

Abstract
Microbial genomes are available at an ever-increasing pace, as cultivation and sequencing become cheaper and obtaining metagenome-assembled genomes (MAGs) becomes more effective. Phylogenetic placement methods to contextualize hundreds of thousands of genomes must thus be efficiently scalable and sensitive from closely related strains to divergent phyla. We present PhyloPhlAn 3.0, an accurate, rapid, and easy-to-use method for large-scale microbial genome characterization and phylogenetic analysis at multiple levels of resolution. PhyloPhlAn 3.0 can assign genomes from isolate sequencing or MAGs to species-level genome bins built from >230,000 publically available sequences. For individual clades of interest, it reconstructs strain-level phylogenies from among the closest species using clade-specific maximally informative markers. At the other extreme of resolution, it scales to large phylogenies comprising >17,000 microbial species. Examples including Staphylococcus aureus isolates, gut metagenomes, and meta-analyses demonstrate the ability of PhyloPhlAn 3.0 to support genomic and metagenomic analyses.
Similar content being viewed by others


Introduction
Genomes from isolate sequencing, metagenomic assembly, and single-cell sequencing are being generated at an increasing pace, and they are all correspondingly increasingly available through public resources. This provides invaluable insights into the overall characterization of microbial diversity affecting the human body and the planet. Phylogenetic and corresponding taxonomic characterization is crucial in microbial genomics, for contextualizing genomes without prior phenotypic information, and for determining their genetic novelty and genotype-phenotype relationships. At the largest scale, reconstructing a complete microbial tree-of-life is fundamental in understanding evolutionary relationships in any context, and in microbial community studies such a reference can be a crucial link between novel sequences and health or environmentally relevant microbes. Regardless of the scale, many current microbial genomic tasks thus include the need to place newly sequenced genomes and metagenomic assembled genomes into the microbial taxonomy and phylogenetically characterize them with respect to the closest relatives. With such a volume of microbial genomes generated at a wide range of qualities and completeness, however, there are no scalable phylogenetic methods that can easily tackle these challenges for investigators studying genomes and metagenomes.
Many methods exist for more targeted microbial genome and metagenome phylogenetics. These, include the first implementation of PhyloPhlAn1, PhyloSift2, ezTree3, GToTree4, and AMPHORA5, among many others for more general genome- and gene-based phylogenetics6,7. Most of these methods are limited in at least one way that prevents their ease of use to link newly sequenced genomes, or metagenomic assemblies, into the tremendous space of already characterized microbial phylogenies. None, for example, allow different genomic regions to be selected to achieve optimal resolution in differing clades. This both degrades performance for some clades and prohibits the same methods from being used for strain-level versus phylum-level placement. None leverage the complete set of >100,000 publicly available microbial genomes and and of >200,000 metagenome-assembled genomes (MAGs) from >10,000 metagenomes, and while GToTree automatically retrieves reference genomes from public resources, it does not provide access to MAGs or phylogenetic markers for species-level clades. While computational methods for genome assembly of isolate sequencing and for quantitative analysis of known features of metagenomic data are now mature and well standardized, comparably convenient and automatic tools for downstream phylogenetic and taxonomic assessment of MAGs and microbial isolate genomes are instead lacking and limiting microbial genomic analyses.
These end-to-end phylogenetic solutions should also be differentiated from algorithms and implementations for individual steps of genome placement (e.g., pplacer8 and SEPP9) and taxonomic assessment. Examples include algorithms for multiple-sequence alignment (MSA) like MUSCLE10, MAFFT11, T-Coffee12, OPAL13, PASTA14, and UPP15 and phylogenetic reconstruction like FastTree16,17, RAxML6, ASTRAL18,19,20, ASTRID21, and IQ-TREE22. Each tool can be separately and sequentially applied providing full step-by-step control on the whole phylogenetic analysis, but doing so requires substantial expertize not only in identifying the right targets, parameters, and steps for computational phylogenetics, but also in understanding how such tools should be interfaced one with the other.
Separate and human-supervised execution of these steps is also impractical when individual studies generate thousands of microbial genomes, or when massive numbers of genomes are retrieved and analyzed in combination. Very efficient algorithms have been proposed, including those based on ty** only a few representative marker genes, such as multilocus sequence ty** (MLST) approach23 or on species-level core genes24. Computational MLST, for instance, can operate rapidly using as few as five to ten loci for each species. However, this comes at the cost of greatly reduced accuracy of phylogenetic placement. Pangenome-based profiling like Roary24 is instead very accurate for phylogenetic modeling at species level but cannot be generalized to higher-level clades. Strain-resolved phylogenies integrating thousands of reference genomes from diverse species—or at least those most closely related to new sequences of interest—result in a more accurate characterization of microbes’ population structure and characteristics, while also more accurately guiding taxonomy. Whole-genome large-scale microbial phylogenies, particularly robust to partial assemblies and able to integrate existing genomes and metagenomic assemblies, are thus an open computational challenge.
We thus present here PhyloPhlAn 3.0, a fully automatic, end-to-end phylogenetic analysis framework for contextualization and characterization of newly assembled microbial isolates and metagenomes. PhyloPhlAn 3.0 can, as needed, retrieve and integrate hundreds of thousands of genomes from public resources, while also incorporating preprocessed information from tens of thousands of metagenomes. It automatically uses species-specific sets of core proteins, stably identified using UniRef90 gene families, to build accurate strain-level phylogenies, while also scaling to tens of thousands of genomes for inferring deep branching and very large size phylogenies. PhyloPhlAn 3.0 is both accurate at the strain and species level and fast when scaling to the whole set of available genomes. Compared to available alternatives such as the genome taxonomy database (GTDB)25, PhyloPhlAn 3.0 is able to automatically perform taxonomic assignment of MAGs based on the NCBI taxonomy and to consider unnamed and uncharacterized species in the genomic contextualization task.
Results
Precise phylogenetic placement of genomes and metagenomes
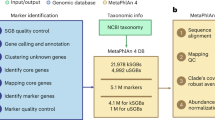
PhyloPhlAn 3.0 provides an easy-to-use and fully automatic method for accurate phylogenetic and taxonomic contextualization of microbial (meta)genomes (Fig. 1). The method can consider combined input sets of microbial genomes from isolate sequencing and of MAGs to produce phylogenies at multiple levels of resolution. Placement of input genomes and MAGs is performed by de novo reconstruction of the phylogeny. For highly resolved phylogenetic trees of related strains, PhyloPhlAn 3.0 uses species-specific core genes from the >18,000 sets of preselected UniRef90 gene families. Instead, for high-diversity genomes, it relies on the 400 most universal markers1,26 with more aggressive alignment trimming options (see “Methods”). Multi-resolution phylogenetic reconstruction is also at the core of the approach to assign taxonomic labels from phylum to species level to input genomes or MAGs, which exploits >150,000 MAGs and >80,000 reference genomes integrated into the PhyloPhlAn 3.0 database. The pipeline thus integrates the large body of available whole-genome microbial data to phylogenetically contextualize input genomes by adopting several methodological advances depending on the characteristics and scale of the specific tasks (see “Methods”). PhyloPhlAn 3.0 is not bound to particular methodological choices for the internal steps: it allows users to choose among multiple tools for sequence map**27,28,23 are highlighted in different colors.
PhyloPhlAn 3.0 can further extend newly generated phylogenies to incorporate one or more existing reference genomes. To illustrate this, we used PhyloPhlAn 3.0 to add 1000 S. aureus reference genomes to the previous tree, automatically selected (from among 7259 total available genomes prioritized based on representativeness, see “Methods”) and retrieved from GenBank38, yielding a larger phylogeny of 1135 genomes (Supplementary Fig. 1). This tree is in close agreement with MLST ty**23, with very small intra-ST phylogenetic distance compared to inter-ST distances (0.0012 vs. 0.1256, respectively), and the resulting genetic context provides a clear interpretation for subspecies structure of the newly sequenced S. aureus isolates in the context of known species diversity (Fig. 2c).
Robust taxonomy assignment for MAGs
In addition to phylogenetic reconstruction, PhyloPhlAn 3.0 can assign a putative taxonomic label to uncharacterized genomes40 if they can be confidently placed in well-labeled phylogenetic clades. Specifically, for each new genome, it identifies the closest SGB from the collection of known and newly defined candidate species spanning 154,723 MAGs and >80,000 isolate genomes41. These span 16,331 SGBs, of which 12,535 have a confident species label based on previous validations alleviating problems of NCBI taxonomic consistency because species labels are assigned to consistently clustered genomes by majority voting (see “Methods”). Following the definition of the SGBs, an input genome is assigned to an SGB (and its associated taxonomy, if any) if the Mash42 average distance to the genomes in the bin is below 5% (see “Methods”), as this threshold has been suggested to be optimal for species definition43,44. If the input cannot be assigned to any SGB, then PhyloPhlAn 3.0 will report the set of closest SGBs (and their average genomic distances). If needed, this procedure is repeated for higher taxonomic clades with genus-level genome bins (GGBs, up to 15% genomic distance) and family-level genome bins (FGBs, up to 30% genomic distance, see “Methods”)41, ultimately providing a more comprehensive taxonomic context for the set of input genomes to guide downstream analyses and complement their phylogenetic placement. Validation on a set of 1520 isolate genomes from the gut microbiome45 assigned an SGB to 1505 genomes (99%) demonstrating that the reference catalog of SGBs covers very well the intestinal microbial diversity including 207 SGBs without a species name. The taxonomic labels inferred by PhyloPhlAn 3.0 were also very consistent (97.7%, Supplementary Data 2) with those assigned at species level in the original work highlighting the consistency of the automatic algorithm.
We used PhyloPhlAn 3.0 to taxonomically place a set of MAGs retrieved from a cohort of 50 rural Ethiopian individuals (see “Data availability”) only used so far to characterize Prevotella copri strains46, as these samples had not been used in the generation of SGBs and are likely to contain substantial unseen phylogenetic diversity. Overall, from the 369 medium- and high-quality input MAGs (see “Methods”), PhyloPhlAn 3.0 provided an assignment to a total of 133 SGBs for 352 MAGs and a closest SGB indication for the remaining 17 MAGs. Twenty-one SGBs were detected in at least 5 samples (Fig. 3A), and the most prevalent SGBs were for Eubacterium rectale (ID 4933 found in 20 samples), an uncultured Dialister species (ID 5809, 18 samples) and an unnamed Succinatimonas species (ID 3677, 18 samples). While PhyloPhlAn 3.0 assigned an SGB to the majority of the genomes using the catalog of SGBs previously compiled through large-scale metagenomic assembly41, a substantial number of these SGBs (39%) lacks taxonomic labels (uSGBs), further highlighting that microbiomes from rural communities contain many organisms that are still very poorly characterized. The few MAGs not assigned to known or unknown SGBs (17) belongs to candidate species that are specific to this cohort and for which none of the >154,000 MAGs from previous metagenomes are within a 5% whole-genome nucleotide similarity (and 14 at >10% genomic distance). This demonstrates that even with a very large reference set of genomes and metagenomes, cohort-specific microbes can be found, classified, and phylogenetically profiled by the proposed approach. This study provides one example of how PhyloPhlAn 3.0 can automatically contextualize tens or hundreds of MAGs with taxonomy relative to characterized isolates or, when unavailable, using consistently cataloged microbial species from thousands of other metagenomes.

a Occurrence of the 20 most prevalent SGBs among 50 previously sequenced Ethiopian gut metagenomes highlights the presence of many previously identified but largely uncharacterized species-level genome bins (uSGBs) and the identification of few additional MAGs (unassigned) that are not recapitulated in any already defined SGB. The presence/absence profiles are clustered using average linkage with Euclidean distances. b Multidimensional scaling ordination using the t-SNE algorithm on phylogenetic distances from PhyloPhlAn 3.0's tree of eight Ethiopian E. coli MAGs (kSGB 10068) integrated with 200 automatically selected E. coli reference genomes using 3246 UniRef90 gene families for phylogenetic reconstruction. c PhyloPhlAn 3.0 phylogeny of Ethiopian MAGs assigned to uSGB ID 19436 including all reference genomes for the closest phyla (589 in total) according to the prokaryotes tree-of-life in Fig. 4. Phylogeny reconstruction used 400 universal markers selected by PhyloPhlAn 3.0 for deep-branching phylogenies. Portions of the tree collapsed are labeled and numbers in parentheses represent the number of genomes in the collapsed subtrees. Uncollapsed phylogeny is available in Supplementary Fig. 4.
Phylogenetic context for taxonomically unassigned genomes
Since PhyloPhlAn 3.0 associates new genomes and MAGs with SGBs even when the latter do not contain previously characterized taxa, this can be used to automatically compare genomes and MAGs with hundreds or thousands of phylogenetically related genome sequences (Fig. 3). In the Ethiopian study, we focused on the prevalent human gut colonizer Escherichia coli (known SGB (kSGB) ID 10068), and on the most prevalent uSGB (ID 19436, 13 MAGs in total) for which the closest reference genomes belonged to the Proteobacteria phylum. Eight E. coli MAGs were constructed from the Ethiopian metagenomes, for which PhyloPhlAn 3.0 retrieved 200 reference genomes and 3246 UniRef90 families pre-calculated as core to the species (3099 of which were retained for phylogenetic reconstruction as they are consistently found in the eight input MAGs, Fig. 3b, Supplementary Fig. 3). This showed the eight Ethiopian input MAGs to be genetically heterogeneous, falling diversely among four different previously defined E. coli phylotypes (see “Methods”) based on PhyloPhlAn 3.0-estimated phylogenetic distances (Fig. 3b, Supplementary Fig. 3). For half of the strains, the placement was confirmed by the phylogroup associated with the MLST types that could be inferred directly on the genomes47, but the phylogenetic placement within the clustered phylotypes provides strong evidence for the assignment of the other four strains as well.
We used PhyloPhlAn 3.0 to place the uncharacterized uSGB 19436 in the context of other reference genomes and MAGs from the human microbiome41 and of all the automatically retrieved species’ representative genomes from the set of closest phyla that are: part of Proteobacteria (class Epsilonproteobacteria, non-monophyletic with the Proteobacteria phylum), Spirochetes, Chlamydiae, Planctomycetes, Candidatus Omintrophica, Lentisphaerae, and Verrucomicrobia, identified as being close to the Epsilonproteobacteria from the tree of life (Fig. 4). PhyloPhlAn 3.0 placed the expanded uSGB 19436 within several very divergent clades taxonomically assigned to the Campylobacter genus (Fig. 3c, Supplementary Fig. 4). The 812 publically available genomes in 108 SGBs assigned to distinct species of Campylobacter, reveal this genus to be extremely wide encompassing substantially more than 30% genetic distance (ANI analysis in Supplementary Fig. 4) which is a diversity usually characterizing whole classes or orders41 suggests that this genus should be revised as also independently confirmed in other taxonomic reorganization efforts25. Although uSGB 19436 is rooted inside these divergent clades, its genetic divergence (Supplementary Fig. 4) is higher than typical family-level divergence and its phylogenetic distance is comparable to the distance between close phyla (Fig. 3c) thus supporting PhyloPhlAn 3.0’s designation of a new species and genus. The new MAGs from the Ethiopian dataset also reinforce the observation that this phylogenetically divergent uSGB 19436 is specific of non-Westernized lifestyles as the previously reconstructed MAGs from this uSGB are all from populations with rural lifestyles in Madagascar41, Peru48, Tanzania49, and Bangladesh50. This analysis thus highlights how PhyloPhlAn 3.0 can be used to expand the phylogenetic diversity of the human microbiome by the simple integration of MAGs from new cohorts in the already large set of microbial genome references considered by the method.

With 17,672 species-dereplicated isolate genomes and MAGs as input (see “Methods“), PhyloPhlAn 3.0 used 400 optimized universal marker sequences to produce a pan-microbial phylogeny in approximately 10 days (~24,000 CPU-hours on 100 parallel cores). The underlying multiple-sequence alignment comprised 4522 amino acid positions from among 1,872,710 in the untrimmed concatenated marker alignments.
PhyloPhlAn can scale to microbial tree-of-life phylogenies
In addition to these small-to-medium examples of phylogenetic reconstruction for individual new genome sets, PhyloPhlAn 3.0 can scale to provide automatic placement of thousands of MAGs within the entire current microbial tree of life (Fig. 4). Specifically, we considered all high-quality microbial isolate genomes included in UniProt51 (87,173 total), >154,000 MAGs from human-associated microbiomes41, and ~8000 MAGs from primarily non-human environments52. These were dereplicated prior to PhyloPhlAn application to one representative per species by hierarchical clustering on genomic distances as estimated by Mash42 with cluster cutoff at 5% intra-cluster nucleotide identity (see “Methods”), resulting in 19,607 clusters. Additional automatic quality control available in PhyloPhlAn 3.0, removed genomes containing less than 100 of PhyloPhlAn’s 400 optimized deep-branching marker genes (“Methods”), resulting in 17,672 representative genomes in the final tree. While Proteobacteria are prevalently found in non-human samples, Actinobacteria are instead mainly associated with human samples. Firmicutes and Bacteroidetes are more equally derived from both human and non-human samples, with some preferences in specific subtrees of the two phyla (Supplementary Fig. 5). Reconstruction of this tree of life required ~24,000 CPU-hours (about ten wall-clock days using 100 cores in parallel), of which more than half were needed by IQ-TREE22 for phylogenetic inference.
The concatenated MSA contained 4522 amino acids out of 1.87 M of total length of the untrimmed concatenated marker sequence alignments. The selection of these most phylogenetically informative positions in the MSA is performed by PhyloPhlAn 3.0 in this aggressive setting for scalability purposes and was validated as we reported elsewhere26 using the trident scoring function53. Although phylogenies spanning all the known bacterial and archaeal phyla using more sites and more extensive computation could be used as a default ref. 26, the automatic PhyloPhlAn 3.0 pipeline provides a convenient way to incorporate new MAGs and update genome sets. This is achieved while maintaining high phylogenetic accuracy, as shown by previous clade-specific analyses focusing on organisms from the human microbiome41, by the overall consistency of the PhyloPhlAn tree with the current reference prokaryotic tree-of-life26 (Supplementary Fig. 6), and by the comparison of the PhyloPhlAn 3.0 approach of using hundreds of universal markers against other prokaryotic tree-of-life phylogenies based on taxonomy or neighbor-joining (Robinson–Foulds distance < 0.3) reported elsewhere26. PhyloPhlAn 3.0 is thus able to efficiently reconstruct extremely large-scale phylogenies, automatically incorporating new isolate genomes, new MAGs, and existing isolate and MAG sequences.
