: implications for evidence-based therapy of delayed wound healing in diabetes. Am. J. Physiol. Endocrinol. Metab. 305, E951–E963 (2013).
Wilkinson, H. N. et al. Elevated local senescence in diabetic wound healing is linked to pathological repair via CXCR2. J. Invest. Dermatol. 139, 1171–1181 (2019).
Bartkova, J. et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 (2005).
Trost, T. M. et al. Premature senescence is a primary fail-safe mechanism of ERBB2-driven tumorigenesis in breast carcinoma cells. Cancer Res. 65, 840–849 (2005).
Alimonti, A. et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Invest. 120, 681–693 (2010).
Tsujimoto, I., Yoshida, A., Yoneda-Kato, N. & Kato, J. Y. Depletion of CSN5 inhibits Ras-mediated tumorigenesis by inducing premature senescence in p53-null cells. FEBS Lett. 586, 4326–4331 (2012).
Yasaei, H. et al. Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumour-suppressor genes drive induced senescence bypass in normal diploid mammalian cells. Oncogene 32, 171–179 (2013).
Guo, X. et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat. Cell Biol. 11, 1451–1457 (2009).
**, X. et al. Inactivation of heat shock factor Hsf4 induces cellular senescence and suppresses tumorigenesis in vivo. Mol. Cancer Res. 10, 523–534 (2012).
Osanai, M. et al. Occludin-mediated premature senescence is a fail-safe mechanism against tumorigenesis in breast carcinoma cells. Cancer Sci. 98, 1027–1034 (2007).
Chen, Y. et al. HBP1-mediated regulation of p21 protein through the Mdm2/p53 and TCF4/EZH2 pathways and its impact on cell senescence and tumorigenesis. J. Biol. Chem. 291, 12688–12705 (2016).
**ong, J., Jiang, P., Zhong, L. & Wang, Y. The novel tumor suppressor gene ZNF24 induces THCA cells senescence by regulating wnt signaling pathway, resulting in inhibition of THCA tumorigenesis and invasion. Front. Oncol. 11, 646511 (2021).
Tront, J. S., Hoffman, B. & Liebermann, D. A. Gadd45a suppresses Ras-driven mammary tumorigenesis by activation of c-Jun NH2-terminal kinase and p38 stress signaling resulting in apoptosis and senescence. Cancer Res. 66, 8448–8454 (2006).
Georgilis, A. et al. PTBP1-mediated alternative splicing regulates the inflammatory secretome and the pro-tumorigenic effects of senescent cells. Cancer Cell 34, 85–102 (2018).
Yang, G. et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl Acad. Sci. USA 103, 16472–16477 (2006).
Alimirah, F. et al. Cellular senescence promotes skin carcinogenesis through p38MAPK and p44/42MAPK signaling. Cancer Res. 80, 3606–3619 (2020).
Alspach, E. et al. p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. 4, 716–729 (2014).
Ruhland, M. K. et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 7, 11762 (2016).
Okamura, K., Suzuki, T. & Nohara, K. Gestational arsenite exposure augments hepatic tumors of C3H mice by promoting senescence in F1 and F2 offspring via different pathways. Toxicol. Appl. Pharmacol. 408, 115259 (2020).
Flanagan, K. C. et al. c-Myb and C/EBPβ regulate OPN and other senescence-associated secretory phenotype factors. Oncotarget 9, 21–36 (2018).
Muñoz-Galván, S. et al. Tumor cell-secreted PLD increases tumor stemness by senescence-mediated communication with microenvironment. Oncogene 38, 1309–1323 (2019).
Guccini, I. et al. Senescence reprogramming by TIMP1 deficiency promotes prostate cancer metastasis. Cancer Cell 39, 68–82 (2021).
Gonçalves, S. et al. COX2 regulates senescence secretome composition and senescence surveillance through PGE2. Cell Rep. 34, 108860 (2021).
Vindrieux, D. et al. Repression of PLA2R1 by c-MYC and HIF-2alpha promotes cancer growth. Oncotarget 5, 1004–1013 (2014).
Yan, W. et al. Mice deficient in poly(C)-binding protein 4 are susceptible to spontaneous tumors through increased expression of ZFP871 that targets p53 for degradation. Genes Dev. 30, 522–534 (2016).
**e, H. et al. Cell-cycle arrest and senescence in TP53-wild type renal carcinoma by enhancer RNA-P53-bound enhancer regions 2 (p53BER2) in a p53-dependent pathway. Cell Death Dis. 12, 1 (2021).
Zhang, Q. et al. Higher expression of XPF is a critical factor in intrinsic chemotherapy resistance of human renal cell carcinoma. Int. J. Cancer 139, 2827–2837 (2016).
Hu, J. et al. Endoglin is essential for the maintenance of self-renewal and chemoresistance in renal cancer stem cells. Stem Cell Rep. 9, 464–477 (2017).
Kamal, Y., Cheng, C., Frost, H. R. & Amos, C. I. Predictors of disease aggressiveness influence outcome from immunotherapy treatment in renal clear cell carcinoma. Oncoimmunology 8, e1500106 (2019).
Sarkisian, C. J. et al. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol. 9, 493–505 (2007).
Ou, H. L. et al. Cellular senescence in cancer: from mechanisms to detection. Mol. Oncol. 15, 2634–2671 (2021).
Rubelt, F. et al. Onset of immune senescence defined by unbiased pyrosequencing of human immunoglobulin mRNA repertoires. PLoS One 7, e49774 (2012).
Zhang, H., Puleston, D. J. & Simon, A. K. Autophagy and immune senescence. Trends Mol. Med. 22, 671–686 (2016).
Sato, Y. & Yanagita, M. Immunology of the ageing kidney. Nat. Rev. Nephrol. 15, 625–640 (2019).
Chiu, Y. L. et al. Emergence of T cell immunosenescence in diabetic chronic kidney disease. Immun. Ageing 17, 31 (2020).
Lioulios, G., Fylaktou, A., Papagianni, A. & Stangou, M. T cell markers recount the course of immunosenescence in healthy individuals and chronic kidney disease. Clin. Immunol. 225, 108685 (2021).
Trzonkowski, P. et al. Immunosenescence increases the rate of acceptance of kidney allotransplants in elderly recipients through exhaustion of CD4+ T-cells. Mech. Ageing Dev. 131, 96–104 (2010).
Rodriguez, I. J. et al. Immunosenescence study of T cells: a systematic review. Front. Immunol. 11, 604591 (2020).
Alonso-Arias, R. et al. NKG2D expression in CD4+ T lymphocytes as a marker of senescence in the aged immune system. Age 33, 591–-605 (2011).
Pellicanò, M. et al. Evidence for less marked potential signs of T-cell immunosenescence in centenarian offspring than in the general age-matched population. J. Gerontol. A Biol. Sci. Med. Sci. 69, 495–504 (2014).
Song, Y. et al. T-cell immunoglobulin and ITIM domain contributes to CD8+ T-cell immunosenescence. Aging Cell 17, e12716 (2018).
Listì, F. et al. A study of serum immunoglobulin levels in elderly persons that provides new insights into B cell immunosenescence. Ann. NY Acad. Sci. 1089, 487–495 (2006).
de Bourcy, C. F. et al. Phylogenetic analysis of the human antibody repertoire reveals quantitative signatures of immune senescence and aging. Proc. Natl Acad. Sci. USA 114, 1105–1110 (2017).
Lescale, C. et al. Reduced EBF expression underlies loss of B-cell potential of hematopoietic progenitors with age. Aging Cell 9, 410–419 (2010).
Ratliff, M., Alter, S., Frasca, D., Blomberg, B. B. & Riley, R. L. In senescence, age-associated B cells secrete TNFα and inhibit survival of B-cell precursors. Aging Cell 12, 303–311 (2013).
Lutz, C. T. & Quinn, L. S. Sarcopenia, obesity, and natural killer cell immune senescence in aging: altered cytokine levels as a common mechanism. Aging 4, 535–546 (2012).
Sadhu, S. et al. Radiation-induced macrophage senescence impairs resolution programs and drives cardiovascular inflammation. J. Immunol. 207, 1812–1823 (2021).
Li, H. et al. Using ROS as a second messenger, NADPH oxidase 2 mediates macrophage senescence via interaction with NF-κB during Pseudomonas aeruginosa infection. Oxid. Med. Cell Longev. 2018, 9741838 (2018).
Zhao, Q. et al. Activating transcription factor 3 involved in Pseudomonas aeruginosa PAO1-induced macrophage senescence. Mol. Immunol. 133, 122–127 (2021).
Wang, H. et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging 12, 9240–9259 (2020).
Behmoaras, J. & Gil, J. Similarities and interplay between senescent cells and macrophages. J. Cell Biol. 220, e202010162 (2021).
Hall, B. M. et al. p16Ink4a and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 9, 1867–1884 (2017).
Tonnessen-Murray, C. A. et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 218, 3827–3844 (2019).
Hickson, L. J., Eirin, A. & Lerman, L. O. Challenges and opportunities for stem cell therapy in patients with chronic kidney disease. Kidney Int. 89, 767–778 (2016).
Malaise, O. et al. Mesenchymal stem cell senescence alleviates their intrinsic and seno-suppressive paracrine properties contributing to osteoarthritis development. Aging 11, 9128–9146 (2019).
Chen, P. M. et al. c-Maf regulates pluripotency genes, proliferation/self-renewal, and lineage commitment in ROS-mediated senescence of human mesenchymal stem cells. Oncotarget 6, 35404–35418 (2015).
Fan, C. et al. TGF‑β induces periodontal ligament stem cell senescence through increase of ROS production. Mol. Med. Rep. 20, 3123–3130 (2019).
Gu, S., Ran, S., Liu, B. & Liang, J. miR-152 induces human dental pulp stem cell senescence by inhibiting SIRT7 expression. FEBS Lett. 590, 1123–1131 (2016).
**ao, Y. Z. et al. Reducing hypothalamic stem cell senescence protects against aging-associated physiological decline. Cell Metab. 31, 534–548 (2020).
Cho, J. et al. Ewing sarcoma gene Ews regulates hematopoietic stem cell senescence. Blood 117, 1156–1166 (2011).
Pi, C. et al. Nicotinamide phosphoribosyltransferase postpones rat bone marrow mesenchymal stem cell senescence by mediating NAD+-Sirt1 signaling. Aging 11, 3505–3522 (2019).
Son, M. J., Kwon, Y., Son, T. & Cho, Y. S. Restoration of mitochondrial NAD+ levels delays stem cell senescence and facilitates reprogramming of aged somatic cells. Stem Cell 34, 2840–2851 (2016).
Zhai, X. Y. et al. Knockdown of SIRT6 enables human bone marrow mesenchymal stem cell senescence. Rejuvenation Res. 19, 373–384 (2016).
Liu, F. et al. NANOG attenuates hair follicle-derived mesenchymal stem cell senescence by upregulating PBX1 and activating AKT signaling. Oxid. Med. Cell Longev. 2019, 4286213 (2019).
Iwata, T. et al. Functional regulatory mechanisms underlying bone marrow mesenchymal stem cell senescence during cell passages. Cell Biochem. Biophys. 79, 321–336 (2021).
Gannon, H. S., Donehower, L. A., Lyle, S. & Jones, S. N. Mdm2-p53 signaling regulates epidermal stem cell senescence and premature aging phenotypes in mouse skin. Dev. Biol. 353, 1–9 (2011).
Dong, X. Y. et al. Downregulation of ROR2 promotes dental pulp stem cell senescence by inhibiting STK4-FOXO1/SMS1 axis in sphingomyelin biosynthesis. Aging Cell 20, e13430 (2021).
Cho, A. et al. An endogenous anti-aging factor, sonic hedgehog, suppresses endometrial stem cell aging through SERPINB2. Mol. Ther. 27, 1286–1298 (2019).
Zhang, D. et al. Autophagy inhibits the mesenchymal stem cell aging induced by D-galactose through ROS/JNK/p38 signalling. Clin. Exp. Pharmacol. Physiol. 47, 466–477 (2020).
Chang, T. C., Hsu, M. F. & Wu, K. K. High glucose induces bone marrow-derived mesenchymal stem cell senescence by upregulating autophagy. PLoS ONE 10, e0126537 (2015).
Conley, S. M. et al. Human obesity induces dysfunction and early senescence in adipose tissue-derived mesenchymal stromal/stem cells. Front. Cell Dev. Biol. 8, 197 (2020).
Hickson, L. J. et al. Diabetic kidney disease alters the transcriptome and function of human adipose-derived mesenchymal stromal cells but maintains immunomodulatory and paracrine activities important for renal repair. Diabetes 70, 1561–1574 (2021).
Davalli, P., Mitic, T., Caporali, A., Lauriola, A. & D’Arca, D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid. Med. Cell Longev. 2016, 3565127 (2016).
Zhang, C. F. et al. Suppression of autophagy dysregulates the antioxidant response and causes premature senescence of melanocytes. J. Invest. Dermatol. 135, 1348–1357 (2015).
Song, X. et al. Autophagy deficient keratinocytes display increased DNA damage, senescence and aberrant lipid composition after oxidative stress in vitro and in vivo. Redox Biol. 11, 219–230 (2017).
**ng, S. et al. Lactose induced redox-dependent senescence and activated Nrf2 pathway. Int. J. Clin. Exp. Pathol. 12, 2034–2045 (2019).
Su, S. et al. Lowering endogenous cathepsin d abundance results in reactive oxygen species accumulation and cell senescence. Mol. Cell Proteom. 16, 1217–1232 (2017).
Volonte, D. & Galbiati, F. Inhibition of thioredoxin reductase 1 by caveolin 1 promotes stress-induced premature senescence. EMBO Rep. 10, 1334–1340 (2009).
Dasari, A., Bartholomew, J. N., Volonte, D. & Galbiati, F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 66, 10805–10814 (2006).
Uraoka, M. et al. Loss of Bcl-2 during the senescence exacerbates the impaired angiogenic functions in endothelial cells by deteriorating the mitochondrial redox state. Hypertension 58, 254–263 (2011).
Sakai, T., Kurokawa, R., Hirano, S. I. & Imai, J. Hydrogen indirectly suppresses increases in hydrogen peroxide in cytoplasmic hydroxyl radical-induced cells and suppresses cellular senescence. Int. J. Mol. Sci. 20, 456 (2019).
Xu, X. et al. Oxidative stress-induced miRNAs modulate AKT signaling and promote cellular senescence in uterine leiomyoma. J. Mol. Med. 96, 1095–1106 (2018).
Kurz, D. J. et al. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell Sci. 117, 2417–2426 (2004).
Barascu, A. et al. Oxydative stress alters nuclear shape through lamins dysregulation: a route to senescence. Nucleus 3, 411–417 (2012).
Chandrasekaran, A. et al. Redox and mTOR-dependent regulation of plasma lamellar calcium influx controls the senescence-associated secretory phenotype. Exp. Biol. Med. 245, 1560–1570 (2020).
McCarthy, D. A., Clark, R. R., Bartling, T. R., Trebak, M. & Melendez, J. A. Redox control of the senescence regulator interleukin-1α and the secretory phenotype. J. Biol. Chem. 288, 32149–32159 (2013).
Mancini, O. K. et al. Oxidative stress-induced senescence mediates inflammatory and fibrotic phenotypes in fibroblasts from systemic sclerosis patients. Rheumatology 61, 1265–1275 (2021).
Bourdens, M. et al. Short exposure to cold atmospheric plasma induces senescence in human skin fibroblasts and adipose mesenchymal stromal cells. Sci. Rep. 9, 8671 (2019).
Tsai, C. H. et al. Up-regulation of cofilin-1 in cell senescence associates with morphological change and p27kip1-mediated growth delay. Aging Cell 20, e13288 (2021).
Oliva, J. L., Caino, M. C., Senderowicz, A. M. & Kazanietz, M. G. S-Phase-specific activation of PKC alpha induces senescence in non-small cell lung cancer cells. J. Biol. Chem. 283, 5466–5476 (2008).
Freund, A., Laberge, R. M., Demaria, M. & Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 23, 2066–2075 (2012).
Yang, H. et al. Reactive oxygen species and nitric oxide induce senescence of rudimentary leaves and the expression profiles of the related genes in Litchi chinensis. Hortic. Res. 5, 23 (2018).
Gewirtz, D. A., Holt, S. E. & Elmore, L. W. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochem. Pharmacol. 76, 947–957 (2008).
Liu, J. et al. Senescence as a consequence of ginsenoside rg1 response on k562 human leukemia cell line. Asian Pac. J. Cancer Prev. 13, 6191–6196 (2012).
Druelle, C. et al. ATF6α regulates morphological changes associated with senescence in human fibroblasts. Oncotarget 7, 67699–67715 (2016).
Chen, Q. M. et al. Involvement of Rb family proteins, focal adhesion proteins and protein synthesis in senescent morphogenesis induced by hydrogen peroxide. J. Cell Sci. 113, 4087–4097 (2000).
Rivas-Chacón, L. D. M. et al. Role of oxidative stress in the senescence pattern of auditory cells in age-related hearing loss. Antioxidants 10, 1497 (2021).
Shaerzadeh, F. et al. Microglia senescence occurs in both substantia nigra and ventral tegmental area. Glia 68, 2228–2245 (2020).
Ridzuan, N., Al Abbar, A., Yip, W. K., Maqbool, M. & Ramasamy, R. Characterization and expression of senescence marker in prolonged passages of rat bone marrow-derived mesenchymal stem cells. Stem Cell Int. 2016, 8487264 (2016).
Berkenkamp, B. et al. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS ONE 9, e88071 (2014).
Cohen, C. et al. Glomerular endothelial cell senescence drives age-related kidney disease through PAI-1. EMBO Mol. Med. 13, e14146 (2021).
Karin, O. & Alon, U. Senescent cell accumulation mechanisms inferred from parabiosis. Geroscience 43, 329–341 (2021).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Wang, A. S. & Dreesen, O. Biomarkers of cellular senescence and skin aging. Front. Genet. 9, 247 (2018).
Netti, G. S. et al. Role of complement in regulating inflammation processes in renal and prostate cancers. Cells 10, 2426 (2021).
Kawagoe, Y. et al. CXCL5-CXCR2 signaling is a senescence-associated secretory phenotype in preimplantation embryos. Aging Cell 19, e13240 (2020).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014).
Mosteiro, L., Pantoja, C., de Martino, A. & Serrano, M. Senescence promotes in vivo reprogramming through p16INK4a and IL-6. Aging Cell 17, e12711 (2018).
Iannello, A., Thompson, T. W., Ardolino, M., Lowe, S. W. & Raulet, D. H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 210, 2057–2069 (2013).
Kang, T. W. et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551 (2011).
Alimbetov, D. et al. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology 17, 305–315 (2016).
Rana, T. et al. PAI-1 regulation of TGF-β1-induced alveolar type II cell senescence, SASP secretion, and SASP-mediated activation of alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 62, 319–330 (2020).
Liu, D. & Hornsby, P. J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67, 3117–3126 (2007).
Barinda, A. J. et al. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat. Commun. 11, 481 (2020).
Zuccolo, E. et al. The microRNA-34a-induced senescence-associated secretory phenotype (SASP) favors vascular smooth muscle cells calcification. Int. J. Mol. Sci. 21, 4454 (2020).
Salotti, J. & Johnson, P. F. Regulation of senescence and the SASP by the transcription factor C/EBPβ. Exp. Gerontol. 128, 110752 (2019).
Takahashi, A. et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun. 9, 1249 (2018).
En, A., Takauji, Y., Ayusawa, D. & Fujii, M. The role of lamin B receptor in the regulation of senescence-associated secretory phenotype (SASP). Exp. Cell Res. 390, 111927 (2020).
Tripathi, U. et al. SARS-CoV-2 causes senescence in human cells and exacerbates the senescence-associated secretory phenotype through TLR-3. Aging 13, 21838–21854 (2021).
Debacq-Chainiaux, F., Erusalimsky, J. D., Campisi, J. & Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4, 1798–1806 (2009).
de Mera-Rodríguez, J. A. et al. Is senescence-associated β-galactosidase a reliable in vivo marker of cellular senescence during embryonic development? Front. Cell Dev. Biol. 9, 623175 (2021).
Severino, J., Allen, R. G., Balin, S., Balin, A. & Cristofalo, V. J. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp. Cell Res. 257, 162–171 (2000).
Palmer, A. et al. Expression of p16 within myenteric neurons of the aged colon: a potential marker of declining function. Front. Neurosci. 15, 747067 (2021).
López-Domínguez, J. A. et al. Cdkn1a transcript variant 2 is a marker of aging and cellular senescence. Aging 13, 13380–13392 (2021).
Boichuck, M., Zorea, J., Elkabets, M., Wolfson, M. & Fraifeld, V. E. c-Met as a new marker of cellular senescence. Aging 11, 2889–2897 (2019).
Kwak, I. H., Kim, H. S., Choi, O. R., Ryu, M. S. & Lim, I. K. Nuclear accumulation of globular actin as a cellular senescence marker. Cancer Res. 64, 572–580 (2004).
Bernadotte, A., Mikhelson, V. M. & Spivak, I. M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 8, 3–11 (2016).
Turinetto, V. & Giachino, C. Multiple facets of histone variant H2AX: a DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 43, 2489–2498 (2015).
Corpet, A. & Stucki, M. Chromatin maintenance and dynamics in senescence: a spotlight on SAHF formation and the epigenome of senescent cells. Chromosoma 123, 423–436 (2014).
Olivieri, F. et al. MiR-146a as marker of senescence-associated pro-inflammatory status in cells involved in vascular remodelling. Age 35, 1157–1172 (2013).
Zhou, R. et al. Cytosolic dsDNA is a novel senescence marker associated with pyroptosis activation. Tissue Cell 72, 101554 (2021).
Hernandez-Segura, A., Nehme, J. & Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 (2018).
Thorin-Trescases, N., Labbé, P., Mury, P., Lambert, M. & Thorin, E. Angptl2 is a marker of cellular senescence: the physiological and pathophysiological impact of Angptl2-related senescence. Int. J. Mol. Sci. 22, 12232 (2021).
Walter, R., Murasko, D. M. & Sierra, F. T-kininogen is a biomarker of senescence in rats. Mech. Ageing Dev. 106, 129–144 (1998).
Gan, W. et al. Age-dependent increases in the oxidative damage of DNA, RNA, and their metabolites in normal and senescence-accelerated mice analyzed by LC-MS/MS: urinary 8-oxoguanosine as a novel biomarker of aging. Free Radic. Biol. Med. 52, 1700–1707 (2012).
Bian, X. et al. Senescence marker activin A is increased in human diabetic kidney disease: association with kidney function and potential implications for therapy. BMJ Open. Diabetes Res. Care 7, e000720 (2019).
Santelli, A. et al. Senescent kidney cells in hypertensive patients release urinary extracellular vesicles. J. Am. Heart Assoc. 8, e012584 (2019).
Tresini, M., Pignolo, R. J., Allen, R. G. & Cristofalo, V. J. Effects of donor age on the expression of a marker of replicative senescence (EPC-1) in human dermal fibroblasts. J. Cell Physiol. 179, 11–17 (1999).
Parish, S. T., Wu, J. E. & Effros, R. B. Sustained CD28 expression delays multiple features of replicative senescence in human CD8 T lymphocytes. J. Clin. Immunol. 30, 798–805 (2010).
Henson, S. M. & Akbar, A. N. KLRG1-more than a marker for T cell senescence. Age 31, 285–291 (2009).
Günther, J. et al. Identification of the activating cytotoxicity receptor NKG2D as a senescence marker in zero-hour kidney biopsies is indicative for clinical outcome. Kidney Int. 91, 1447–1463 (2017).
Snyder, L. M. et al. Irreversible spectrin-haemoglobin crosslinking in vivo: a marker for red cell senescence. Br. J. Haematol. 53, 379–384 (1983).
Blanco, F. J. & Bernabéu, C. The splicing factor SRSF1 as a marker for endothelial senescence. Front. Physiol. 3, 54 (2012).
Bascones-Martínez, A. et al. Differences in the expression of five senescence markers in oral cancer, oral leukoplakia and control samples in humans. Oncol. Lett. 3, 1319–1325 (2012).
Bertolo, A., Baur, M., Guerrero, J., Pötzel, T. & Stoyanov, J. Autofluorescence is a reliable in vitro marker of cellular senescence in human mesenchymal stromal cells. Sci. Rep. 9, 2074 (2019).
Yamagishi, S. et al. Upregulation of cannabinoid receptor type 2, but not TSPO, in senescence-accelerated neuroinflammation in mice: a positron emission tomography study. J. Neuroinflammation 16, 208 (2019).
Khandjian, E. W., Salomon, C., Léonard, N., Tremblay, S. & Türler, H. Fibronectin gene expression in proliferating, quiescent, and SV40-infected mouse kidney cells. Exp. Cell Res. 202, 464–470 (1992).
Chen, C. et al. Lipoxin A4 restores septic renal function via blocking crosstalk between inflammation and premature senescence. Front. Immunol. 12, 637753 (2021).
Bae, E. et al. Paricalcitol attenuates contrast-induced acute kidney injury by regulating mitophagy and senescence. Oxid. Med. Cell Longev. 2020, 7627934 (2020).
Khan, S., Loi, V. & Rosner, M. H. Drug-induced kidney injury in the elderly. Drugs Aging 34, 729–741 (2017).
Marquez-Exposito, L. et al. Acute kidney injury is aggravated in aged mice by the exacerbation of proinflammatory processes. Front. Pharmacol. 12, 662020 (2021).
Li, Y. & Lerman, L. O. Cellular senescence: a new player in kidney injury. Hypertension 76, 1069–1075 (2020).
Johnson, A. C. & Zager, R. A. Plasma and urinary p21: potential biomarkers of AKI and renal aging. Am. J. Physiol. Renal Physiol. 315, F1329–F1335 (2018).
Castellano, G. et al. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/βcatenin signaling after ischemia/reperfusion injury. Aging 11, 4382–4406 (2019).
Rodrigues, C. E. et al. Human umbilical cord-derived mesenchymal stromal cells protect against premature renal senescence resulting from oxidative stress in rats with acute kidney injury. Stem Cell Res. Ther. 8, 19 (2017).
Westhoff, J. H. et al. Telomere shortening reduces regenerative capacity after acute kidney injury. J. Am. Soc. Nephrol. 21, 327–336 (2010).
Cheng, H., Fan, X., Lawson, W. E., Paueksakon, P. & Harris, R. C. Telomerase deficiency delays renal recovery in mice after ischemia-reperfusion injury by impairing autophagy. Kidney Int. 88, 85–94 (2015).
Sari, F. T., Sari, F. T., Sari, F. T., Arfian, N. & Sari, D. C. R. Effect of kidney ischemia/reperfusion injury on proliferation, apoptosis, and cellular senescence in acute kidney injury in mice. Med. J. Malays. 75, 20–23 (2020).
Jia, Y. et al. Nicotinamide mononucleotide attenuates renal interstitial fibrosis after AKI by suppressing tubular DNA damage and senescence. Front. Physiol. 12, 649547 (2021).
DiRocco, D. P. et al. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am. J. Physiol. Renal Physiol. 306, F379–F388 (2014).
Kim, S. R. et al. Progressive cellular senescence mediates renal dysfunction in ischemic nephropathy. J. Am. Soc. Nephrol. 32, 1987–2004 (2021).
Schroth, J., Thiemermann, C. & Henson, S. M. Senescence and the aging immune system as major drivers of chronic kidney disease. Front. Cell Dev. Biol. 8, 564461 (2020).
Carracedo, J. et al. Mechanisms of cardiovascular disorders in patients with chronic kidney disease: a process related to accelerated senescence. Front. Cell Dev. Biol. 8, 185 (2020).
Crépin, T. et al. Uraemia-induced immune senescence and clinical outcomes in chronic kidney disease patients. Nephrol. Dial. Transpl. 35, 624–632 (2020).
Sosa, P. et al. Hyperphosphatemia promotes senescence of myoblasts by impairing autophagy through Ilk overexpression, a possible mechanism involved in sarcopenia. Aging Dis. 9, 769–784 (2018).
Olmos, G. et al. Hyperphosphatemia induces senescence in human endothelial cells by increasing endothelin-1 production. Aging Cell 16, 1300–1312 (2017).
Troyano, N. et al. Hyperphosphatemia induces cellular senescence in human aorta smooth muscle cells through integrin linked kinase (ILK) up-regulation. Mech. Ageing Dev. 152, 43–55 (2015).
Okada, A. et al. D-serine, a novel uremic toxin, induces senescence in human renal tubular cells via GCN2 activation. Sci. Rep. 7, 11168 (2017).
Dong, D. et al. Alleviation of senescence and epithelial-mesenchymal transition in aging kidney by short-term caloric restriction and caloric restriction mimetics via modulation of AMPK/mTOR signaling. Oncotarget 8, 16109–16121 (2017).
Gong, W. et al. Brahma-related gene-1 promotes tubular senescence and renal fibrosis through Wnt/β-catenin/autophagy axis. Clin. Sci. 135, 1873–1895 (2021).
Luo, C. et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J. Am. Soc. Nephrol. 29, 1238–1256 (2018).
Zhou, L., Li, Y., Zhou, D., Tan, R. J. & Liu, Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/β-catenin signaling. J. Am. Soc. Nephrol. 24, 771–785 (2013).
Li, C., Shen, Y., Huang, L., Liu, C. & Wang, J. Senolytic therapy ameliorates renal fibrosis postacute kidney injury by alleviating renal senescence. FASEB J. 35, e21229 (2021).
Zhang, L. et al. C/EBPα deficiency in podocytes aggravates podocyte senescence and kidney injury in aging mice. Cell Death Dis. 10, 684 (2019).
Juvet, C. et al. Renal programming by transient postnatal overfeeding: the role of senescence pathways. Front. Physiol. 11, 511 (2020).
Liu, J. et al. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am. J. Physiol. Cell Physiol. 308, C621–C630 (2015).
Yu, S. et al. M1 macrophages accelerate renal glomerular endothelial cell senescence through reactive oxygen species accumulation in streptozotocin-induced diabetic mice. Int. Immunopharmacol. 81, 106294 (2020).
Shi, M. et al. The RAGE/STAT5/autophagy axis regulates senescence in mesangial cells. Cell Signal. 62, 109334 (2019).
Kim, S. R., Eirin, A., Zhang, X., Lerman, A. & Lerman, L. O. Mitochondrial protection partly mitigates kidney cellular senescence in swine atherosclerotic renal artery stenosis. Cell Physiol. Biochem. 52, 617–632 (2019).
Chen, X. J. et al. Renovascular disease induces senescence in renal scattered tubular-like cells and impairs their reparative potency. Hypertension 77, 507–518 (2021).
Liu, J. et al. Accelerated senescence of renal tubular epithelial cells is associated with disease progression of patients with immunoglobulin A (IgA) nephropathy. Transl. Res. 159, 454–463 (2012).
Tilman, G. et al. High p16INK4a, a marker of cellular senescence, is associated with renal injury, impairment and outcome in lupus nephritis. RMD Open 7, e001844 (2021).
Kim, S. R. et al. Increased renal cellular senescence in murine high-fat diet: effect of the senolytic drug quercetin. Transl Res. 213, 112–123 (2019).
Sis, B. et al. Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int. 71, 218–226 (2007).
Kim, D. Y., Lee, M. & Kim, E. J. Involvement of Klotho, TNF‑α; and ADAMs in radiation‑induced senescence of renal epithelial cells. Mol. Med. Rep. 23, 22 (2021).
Eleftheriadis, T., Pissas, G., Filippidis, G., Liakopoulos, V. & Stefanidis, I. The role of indoleamine 2,3-dioxygenase in renal tubular epithelial cells senescence under anoxia or reoxygenation. Biomolecules 11, 1522 (2021).
Troyano-Suárez, N. et al. Glucose oxidase induces cellular senescence in immortal renal cells through ILK by downregulating Klotho gene expression. Oxid. Med. Cell Longev. 2015, 416738 (2015).
Ferlicot, S. et al. The role of replicative senescence in chronic allograft nephropathy. Hum. Pathol. 34, 924–928 (2003).
Pesce, F. et al. DelCFHR3-1 influences graft survival in transplant patients with IgA nephropathy via complement-mediated cellular senescence. Am. J. Transpl. 21, 838–845 (2021).
Lee, D. H., Wolstein, J. M., Pudasaini, B. & Plotkin, M. INK4a deletion results in improved kidney regeneration and decreased capillary rarefaction after ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 302, F183–F191 (2012).
Baar, M. P. et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147 (2017).
Kim, S. G., Sung, J. Y., Kim, J. R. & Choi, H. C. Quercetin-induced apoptosis ameliorates vascular smooth muscle cell senescence through AMP-activated protein kinase signaling pathway. Korean J. Physiol. Pharmacol. 24, 69–79 (2020).
Jiang, Y. H., Jiang, L. Y., Wang, Y. C., Ma, D. F. & Li, X. Quercetin attenuates atherosclerosis via modulating oxidized LDL-induced endothelial cellular senescence. Front. Pharmacol. 11, 512 (2020).
Liu, T. et al. Quercetin alleviates kidney fibrosis by reducing renal tubular epithelial cell senescence through the SIRT1/PINK1/mitophagy axis. Life Sci. 257, 118116 (2020).
Shao, Z. et al. Senolytic agent quercetin ameliorates intervertebral disc degeneration via the Nrf2/NF-κB axis. Osteoarthritis Cartilage 29, 413–422 (2021).
Yu, S. et al. Quercetin reverses cardiac systolic dysfunction in mice fed with a high-fat diet: role of angiogenesis. Oxid. Med. Cell Longev. 2021, 8875729 (2021).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018).
Zhu, Y. et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 9, 955–963 (2017).
Xu, Q. et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat. Metab. 3, 1706–1726 (2021).
Wang, Z. et al. Ginsenoside Rg1 prevents bone marrow mesenchymal stem cell senescence via NRF2 and PI3K/Akt signaling. Free Radic. Biol. Med. 174, 182–194 (2021).
Yokozawa, T., Satoh, A. & Cho, E. J. Ginsenoside-Rd attenuates oxidative damage related to aging in senescence-accelerated mice. J. Pharm. Pharmacol. 56, 107–113 (2004).
Hou, J., Cui, C., Kim, S., Sung, C. & Choi, C. Ginsenoside F1 suppresses astrocytic senescence-associated secretory phenotype. Chem. Biol. Interact. 283, 75–83 (2018).
Ogrodnik, M. et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 20, e13296 (2021).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Zhu, Y. et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 (2016).
Adams, J. M. & Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 25, 27–36 (2018).
Mylonas, K. J. et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci. Transl Med. 13, eabb0203 (2021).
Fuhrmann-Stroissnigg, H. et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 8, 422 (2017).
Rinaldi, S. et al. Stem cell senescence. Effects of REAC technology on telomerase-independent and telomerase-dependent pathways. Sci. Rep. 4, 6373 (2014).
Poblocka, M. et al. Targeted clearance of senescent cells using an antibody-drug conjugate against a specific membrane marker. Sci. Rep. 11, 20358 (2021).
Arora, S. et al. Invariant natural killer T cells coordinate removal of senescent cells. Med 2, 938–950 (2021).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020).
Mendelsohn, A. R. & Larrick, J. W. Antiaging vaccines targeting senescent cells. Rejuvenation Res. 25, 39–45 (2022).
Soukas, A. A., Hao, H. & Wu, L. Metformin as anti-aging therapy: is it for everyone? Trends Endocrinol. Metab. 30, 745–755 (2019).
Moiseeva, O. et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 12, 489–498 (2013).
Zhang, E. et al. Metformin and resveratrol inhibited high glucose-induced metabolic memory of endothelial senescence through SIRT1/p300/p53/p21 pathway. PLoS ONE 10, e0143814 (2015).
Menendez, J. A. et al. Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging 3, 1063–1077 (2011).
Xu, M. et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl Acad. Sci. USA 112, E6301–E6310 (2015).
Sasaki, N., Itakura, Y. & Toyoda, M. Rapamycin promotes endothelial-mesenchymal transition during stress-induced premature senescence through the activation of autophagy. Cell Commun. Signal. 18, 43 (2020).
Wang, R. et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 16, 564–574 (2017).
Correia-Melo, C. et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742 (2016).
Menicacci, B. et al. Chronic resveratrol treatment inhibits MRC5 fibroblast SASP-related protumoral effects on melanoma cells. J. Gerontol. A Biol. Sci. Med. Sci. 72, 1187–1195 (2017).
Zhang, B. et al. KDM4 orchestrates epigenomic remodeling of senescent cells and potentiates the senescence-associated secretory phenotype. Nat. Aging 1, 454–472 (2021).
Pazolli, E. et al. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 72, 2251–2261 (2012).
Yu, S. et al. Melatonin regulates PARP1 to control the senescence-associated secretory phenotype (SASP) in human fetal lung fibroblast cells. J. Pineal Res. 63, 12405 (2017).
Yoo, Y. M., Jang, S. K., Kim, G. H., Park, J. Y. & Joo, S. S. Pharmacological advantages of melatonin in immunosenescence by improving activity of T lymphocytes. J. Biomed. Res. 30, 314–321 (2016).
Rodríguez, M. I. et al. Chronic melatonin treatment prevents age-dependent cardiac mitochondrial dysfunction in senescence-accelerated mice. Free Radic. Res. 41, 15–24 (2007).
Han, Y. S., Kim, S. M. & Lee, J. H. Melatonin protects chronic kidney disease mesenchymal stem cells against senescence via PrPC-dependent enhancement of the mitochondrial function. J. Pineal Res. 66, e12535 (2019).
Rais, M., Wilson, R. M., Urbanski, H. F. & Messaoudi, I. Androgen supplementation improves some but not all aspects of immune senescence in aged male macaques. Geroscience 39, 373–384 (2017).
Toillon, R. A. et al. Estrogens decrease gamma-ray-induced senescence and maintain cell cycle progression in breast cancer cells independently of p53. Int. J. Radiat. Oncol. Biol. Phys. 67, 1187–1200 (2007).
Engelmann, F. et al. Accelerated immune senescence and reduced response to vaccination in ovariectomized female rhesus macaques. Age 33, 275–289 (2011).
Poulsen, R. C. et al. Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: in vivo and in vitro evidence. Ann. Rheum. Dis. 73, 1405–1413 (2014).
Zhang, M. et al. Bone marrow mesenchymal stem cell transplantation retards the natural senescence of rat hearts. Stem Cell Transl Med. 4, 494–502 (2015).
Zhang, Y. et al. Rat induced pluripotent stem cells protect H9C2 cells from cellular senescence via a paracrine mechanism. Cardiology 128, 43–50 (2014).
Kim, S. R. et al. Increased cellular senescence in the murine and human stenotic kidney: effect of mesenchymal stem cells. J. Cell Physiol. 236, 1332–1344 (2021).
Cheng, T., Ding, S., Liu, S., Li, Y. & Sun, L. Human umbilical cord-derived mesenchymal stem cell therapy ameliorates lupus through increasing CD4+ T cell senescence via MiR-199a-5p/Sirt1/p53 axis. Theranostics 11, 893–905 (2021).
**ao, X. et al. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal. Transduct. Target. Ther. 6, 354 (2021).
Zhang, L. et al. Selective intrarenal delivery of mesenchymal stem cell-derived extracellular vesicles attenuates myocardial injury in experimental metabolic renovascular disease. Basic. Res. Cardiol. 115, 16 (2020).
Dong, J. et al. Dental pulp stem cell-derived small extracellular vesicle in irradiation-induced senescence. Biochem. Biophys. Res. Commun. 575, 28–35 (2021).
Lei, J. et al. Exosomes from antler stem cells alleviate mesenchymal stem cell senescence and osteoarthritis. Protein Cell 13, 220–226 (2021).
Carroll, J. E. et al. Partial sleep deprivation activates the DNA damage response (DDR) and the senescence-associated secretory phenotype (SASP) in aged adult humans. Brain Behav. Immun. 51, 223–229 (2016).
Sun, L. et al. Alterations in mitochondrial biogenesis and respiratory activity, inflammation of the senescence-associated secretory phenotype, and lipolysis in the perirenal fat and liver of rats following lifelong exercise and detraining. FASEB J. 35, e21890 (2021).
Tylutka, A., Morawin, B., Gramacki, A. & Zembron-Lacny, A. Lifestyle exercise attenuates immunosenescence; flow cytometry analysis. BMC Geriatr. 21, 200 (2021).
Bianchi, A. et al. Moderate exercise inhibits age-related inflammation, liver steatosis, senescence, and tumorigenesis. J. Immunol. 206, 904–916 (2021).
Yzydorczyk, C. et al. Transient postnatal overfeeding causes liver stress-induced premature senescence in adult mice. Sci. Rep. 7, 12911 (2017).
Greeley, E. H., Spitznagel, E., Lawler, D. F., Kealy, R. D. & Segre, M. Modulation of canine immunosenescence by life-long caloric restriction. Vet. Immunol. Immunopathol. 111, 287–299 (2006).
List, E. O. et al. Diet-induced weight loss is sufficient to reduce senescent cell number in white adipose tissue of weight-cycled mice. Nutr. Healthy Aging 4, 95–99 (2016).
Mafra, D. et al. Food as medicine: targeting the uraemic phenotype in chronic kidney disease. Nat. Rev. Nephrol. 17, 153–171 (2021).
Shiels, P. G. et al. Manipulating the exposome to enable better ageing. Biochem. J. 478, 2889–2898 (2021).
Lee, Y. I. & Kim, E. Synergistic effect of 300 μm needle-depth fractional microneedling radiofrequency on the treatment of senescence-induced aging hyperpigmentation of the skin. Int. J. Mol. Sci. 22, 7480 (2021).
Hickson, L. J. et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456 (2019).
Torres, B. et al. Impact of switching to raltegravir and/or adding losartan in lymphoid tissue fibrosis and inflammation in people living with HIV. A randomized clinical trial. HIV Med. 22, 674–681 (2021).
Wong, G. C. L. et al. Horticultural therapy reduces biomarkers of immunosenescence and inflammaging in community-dwelling older adults: a feasibility pilot randomized controlled trial. J. Gerontol. A Biol. Sci. Med. Sci. 76, 307–317 (2021).
Sharma, A. K. et al. The senolytic drug navitoclax (ABT-263) causes trabecular bone loss and impaired osteoprogenitor function in aged mice. Front. Cell Dev. Biol. 8, 354 (2020).
Puglisi, M. et al. A phase I study of the safety, pharmacokinetics and efficacy of navitoclax plus docetaxel in patients with advanced solid tumors. Future Oncol. 17, 2747–2758 (2021).
Rodríguez, J. A. et al. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat. Ecol. Evol. 1, 55 (2017).
Lei, J., Jiang, X., Li, W. & Ren, J. Exosomes from antler stem cells alleviate mesenchymal stem cell senescence and osteoarthritis. Protein Cell 13, 220–226 (2022).
Acknowledgements
The authors’ work was supported by National Institutes of Health grant numbers DK120292, DK122734, AG062104, AG013925 and AG61456, the Connor Fund, Robert P. and Arlene R. Kogod, Robert J. and Theresa W. Ryan, and the Noaber Foundation.
Author information
Authors and Affiliations
Contributions
W.H. researched the data for the article and wrote the text. W.H. and L.O.L contributed substantially to discussion of the content. All authors reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
L.O.L. is an adviser to AstraZeneca, CureSpec, Beren, Ribocure Pharmaceuticals and Butterfly Biosciences. Patents on senolytic drugs and their uses are held by the Mayo Clinic. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic Conflict of Interest policies. The other authors declare no conflicts of interest.
Peer review
Peer review information
Nature Reviews Nephrology thanks Giuseppe Castellano and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- DREAM complex
-
A multisubunit complex formed by the assembly of p130 and p107 with their dimerization partner, E2F4/5, and a multi-vulva class-B core complex.
- Somatic hypermutation
-
A programmed process of adaptation to new foreign elements (such as microbes) whereby changes are introduced to the nucleotide sequences of immunoglobulin genes during B cell development.
- Efferocytosis
-
The process by which apoptotic cells are removed by phagocytic cells.
- Ramified
-
Used to describe cells that have long branch-like processes.
- Geroprotective protein
-
A protein that has anti-ageing effects.
- Radio-electric asymmetric conveyer technology
-
A technology delivering very low-intensity radio-electric emission to generate microcurrents in tissues or culture media and thereby produce biological effects.
- Metabolic memory
-
The durable effect of prior hyperglycaemia on the initiation and progression of diabetic complications.
- Tenocytes
-
Elongated fibroblasts and fibrocytes that reside between collagen fibres.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, W., Hickson, L.J., Eirin, A. et al. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol 18, 611–627 (2022). https://doi.org/10.1038/s41581-022-00601-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41581-022-00601-z
- Springer Nature Limited
This article is cited by
-
Tobacco smoke condensate-induced senescence in endothelial cells was ameliorated by colchicine treatment via suppression of NF-κB and MAPKs P38 and ERK pathways activation
Cell Communication and Signaling (2024)
-
DEC1 is involved in circadian rhythm disruption-exacerbated pulmonary fibrosis
Cell Communication and Signaling (2024)
-
YME1L-mediated mitophagy protects renal tubular cells against cellular senescence under diabetic conditions
Biological Research (2024)
-
Macrophage-derived exosomes promote telomere fragility and senescence in tubular epithelial cells by delivering miR-155
Cell Communication and Signaling (2024)
-
Survey on large language model annotation of cellular senescence from figures in review articles
Genomics & Informatics (2024)