
Abstract
Stainless steel ASTM 316 L is often used in the food industry as contact material with protein-rich dairy products. It has to be welded at some locations for many of these contact materials. This study aimed at i) determining any combined effects of the presence of whey protein (WP) and welding-induced weaknesses on corrosion and metal release, and ii) determining the appropriate welding procedure and filler metal (316 L, 309 L, 312). All weld metals (WMs) showed a higher pitting corrosion susceptibility as compared to the base metal (BM). Under induced friction (stirring), WP significantly enhanced the metal release from all materials, which was accelerated between 1 and 3 days of exposure. Post-imaging indicated pits. 312-WM released significantly higher amounts of metals as compared to the BM and the other WMs. This study indicated that the presence of WP, friction, and weldment-induced corrosion susceptibilities could synergistically cause metal release and corrosion of food contact materials.
Similar content being viewed by others


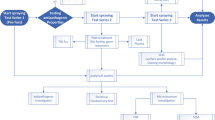
Introduction
Non-corroding, easily cleanable, and safe materials for the production, transportation, and storage of food and beverages are vital for the achievement of food security and improved nutrition1, which are sustainability goals of the United Nations. Food production is increasing as a consequence of the world’s growing population1. Food and beverages, and even more their production, can be very corrosive due to high salt, protein, acid or base contents, high temperatures, wear, and frequent cleaning2,3,4. In addition to material failure, the release of corrosion products, such as metal ions, metal-binding proteins, and metal or metal oxide particles, is of great concern for the food quality2,3,4.
Stainless steels are frequently used in the food industry due to their corrosion resistance, mechanical properties, and appropriate lifetime2. Among all stainless steel grades, AISI 316 L is a popular grade in dairy industries4,5. It is an austenitic (face-centered cubic, non-ferromagnetic) grade with about 16–18 wt.% chromium (Cr), which is responsible for the formation of a thin (1–3 nm), passive surface oxide6. It also contains about 10 wt.% nickel (Ni), which is not present in its surface oxide, but enriched beneath the oxide6,7,8. It contains about 2.5 wt.% molybdenum (Mo), which is responsible for its high resistance against localized corrosion (pitting or crevice corrosion)9. The low amount of carbon in the grade 316 L ensures that the formation of Cr carbides is minimized10.
Milk and its products are significant sources of protein11. Bovine milk contains two types of protein types: casein and whey protein (WP). The former makes up about 80%, and the latter about 20% of its total protein12,13. WP possesses greater functional, biological, and nutritional values, and is more rapidly assimilated by the human body than casein14,15. WP is a mixture of α-lactalbumin, β-lactoglobulin, lactoferrin, immunoglobulins, serum albumin, glycomacropeptides, and enzymes12,13. There is a globally increased protein and WP demand, among others for protein food supplements, bakery, snacks, confectionery, and beverages1,16.
Milk proteins and their adsorption on food contact material surfaces can influence the corrosion and metal release rates17,18,19. It has been shown that metal release from stainless steel 316 L increases strongly under stirring (wear) conditions in the presence of WP in a synergistic way due to the complexation ability of WP under neutral pH conditions18. In addition, the presence of proteins is important for the formation of bacterial biofilms on these surfaces4,20,21,22. For food security, bacterial biofilms need to be removed by regular and frequent cleaning procedures under potentially corrosive conditions, such as oxidative or alkaline chemical environments, high pressure, and elevated temperature4,23.
Food contact materials require welding for the assembling of equipment24, the construction of transmission tanks25, or equipment repair. Gas Tungsten Arc Welding (GTAW) is a common welding method for stainless steel26,27. From a corrosion perspective, welding poses a risk due to a combination of microstructure and residual stress effects induced by a temperature gradient and varying cooling rates representing nonequilibrium solidification28, affecting a variety of possible corrosion types including intergranular corrosion due to carbide precipitations and potential differences (galvanic effects) between the weld metal (WM) and base metal (BM) or along the heat-affected zone (HAZ)29,30,31.
To the best of our knowledge, there is no investigation on the corrosion behavior and metal release of stainless steel weldments in WP-containing solutions. This study aims at (i) determining any combined effects of the presence of WP and welding-induced weaknesses on corrosion and metal release, and (ii) determining the appropriate welding procedure and filler metal to use for joining and repairing processing and transportation equipment made of 316 L in the dairy industry.
Results
Microstructural analysis and microhardness
The microstructure of the BM 316 L consisted of equiaxed austenitic grains with some annealing twins, as expected18,32, Fig. 1a (light optical microscope - LOM), while a varying fraction of ferritic phases is also found for the WMs, Fig. 1b–d. This was also confirmed by X-ray diffraction (XRD), Supplementary Fig. 1, with only austenite (γ) for the BM 316 L, and both γ and delta ferrite (δ) for the WMs. Note that the employed XRD cannot detect less than 5% of volume fraction of a phase, which means that small amounts of δ could still be present in the BM specimen. Both LOM and XRD measurements clearly show that the volume fraction of γ and δ phases and their morphology vary among the different WMs. Energy dispersive X-ray spectroscopy (EDS) analysis revealed an increased Ni and increased Cr/molybdenum (Mo) contents in the γ (inter-dendritic) and δ (dendritic) phases of the WMs, respectively, Fig. 2 and Supplementary Table 1. The varying δ to γ ratios in the WMs can be understood from the solidification modes, which are influenced by their Creq ∕ Nieq ratio, see Supplementary equations 1–6 in the supplementary methods. The 316L-WM and 309L-WM solidify in the ferrite-austenitic (FA) mode, which means that δ dendrites are first solidified from the melt in the melting zone, followed by the formation of the γ phase in the inter-dendritic regions after peritectic and eutectic reactions between the primary ferrite phase and the melt28. The ferrite morphology in the FA mode can be skeletal ferrite and lathy ferrite (see Fig. 1b, c)28. Differences in cooling rates in the early and later stages of solidification cause these different morphologies33. 312-WM, in contrast, exhibits acicular ferrite, widmanstatten, and intergranular austenitic microstructure. This is caused by the higher Creq ∕ Nieq ratio (2.5 as compared to <1.7, Supplementary Table 2) and a ferrite mode (F) solidification mode, Figs. 1d and 2e, f32. In the ferrite (F) mode, the ferrite phase is the only phase that solidified from the melt and exists as a single phase up to the solidus temperature (Supplementary Equation 4). The γ phase is first formed subsequently through incomplete transformation in the solid state34. The absence of the sigma (σ) phase in the microstructure of all three WMs indicates appropriate welding parameters. The fraction of δ ferrite in the WMs was quantified by a ferrite scope to 6.7%, 8.4%, and 19.3% (volume fraction) for the 316L-WM, 309L-WM, and 312-WM, respectively. Figure 3 shows the microstructure at the BM/WM interface for all three WMs, along with line EDS scans. It can clearly be seen that the ferritic fraction increases throughout the HAZ towards the WM zone in all cases. There was no significant difference in composition for the 316L-WM and 309L-WM specimens in the different zones, however 312-WM exhibits a higher Cr content in the weld zone as compared to the BM zone. 316L-WM and 309L-WM showed an epitaxial growth in the transition area, however, 312-WM did not reveal any epitaxial growth, Fig. 3. An epitaxial structure prevents the joint zone between the HAZ and the WM from becoming a stress-focused zone.

LOM micrograph of electro-etched (a) AISI 316-BM. b 316L-WM. c 309L-WM. d 312-WM.

SEM micrograph and corresponding ImageJ for (a, b) 316L-WM, (c, d) 309L-WM and (e, f) 312-WM. Right: relative composition in wt.% of Fe, Cr, Ni, and Mo in the marked areas 1 (δ) and 2 (γ) for each of the WMs. Corresponding compositional data are in Supplementary Table 1.

LOM micrograph and EDS line analysis at the interface of base metal and welds made by (a, b) 316 L, (c, d) 309 L, and (e, f) 312 filler metal.
Microhardness profiles for the WMs showed continuous increasing trends of hardness from the BM through the HAZ and to the WM, Fig. 4. The increasing hardness to the welding zone is explained by the precipitation of the ferrite phase in the austenite grain boundaries, and its role in restricting the grain growth35,36. 312-WM, which had acicular ferrite, widmanstatten, and intergranular austenite, had clearly a higher hardness in the welding zone than 309L-WM and 316L-WM, which had skeletal/lathy ferrite and intergranular austenite, Fig. 4.

Microhardness profile showing hardness for BM, HAZ, and WM of the different weldments. The connecting lines are only guidance for the eye. The vertical dashed lines mark the edges of the HAZ.
Corrosion behavior
Electrochemical impedance spectroscopy (EIS) measurements were conducted to compare the barrier layer characteristics of the oxide layer of BM and weldments in both solutions (phosphate buffered saline, PBS, and PBS + 10 g L−1 WP). Before the measurements, the open circuit potential (OCP) was recorded for one hour. In PBS, there was no statistically significant difference among the specimens in terms of OCP, which was either relatively stable or increasing with time, Supplementary Fig. 2. 312-WM changed the rank from highest OCP among the specimens in PBS to the lowest OCP in PBS + WP, in which it had a significantly lower OCP than 316L-WM. 316L-WM, on the other hand, showed the lowest OCP in PBS and the highest in PBS + WP, Supplementary Fig. 2. The OCP values after 1 h varied from −0.3 to −0.15 VAg/AgCl. Figure 5 depicts representative Nyquist and Bode plots of the different materials. A capacitive arc is shown in the Nyquist plots for all the specimens (Fig. 5a, d). The larger the capacitive semicircle, the higher the corrosion resistance under the experimental conditions. The Bode plots measured in the high-frequency range (from 104 to 105 Hz in Fig. 5b, e) are generally related to the electrolyte resistance, and high negative phase angle values at medium frequencies (102 to 10−2 Hz), between 60 and 80 degrees, indicate a capacitive surface layer. A single unit time constant, corresponding to the oxide layer, can be identified from the EIS measurements. Therefore, the EIS spectra were fitted using the Randles cell equivalent model (Fig. 5c). This equivalent electrical circuit (EEC) model is appropriate for stainless steel EIS plots recorded in solutions of relevance for this study18,37. Supplementary Table 3 shows the average and standard deviation values of the solution resistance (Rs), the constant phase element (CPE), the charge transfer resistance (Rct), the exponent n, and the fitting error indicator χ2. The latter (<0.0053 in all cases) indicates a good agreement between the measured data and the theoretical values, with a better agreement in the solution without proteins (<0.0017). The differences between the different materials were minor in PBS. The only significant (p < 0.05) difference in PBS was an about 1.6-fold decreased CPE and an about 2.7-fold increased Rct of 312-WM when compared to 316L-WM, in agreement with the higher OCP (indicating more passive conditions) of 312-WM in PBS, Supplementary Fig. 2. There were no significant differences among the different materials in PBS + WP, except a significantly higher (2-3-fold) CPE for the BM when compared to the weld metals, but no difference in the Rct. When comparing the different solutions, there was a lower CPE for BM and a higher CPE for 316L-WM in the presence, as compared to the absence, of WP, however, the difference was less than 2.6-fold. The only significant difference in the Rct in the presence of WP as compared to PBS alone was seen for 312-WM, with an increased (3.1-fold) Rct in the presence of WP. The exponent n indicated passive conditions in all cases (0.81-0.91) with small differences among materials and solutions. Only two (out of twelve) comparisons resulted in statistically significant (p < 0.05) differences when comparing n for the different materials in the two solutions. In PBS, the exponent n was larger for 312-WM (0.89) when compared to BM (0.81). In the presence of WP, the exponent n was larger for 316L-WM (0.91) than for BM (0.83). Except for 316L-WM (increased n in the presence of WP), there was no significant difference for the exponent n in the different solutions. In all, the difference between the different solutions and investigated materials is relatively small or non-significant at open circuit potential after 1 h pre-exposure under static (non-stirring) conditions.

Representative Nyquist (a, d) and Bode (b, e) plots of BM and weldments after 1 h immersion at 25 °C in PBS (a, b) and PBS + 10 g L-1 WP (d, e). c Data fitted with one constant equivalent electrical circuit (Randles).
A comparison of the pitting corrosion susceptibility between BM and the welded specimens after 1 h of immersion in both solutions using potentiodynamic polarization is shown in Fig. 6 and Supplementary Table 4. Figure 6 shows representative potentiodynamic curves along with post-polarization optical images (confirming the absence or presence of pits). A low passive current density (<5.2 µA cm−2) is measured in all cases. There was no significant difference in the passive current density among the materials or in the different fluids, except a slightly higher passive current density of the BM when compared to 309L-WM in PBS (p < 0.05). There was no significant difference (p > 0.05) in the corrosion potential (Ecorr) between the different materials in PBS. In the presence of WP, the corrosion potential was significantly (p < 0.05) lower for both BM and 312-WM compared to the 316L-WM. Also, the Ecorr of 312-WM was significantly lower in PBS + WP when compared with PBS alone. All weld metals showed pitting, while the BM did not. The trans-passive breakdown potential of BM in both solutions was significantly (p < 0.05) higher than the pitting potential of all weld metals. In PBS, 309L-WM had a significantly (p < 0.05) lower pitting potential (higher pitting susceptibility) when compared with the other two weld metals, but there was no such difference in PBS + WP. 309L-WM increased its pitting potential significantly in the presence of WP. In all, the welding resulted in higher pitting susceptibility, but minor or no significant differences under passive conditions. The presence of proteins rather suppressed the pitting susceptibility.

a Representative potentiodynamic polarization curves for BM and weldments in PBS. b PBS + 10 g L−1 WP corresponding post-polarization LOM images.
Metal release and visible surface changes
Figure 7 represents the released amounts, normalized on geometrical surface area, of Fe, Cr, and Ni into PBS and PBS + 10 g L−1 WP after 24 h and 72 h of immersion under stirred conditions for the different investigated materials. Any statistically significant differences between the solutions and between the different materials in PBS + WP are also indicated in Fig. 7 and individual elemental release data is shown in Supplementary Fig. 3. All three investigated elements were detected in all solution samples. On average, 67 wt% of the total release was Fe, 16 wt% of the total release was Cr, and 8.4 wt% of the total release was Ni. This is close to the nominal composition (Supplementary Table 2). The presence of WP increased the metal release significantly for two out of four materials after 24 h, and for all materials after 72 h. The release rate increased between 24 and 72 h of exposure in the presence of WP, but not in the absence of it. The amount of released metals increased about 7-fold for a 3-fold increase in time, which means that the release rate increased more than 2-fold in PBS + WP. Instead, in PBS, it decreased up to 2-fold despite the stirring conditions. The 312-WM was consistently releasing the highest amounts of metals. There was no statistically significant difference between the other three materials.

Released average amounts of Fe, Cr, and Ni from the BM, 316L-WM, 309L-WM, and 312-WM triplicate coupons into PBS (adjusted to pH 6.8) and PBS with 10 g L-1 WP (PBS + WP) after 24 h (a) and 72 h (b) under stirring conditions with a magnetic stir bar at room temperature. Any significant difference between the solutions and between the different materials in PBS + WP are indicated by asterisks: *p < 0.05; **p < 0.01; ***p < 0.001, for the respective elements. For details on the elemental release, see Supplementary Fig. 3.
In post-exposure LOM examination, some pits on the metal surface were found for all welded specimens after 72 h of exposure in both solutions (Supplementary Fig. 4). Also, ‘polished’ areas were observed after stirring exposure to PBS + 10 g L−1 WP, but not PBS alone (Supplementary Fig. 4).
Discussion
The pitting corrosion resistance of austenitic stainless steel weldments caused by solidification segregation and partitioning of alloying elements is often lower than that of BM even with matching composition37. The microstructure of each WM in this work contained a dual-phase (γ+δ). The chemical composition inhomogeneity at the γ/δ interface can aggravate localized corrosion due to depletion of Cr and Mo in the austenitic phase (see Fig. 2)38. In the absence of WP, 309L-WM showed the lowest pitting corrosion resistance, which we speculate is related to its lowest Mo content39. There was no significant difference between 316L-WM and 312-WM in terms of pitting corrosion resistance in both solutions, despite the different δ ferrite and Mo contents.
In the presence of WP, we have previously hypothesized that there is a synergistic effect of abrasion (under stirring conditions) and complexation, indicated by the polished areas in Supplementary Fig. 4 and by our previous study18 on rolled 316 L, in which a similar mechanism was found for WP and for citrate (another complexing agent without the lubricating properties of proteins). This previously hypothesized mechanism was further confirmed in this study, and interestingly, an accelerating metal release rate with time was found between 24 and 72 h.
Similar to our previous study, we found the surface abrasion mechanism in PBS to be abrasive wear (Supplementary Fig. 4), with both two-body and three-body abrasive wear, causing the formation of debris and scratches on the surface. The roughness of the coupons stirred in PBS solution increased with time, resulting in a reduced contact area between the magnetic stir bar and the surface of the coupons. In contrast, the wear mechanism in PBS + 10 g L−1 WP seems to be predominantly fatigue wear, along with some two-body and three-body wear, similar to our previous findings in 80 g L−1 WP18. The greater hardness of 312-WM did not result in a decreased metal release or decreased wear under stirring conditions in the presence of WP. Most probably, the other degradation mechanisms (complexation and pitting corrosion) were increased due to the increased ferrite content and low Mo content in 312-WM.
Although there were some visible pits for the WMs as compared to the BM after the stirred exposure for up to 3 days, it is not possible to reveal their status (active, metastable, re-passivated pits and/or pits caused by abrasion) from the optical images alone. However, the accelerated metal release between 1 and 3 days in PBS + WP, along with a greater pitting corrosion susceptibility for the WMs as compared to the BM, indicate that localized corrosion could be involved in this accelerated metal release process. Also, the metal release in approximate proportion to the bulk composition is another indication of active/localized corrosion processes3.
The components of WP are similar to the components of other proteins, especially albumin and globulin proteins, which can increase metal release by a complexation mechanism18,40. In the present work, the WP was present in PBS, which contains salt. This resulted also in pitting corrosion even at open circuit potential under prolonged stirring. However, the pitting corrosion and metal release under 24 h and 72 h stirring did not correlate with the potentiodynamic polarization investigations under static conditions and after 1 h at open-circuit potential. In the metal release investigations, 312-WM clearly released most metals, followed by BM, and all coupons showed some pits. In the potentiodynamic polarization tests, all WMs showed increased susceptibility to pitting, while the BM did not. This indicates that microstructure is of lower importance to metal release under stirring conditions at open circuit potential, as compared to pitting corrosion in an accelerated corrosion test. One explanation is that the pitting potential in static (no friction) conditions is primarily determined by bulk factors, such as alloying elements and microstructure41, while metal release at open circuit potential under stirring in the presence of WP is governed by many different possible factors, such as complexation, metastable pitting corrosion, and tribological factors3. Another possible explanation for the lower or similar metal release of 309L-WM and 316L-WM, compared with BM, is the larger hardness of the welds, which could have locally increased their wear resistance.
This study cannot answer on specific mechanistic questions, such as the predominating metal release mechanism and how the welding parameters influence those. However, it provides an insight in combined mechanisms and points towards WP as a strongly accelerating factor for some release mechanisms. As obvious from the pits and accelerated metal release after the 3 days exposure at open circuit potential under stirring, these release mechanisms have an important implication to corrosion safety and food safety. Future studies should investigate whether standardized testing of welded food contact materials in the dairy industry needs to be adjusted to account for the effect of proteins. For example, neither the European guideline for testing metal release from food contact materials42 nor the ISO 4531:2018 standard test43 suggest proteins or friction. While citric acid, which is suggested in these testing protocols (along with acetic acid), could theoretically be a good simulant for proteins in terms of complexation ability, the pH (2.4) of the food simulant does not allow for complexation with citric acid due to its full protonation at this pH value44.
In summary, the BM 316 L microstructure was austenitic, while the microstructure of the WMs contained increasing (316L-WM < 309L-WM < 312-WM) amounts of δ-ferrite. 312-WM was harder than 309L-WM and 316L-WM. Under static conditions after 1 h pre-exposure under open circuit potential, followed by potentiodynamic polarization, all WMs showed a higher pitting corrosion susceptibility as compared to the BM. This pitting susceptibility was slightly suppressed in the presence of WP. Under induced friction from a magnetic stir bar and open circuit potential, the presence of WP significantly enhanced the metal release from all materials. The proteins further induced an accelerated metal release between 1 and 3 days of exposure, compared with a reduced metal release rate in the reference solution without WP. Post-imaging indicated pits. The release was proportionally to the bulk composition, indicating active/localized corrosion. 312-WM released significantly higher amounts of metals as compared to the BM and the other WMs. This study indicates that the presence of whey proteins, friction, and weldment-induced corrosion susceptibilities could synergistically cause a greater metal release and corrosion of food contact materials.
Methods
Material
The nominal compositions, as provided by suppliers, of the BM and three different filler metals is shown in Supplementary Table 2. The BM, a sheet with 2 mm thickness, was supplied via the International Stainless Steel Forum and investigated/characterized previously18,45. The filler metals were supplied by ESAB, Gothenburg, Sweden, and had a diameter of 2.15 mm. The BM (rolled sheet, 2 mm thickness, 200 mm × 100 mm) was cut using a Wire-cut Electrical Discharge Machining (WEDM). The WP isolate (Lacprodan DI-9224) had a total protein content of at least 92% and was obtained from Arla Food Ingredient, Denmark, and similar to the one we investigated in previous studies17,18.
Welding procedure
Two weld specimens were prepared from the BM (ASTM 316 L) and each of the three filler metals by using the GTAW process, denoted 316L-WM, 309L-WM, and 312-WM, respectively. Prior to welding, surface contaminants were mechanically and chemically removed by wire-brushing and wi** with acetone, respectively. Pre/post-heat treatments were not applied. Visible and ultrasonic inspections during and after welding confirmed the absence of geometrical, surface, and under-surface defects. The shielding gas (100% argon) was purged from the front and back, to prevent the penetration of gases from the atmosphere to the weld zone. All specimens were welded using identical conditions, including a heat input of 0.456 kJ mm−1. More details are given in the supplementary information (Supplementary Table 5 and Supplementary Equation 7).
Microstructural characterization
Specimens for microstructural examination were cut using WEDM and abraded mechanically with a series of SiC papers (80-1200 mesh) and then polished with 1 μm and finally 0.3 μm Al2O3 powder suspension. Then, they were electro-etched for 60–90 s in 100 g L−1 oxalic acid at a voltage of 6 V at room temperature. The different zones of the specimens (Welding zone: WM, heat-affected zone: HAZ, base metal: BM) were investigated using LOM and scanning electron microscopy (SEM), using a Philips-XL30 instrument with an EDS. For qualitative phase analysis, XRD was performed in the weldment and BM using an X Prt-MPD System with Cu-Kα target at 40 kV and 30 mA and the X-pert software. Once we identified all magnetizable phases by XRD (only δ-ferrite present), we used an Aka Scan ferrite scope (MQ51H4) to quantitatively (detection limit about 0.1 vol-%) measure the δ-ferrite content, reporting a five-reading average. The hardness of the weld joints was determined by a micro-hardness tester equipped with a Vickers diamond indenter at a load of 100 g and a dwell time of 10 s. This test was performed along the transverse direction parallel to the surface that includes the different zones (BM, HAZ, and WM) at both sides of the weld.
Corrosion and metal release
Solutions
The solvent in this study was double distilled water (18.2 MΩ cm resistivity, Heidolph instrument). Two solutions were prepared from analytical grade reagents with and without 10 g L−1 WP. The chemical composition of PBS was 1.36 g L−1 KH2PO4, 1.28 g L−1 Na2HPO4, and 8.77 g L−1 NaCl, adjusted to pH 6.8 with NaOH. This pH is relevant for simulated milk and whey protein solutions, and PBS at pH 6.8 (without WP) has previously been found to be a best-case, non-complexing, reference solution18. PBS + 10 g L−1 WP was prepared one day (24 ± 1 h) before each exposure and stored in a refrigerator at 4 °C temperature.
Metal release tests
Coupons (10 mm × 10 mm × 2 mm) were cut using WEDM and abraded with 1200 grit SiC paper on both sides. The edges and back side were sealed, and the total exposed surface area to the solution was 1 cm2. After grinding, coupons were ultrasonically degreased in acetone for 15 min and dried with nitrogen gas at room temperature, followed by storage in a desiccator (<10% relative humidity) at room temperature for 24 ± 1 h prior to exposure to the solution. This surface preparation ensures the formation of a comparable surface oxide46.
All containers and material in contact with the solution were acid-washed (10% HNO3 for at least 24 h, followed by four times rinsing with double distilled water) in order to desorb and dissolve any metal species from the walls, so that they could be rinsed away, and background contamination could be kept minimal47.
Independent samples of triplicate coupons and one blank (no coupon) sample were exposed in parallel, under stirring conditions (with a magnetic stir bar) to 5 mL of PBS and PBS + 10 g L−1 WP at room temperature for 24 h and 72 h. The magnetic stirrer was adjusted to 300 rpm at room temperature. The magnetic bars were smaller than the diameter of the cylinders and were stirred easily without touching the container during the test run. They were placed on top of the coupons, providing friction. After exposure, all coupons were rinsed with 1 mL double distilled water (the rinsing water was wasted) and dried with nitrogen gas at room temperature. The coupons were further examined by LOM, and the solution samples were prepared for solution analysis.
Inductively coupled plasma optical emission spectroscopy (ICP-OES) using a Perkin Elmer Optima 7300DV measured the solution concentrations of Fe, Cr, and Ni released from coupons to both solutions under stirring conditions. Prior to analysis, the solution samples were prepared by acidification (PBS samples) and digestion (PBS + 10 g L−1 WP). For digestion, 0.25 mL of 65% HNO3 was added to 5 mL of solution sample, and then the mixture was boiled gently over a water bath (90 °C) for 1–2 h or until a clear solution was obtained. Later, 0.125 mL of 65 % HNO3 was added, followed by further heating until total digestion. PerkinElmer NIST® quality control standards for ICP, Part No. N9300281, were used as the stock standards for preparing working standards48. The metal concentrations, in μg L−1 were determined based on three replicate readings for each sample. The limits of detection (in both solutions) were 11.5 μg Fe L−1, 1.77 μg Cr L−1, 2.46 μg Ni L−1. All sample concentrations exceeded the limits of detection in this study. The corresponding blank concentrations, if detectable, were subtracted from the triplicate average of sample concentrations. Released amounts of metals (μg cm−2) were calculated by multiplying the blank and dilution factor (DF)-corrected concentration (µg L−1) by the exposure volume V (0.005 L) divided by the exposed coupon area A (1 cm2), as shown in Eq. (1). The number of replicates (n) was 3. The DF is a unitless factor determined from the final volume after digestion divided by the initial sample volume, for example it is 5, if 1 mL of solution sample is diluted with 4 mL of acid or water.
Electrochemical measurements
Coupons (7 mm × 6 mm × 2 mm) were abraded mechanically with 1200 grit SiC paper on both sides, mounted in epoxy, ultrasonically cleaned for 15 min in acetone, dried with nitrogen gas at room temperature, and stored in the desiccator for 24 ± 1 h. The electrochemical measurements were accomplished by the Ivium Potentiostat System and a three-electrode cell with Ag/AgCl sat. KCl as a reference electrode, Pt wire as a counter electrode, and the coupon as working electrode. First, the OCP was measured for 60 min. Then, EIS was conducted with an amplitude of 10 mVrms and a frequency range of 10,000–0.01 Hz. Last, potentiodynamic anodic polarization tests were performed from −0.250 V vs. OCP to 1.6 V or 0.02 A.
To determine the resistance and capacitance of the oxide (passive) layer, EIS experimental data was analyzed with the Z-View software. Measurement plots were fitted with an electrical equivalent circuit model, as specified in the result section. Rs is the solution resistance between the reference and the working electrode. Rp is the polarization resistance, and CPE is the constant phase element, which is related to the passive layer. More details are given in the Supplementary methods and Supplementary Equation 8.
Statistical evaluation
Student’s t-test for unpaired data with unequal variance was used (KaleidaGraph v. 4.0) to determine any statistical significant differences between two sets of data with at least three independent specimens in each set. Differences were counted as statistically significant for probabilities (p) of less than 0.05 of being equal.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Henchion, M., Hayes, M., Mullen, A. M., Fenelon, M. & Tiwari, B. Future protein supply and demand: strategies and factors influencing a sustainable equilibrium. Foods 6, 53 (2017).
Santamaria, M., Tranchida, G. & Di Franco, F. Corrosion resistance of passive films on different stainless steel grades in food and beverage industry. Corros. Sci. 173, 108778 (2020).
Hedberg, Y. S. & Odnevall Wallinder, I. Metal release from stainless steel in biological environments: A review. Biointerphases. 11, 018901-1 - 018901-17 (2016).
Jellesen, M. S., Rasmussen, A. A. & Hilbert, L. R. A review of metal release in the food industry. Mater. Corros. 57, 387–393 (2006).
Jullien, C., Bénézech, T., Carpentier, B., Lebret, V. & Faille, C. Identification of surface characteristics relevant to the hygienic status of stainless steel for the food industry. J. Food Eng. 56, 77–87 (2003).
Olsson, C.-O. A. & Landolt, D. Passive films on stainless steels - chemistry, structure and growth. Electrochim. Acta. 48, 1093–1104 (2003).
Olefjord, I. & Wegrelius, L. Surface analysis of passive state. Corros. Sci. 31, 89–98 (1990).
Hanawa, T., Hiromoto, S., Yamamoto, A., Kuroda, D. & Asami, K. XPS characterization of the surface oxide film of 316L stainless steel samples that were located in quasi-biological environments. Mater. T. JIM. 43, 3088–3092 (2002).
Ameer, M., Fekry, A. M. & Heakal, F. E.-T. Electrochemical behaviour of passive films on molybdenum-containing austenitic stainless steels in aqueous solutions. Electrochim. Acta. 50, 43–49 (2004).
Lunarska, E., Szklarska-Smialowska, Z. & Janik-Czachor, M. Susceptibility of Cr-Ni-Mn stainless steels to pitting in chloride solutions. Corrosion. 31, 231–234 (2013).
Delgado, C., Rosegrant, M., Steinfeld, H., Ehui, S. & Courbois, C. Livestock to 2020: The next food revolution. Outlook Agric. 30, 27–29 (2001).
Rice, B. H., Cifelli, C. J., Pikosky, M. A. & Miller, G. D. Dairy components and risk factors for cardiometabolic syndrome: recent evidence and opportunities for future research. Adv. Nutr. 2, 396–407 (2011).
Minorova, A., Romanchuk, I., Zhukova, Y., Krushelnytska, N. & Vezhlivtseva, S. Protein composition and technological properties of milk whey concentrates. Agric Sci. Pr. 4, 52–58 (2017).
Boirie, Y. et al. Slow and fast dietary proteins differently modulate postprandial protein accretion. Proc. Natl. Acad. Sci. 94, 14930–14935 (1997).
Pennings, B. et al. Whey protein stimulates postprandial muscle protein accretion more effectively than do casein and casein hydrolysate in older men. Am. J. Clin. Nutr. 93, 997–1005 (2011).
Kuntz, L. A. Concentrating on whey protein isolate. Proteins 20, https://college.agrilife.org/talcottlab/wp-content/uploads/sites/108/2019/01/FPD-Whey-Protein-Isolates.pdf (2010).
Atapour, M., Odnevall Wallinder, I. & Hedberg, Y. Stainless steel in simulated milk and whey protein solutions – Influence of grade on corrosion and metal release. Electrochim. Acta. 331, 135428 (2020).
Atapour, M. et al. Metal release from stainless steel 316L in whey protein - and simulated milk solutions under static and stirring conditions. Food Control. 101, 163–172 (2019).
Rabizadeh, T. & Asl, S. K. Casein as a natural protein to inhibit the corrosion of mild steel in HCl solution. J. Mol. Liq. 276, 694–704 (2019).
Marques, S. C. et al. Formation of biofilms by Staphylococcus aureus on stainless steel and glass surfaces and its resistance to some selected chemical sanitizers. Braz. J. Microbiol. 38, 538–543 (2007).
Gupta, S. & Anand, S. Induction of pitting corrosion on stainless steel (grades 304 and 316) used in dairy industry by biofilms of common sporeformers. Int. J. Dairy Technol. 71, 519–531 (2018).
Gao, C. et al. Effect of preparation conditions on protein secondary structure and biofilm formation of kafirin. J. Agr. Food Chem. 53, 306–312 (2005).
Memisi, N., Moracanin, S. V., Milijasevic, M., Babic, J. & Djukic, D. CIP cleaning processes in the dairy industry. Proc. Food Sci. 5, 184–186 (2015).
Unnikrishnan, R. et al. Effect of heat input on the microstructure, residual stresses and corrosion resistance of 304L austenitic stainless steel weldments. Mater. Character 93, 10–23 (2014).
Wardana, R. W., & Warinsiriruk, E. Data envelopment analysis (DEA) and TOPSIS to select welding process at storage tank in the food and beverage industry, in 2018 3rd Technology Innovation Management and Engineering Science International Conference 1-5 (IEEE: 2018).
Norrish, J., Advanced welding processes (Elsevier: Bristol, Philadelphia, 2006).
Kutelu, B. J., Seidu, S. O., Eghabor, G. I. & Ibitoye, A. I. Review of GTAW welding parameters. J. Min. Mater. Char Eng. 6, 541 (2018).
Lippold, J. C. Welding metallurgy and weldability (John Wiley & Sons: Hoboken, New Jersey, 2014).
Kumar, M., Sharma, A., & Shahi, A. A sensitization studies on the metallurgical and corrosion behavior of AISI 304 SS welds. In Advances in Manufacturing Processes 257–265 (Springer: 2019).
Bilmes, P., Llorente, C., Méndez, C. & Gervasi, C. Microstructure, heat treatment and pitting corrosion of 13CrNiMo plate and weld metals. Corros. Sci. 51, 876–881 (2009).
Davis, J. R., Corrosion of weldments (ASM International: Materials Park, OH, 2006).
Chuaiphan, W. & Srijaroenpramong, L. Microstructure, mechanical properties and pitting corrosion of TIG weld joints alternative low-cost austenitic stainless steel grade 216. J. Adv. Join. Proc. 2, 100027 (2020).
Kou, S. Welding metallurgy. 2nd ed. (John Wiley & Sons, Inc.: New Jersey, USA, 2003).
Inoue, H. & Koseki, T. Solidification mechanism of austenitic stainless steels solidified with primary ferrite. Acta Mater. 124, 430–436 (2017).
Mirshekari, G., Tavakoli, E., Atapour, M. & Sadeghian, B. Microstructure and corrosion behavior of multipass gas tungsten arc welded 304L stainless steel. Mater. Des. 55, 905–911 (2014).
Wessman, S. Evaluation of the WRC 1992 diagram using computational thermodynamics. Weld. World 57, 305–313 (2013).
Rajani, H. Z., Torkamani, H., Sharbati, M. & Raygan, S. Corrosion resistance improvement in Gas Tungsten Arc Welded 316L stainless steel joints through controlled preheat treatment. Mater. Des. 34, 51–57 (2012).
Marshall, P. & Gooch, T. Effect of composition on corrosion resistance of high-alloy austenitic stainless steel weld metals. Corrosion 49, 514–526 (1993).
Tokita, S., Kadoi, K., Aoki, S. & Inoue, H. Relationship between the microstructure and local corrosion properties of weld metal in austenitic stainless steels. Corros. Sci. 175, 108867 (2020).
Hedberg, Y. S. Role of proteins in the degradation of relatively inert alloys in the human body. npj Mater. Deg. 2, 26 (2018).
Li, T., Wu, J. & Frankel, G. S. Localized corrosion: Passive film breakdown vs. Pit growth stability, Part VI: Pit dissolution kinetics of different alloys and a model for pitting and repassivation potentials. Corros. Sci. 182, 109277 (2021).
Keitel, S., Metals and alloys used in food contact materials and articles, a practical guide for manufacturers and regulators. ISBN: 978-92-871-7703-2 (Council of Europe: Strasbourg, France, 2013).
ISO 4531:2018, Vitreous and porcelain enamels — Release from enamelled articles in contact with food — Methods of test and limits. International Standard Organization (2018).
Mazinanian, N. & Hedberg, Y. S. Metal release mechanisms for passive stainless steel in citric acid at weakly acidic pH. J. Electrochem. Soc. 163, C686–C693 (2016).
Mazinanian, N., Herting, G., Odnevall Wallinder, I. & Hedberg, Y. Metal release and corrosion resistance of different stainless steel grades in simulated food contact. Corrosion 72, 775–790 (2016).
Herting, G., Odnevall Wallinder, I. & Leygraf, C. Metal release rate from AISI 316L stainless steel and pure Fe, Cr and Ni into a synthetic biological medium- a comparison. J. Environ. Monit. 10, 1092–1098 (2008).
Namieśnik, J. Trace analysis - Challenges and problems. Crit. Rev. Anal. Chem. 32, 271–300 (2002).
Sarojam, P. Analysis of Trace Metals in Surface and Bottled Water with the Optima 7300 DV ICP-OES, 1–7 (2010).
Acknowledgements
This work has been supported by the Canada Research Chairs program (Grant No. 950 – 233099), the Wolfe-Western Fellowship (Grant No. 2020), the Natural Sciences and Engineering Research Council of Canada (Grant Nos. DGDND-2021-03997 and RGPIN-2021-03997), and faculty grants from the University of Western Ontario, Canada, and the Isfahan University of Technology, IRAN (Invited Collaborative Research Program (ICRP)).
Author information
Authors and Affiliations
Contributions
S.V. drafted the paper and conducted the experimental work under the supervision of M.A. All authors evaluated and visualized the results. M.A. and Y.S.H. reviewed and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Varmaziar, S., Atapour, M. & Hedberg, Y.S. Corrosion and metal release characterization of stainless steel 316L weld zones in whey protein solution. npj Mater Degrad 6, 19 (2022). https://doi.org/10.1038/s41529-022-00231-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-022-00231-7
- Springer Nature Limited
This article is cited by
-
Investigating the impact of post-weld cleaning and passivation on the performance of austenitic stainless steel using an EIS paste-electrolyte cell
Journal of Solid State Electrochemistry (2024)
-
3D Printing of high melting iron alloys using metal-fused deposition modeling: a comprehensive review
The International Journal of Advanced Manufacturing Technology (2023)