
Abstract
Diabetic kidney disease, known as a glomerular disease, arises from a metabolic disorder impairing renal cell function. Mitochondria, crucial organelles, play a key role in substance metabolism via oxidative phosphorylation to generate ATP. Cells undergo metabolic reprogramming as a compensatory mechanism to fulfill energy needs for survival and growth, attracting scholarly attention in recent years. Studies indicate that mitochondrial metabolic reprogramming significantly influences the pathophysiological progression of DKD. Alterations in kidney metabolism lead to abnormal expression of signaling molecules and activation of pathways, inducing oxidative stress-related cellular damage, inflammatory responses, apoptosis, and autophagy irregularities, culminating in renal fibrosis and insufficiency. This review delves into the impact of mitochondrial metabolic reprogramming on DKD pathogenesis, emphasizing the regulation of metabolic regulators and downstream signaling pathways. Therapeutic interventions targeting renal metabolic reprogramming can potentially delay DKD progression. The findings underscore the importance of focusing on metabolic reprogramming to develop safer and more effective therapeutic approaches.
Similar content being viewed by others
Facts
-
Metabolic reprogramming refers to the changes in cellular metabolic pathways that are necessary to support cell growth and proliferation.
-
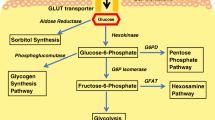
Metabolic reprogramming is characterized by mitochondrial biosynthesis dysfunction, increased glycolysis, and abnormal lipid and amino acid metabolism.
-
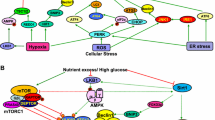
Oxidative stress, inflammatory damage, abnormal autophagy and apoptosis caused by metabolic disorders contribute to the development of DKD and ultimately renal fibrosis.
-
While current conventional treatments for DKD can slow down disease progression to some extent, they come with adverse effects, increased risk of cardiovascular disease, and eventual loss of renal function.
Open questions
-
Renal cells exhibit distinct physiological characteristics and functions. Is the mechanism of metabolic reprogramming consistent across these diverse cell types?
-
DKD is a chronic pathological process, does the metabolic disorders of renal cells change as the disease progresses?
-
Could it be a promising and safe therapeutic approach for managing DKD by regulating metabolic reprogramming and restoring normal mitochondrial function?
Introduction
It is estimated that approximately 40% of diabetics can develop diabetic kidney disease (DKD), which is responsible for end-stage renal disease (ESRD) worldwide [1]. Metabolic disorders, such as hyperglycemia and hyperlipidemia, can damage renal cells through oxidative stress-mediated injury, inflammatory response, apoptosis, and autophagy, leading to renal tubulointerstitial inflammation and fibrosis, glomerular hypertrophy, glomerulosclerosis and ultimately renal insufficiency [2]. Metabolic reprogramming is a mechanism by which cells change metabolic patterns to meet energy needs for cell survival and growth [3]. Recently, it has been found that metabolic reprogramming was triggered by impaired mitochondrial biogenesis in a high-glucose environment and played a crucial role in the pathogenesis of DKD [4,102].
Recent studies have shown that SGLT2 inhibitor drugs play a role in regulating amino acid metabolism in DKD. Kogot et al. discovered in mice that dapagliflozin can reduce renal fibrosis by suppressing the abnormal expression of collagen and amino acid transport proteins via mTORC1 inhibition [103]. Lu et al. conducted a 12-week study administering empagliflozin to db/db mice, resulting in decreased levels of KYN and increased expression of acetyl-CoA and NAD+ in the kidneys. The researchers proposed that empagliflozin may activate the crucial enzyme for NAD+ synthesis, potentially restoring the tryptophan metabolic pathway in DKD [104].
Glucagon-like peptide-1 receptor (GLP-1R) agonist
Glucagon-like peptide-1(GLP-1), an enteroglucagon hormone produced by intestinal L-cells, activates the Glucagon-like peptide-1 receptor (GLP-1R) to enhance insulin secretion and inhibit glucagon secretion in a glucose concentration-dependent manner. Additionally, it can delay gastric emptying and reduce food intake through central appetite suppression, ultimately leading to lower blood glucose levels. Recent studies have also shown the presence of GLP-1R in kidney cells, where chronic high glucose stimulation may lead to GLP-1R ubiquitination and subsequent kidney cell damage [105].
Recent studies have shown that GLP-1 agonists play a role in regulating lipid metabolism. Some meta-analysis by Yao and Sun et al. revealed that GLP-1 agonists have notable lipid-lowering effects compared to glucose-lowering drugs like insulin, including reducing LDL, cholesterol, and triglycerides [106]. It has been proposed that GLP-1 agonists may hinder lipase secretion in the intestinal lumen, thereby slowing gastric emptying and inhibiting fat absorption [107]. Exendin-4, as a GLP-1R agonist, inhibits ERK1/2 by activating the PI3K/AKT signaling pathway. In addition, it increases the phosphorylation of AMPK and ACC and promotes the expression of PPAR-α and CPT1, which not only promotes lipolysis and inhibits adipogenesis, but also exerts an anti-inflammatory effect in glomerular endothelial cells [108]. Further research is needed to fully understand the mechanisms of GLP-1 agonists in lipid absorption and metabolism.
Traditional Chinese medicines
Traditional Chinese medicine (TCM) has a long history in treating DKD, either alone or in combination with conventional drugs like ACEI/ARB’s, showing positive therapeutic effects [109, 110]. Recent studies have demonstrated that biologically active phytochemicals in TCM can modulate metabolic reprogramming in DKD with minimal adverse effects in clinical settings [111]. For example, the Zhenqing recipe, containing Fructus Ligustri Lucidi, Eclipta Prostrata, and Dioscorea opposite, was found to inhibit the expression of SREBP-1C and its target genes, ACC and FAS, in diabetic rats, leading to a significant reduction in triglycerides and cholesterol levels [112]. Morroniside, an iridoid glycoside from Cornus officinalis Sieb, was shown by Gao et al. to up-regulate the expression of PGC-1α, LXR, ABCA1, and ApoE, promoting intracellular cholesterol efflux in the kidneys of diabetic mice [113]. Qin et al. discovered that berberine has the ability to activate the PGC-1α/LXR signaling pathway in db/db mouse foot cells. This activation restores mitochondrial homeostasis, normalizes fatty acid oxidation, reverses metabolic disorders, and repairs cellular damage [114]. The precise targeting mechanism of Chinese herbs for DKD requires further investigation. Current research indicates that various biologically active plant components found in Chinese herbs show promise in treating DKD effectively. However, a deeper understanding of the therapeutic mechanisms of these components is necessary to achieve precise treatment of DKD using Chinese herbs.
Emerging drugs
With the advancing comprehension of mitochondrial metabolic reprogramming, there is a growing number of targeted therapeutic agents being uncovered. Recent studies have shown that artemether treatment in diabetic mice led to decreased levels of branched-chain amino acids and citrulline, indicating a potential alteration in amino acid metabolism in DKD [115]. Additionally, artemether was found to upregulate the expression of PGC-1α and MPC in the renal cortex of diabetic mice, enhancing pyruvate oxidation in the mitochondrial matrix and regulating mitochondrial function to ameliorate renal injury [116]. Clinical trials focusing on targeted drug therapies for metabolic reprogramming have been emerging. For instance, fenofibrate (NCT03869931), a PPARα agonist, has been shown to restore fatty acid oxidation and is commonly used for hypertriglyceridemia treatment. Annelie et al. discovered that inhibition of VEGF-B signaling reduced lipid accumulation in the kidney, mitigated renal lipotoxicity, and prevented renal dysfunction [117]. The effectiveness of 2H10 (NCT04419467), a monoclonal antibody targeting VEGF-B, is currently being investigated in patients with DKD. Furthermore, Dorzagliatin (NCT06222476), a glucokinase activator serving as a novel hypoglycemic agent, aids in enhancing the body’s intrinsic capacity to regulate glucose levels.
The treatment program for DKD is now more advanced, incorporating lifestyle improvements alongside symptomatic medications to regulate blood glucose, blood lipids, and blood pressure. In cases of end-stage DKD, renal replacement therapy is the only option. With the discovery of metabolic reprogramming in DKD and its impact on kidney damage, there is potential for drugs targeting metabolic reprogramming to halt disease progression at a pathogenic level.
Conclusions
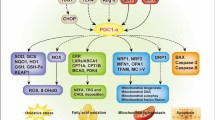
DKD is a glomerular disease that results from diabetes mellitus, and is classified as one of its microvascular complications. The increased sugar concentration in the blood causes hyperperfusion and hyperfiltration of the glomeruli, as well as excessive tubular reabsorption, leading to an increase in renal oxygen consumption. This altered hemodynamics in the kidney, along with activation of the local RAS system, can lead to vascular lesions and a reduction in blood vessels. Stimulation by high glucose and hypoxia can cause mitochondrial dysfunction in renal cells. This can lead to abnormal expression of regulatory factors, such as HIF-α and SIRT3, which can affect a range of signaling pathways and stimulate the onset of abnormal glycolysis and amino acid metabolism. Additionally, this can inhibit the FAO response, which can enhance lipotoxicity. The production of large amounts of ROS and inflammatory factors can result in oxidative stress damage, inflammatory damage, autophagy, and apoptosis in renal cells (Table 2). Ultimately, this can lead to renal fibrosis and renal dysfunction (Fig. 2).

In a healthy kidney, the glomerular filtration barrier is formed by intact glomerular endothelial cells, basement membranes, and podocytes. This barrier effectively prevents the filtration of macromolecules like plasma albumin from the blood. The contractile action of mesangial cells regulate the tubular diameter of the afferent and efferent arterioles, thereby controlling the glomerular filtration rate. Additionally, mesangial cells support the position of vascular collaterals. Tubular epithelial cells play a role in regulating the formation of primary urine through functions like reabsorption, secretion, and excretion. However, hyperglycemia-induced changes in kidney hemodynamics, activation of the RASS, generation of AGEs leading to mitochondrial metabolic reprogramming, and excessive release of ROS trigger a cascade of signaling pathways, cytokines, and transcription factors. These factors contribute to the development of various pathological responses in renal cells, including inflammation, autophagy, apoptosis, and fibrosis.
DKD is a condition in which high blood glucose levels lead to a series of kidney damages. Lowering blood glucose levels alone cannot prevent the development of DKD. The pathogenesis of DKD is complex, as it involves extensive damage caused by the reprogramming of mitochondrial metabolism in kidney cells. Therefore, treatment for DKD should focus on correcting mitochondrial metabolic reprogramming and restoring normal glucose and fatty acid metabolism, in addition to controlling blood glucose levels. Understanding the role of mitochondrial metabolic reprogramming in various cells during the development of DKD is crucial for comprehending the disease’s pathogenesis and develo** more effective nephroprotective therapies.
References
International Diabetes Federation. IDF Diabetes Atlas Reports. 2023. https://diabetesatlas.org/atlas/diabetes-and-kidney-disease/.
Oshima M, Shimizu M, Yamanouchi M, Toyama T, Hara A, Furuichi K, et al. Trajectories of kidney function in diabetes: a clinicopathological update. Nat Rev Nephrol. 2021;17:740–50.
Carvalho-Santos Z, Cardoso-Figueiredo R, Elias AP, Tastekin I, Baltazar C, Ribeiro C. Cellular metabolic reprogramming controls sugar appetite in Drosophila. Nat Metab. 2020;2:958–73.
Hou Y, Tan E, Shi H, Ren X, Wan X, Wu W, et al. Mitochondrial oxidative damage reprograms lipid metabolism of renal tubular epithelial cells in the diabetic kidney. Cell Mol Life Sci. 2024;81:23.
Linnan B, Yanzhe W, Ling Z, Yuyuan L, Sijia C, **nmiao X, et al. In situ metabolomics of metabolic reprogramming involved in a mouse model of type 2 diabetic kidney disease. Front Physiol. 2021;12:779683.
Song C, Wang S, Fu Z, Chi K, Geng X, Liu C, et al. IGFBP5 promotes diabetic kidney disease progression by enhancing PFKFB3-mediated endothelial glycolysis. Cell Death Dis. 2022;13:340.
Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. 2020;11:390.
Zhu H, Bai M, **e X, Wang J, Weng C, Dai H, et al. Impaired amino acid metabolism and its correlation with diabetic kidney disease progression in type 2 diabetes mellitus. Nutrients. 2022;14:3345.
Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, et al. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomised, placebo-controlled trial. Lancet. 2019;394:131–8.
Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019;380:2295–306.
Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20:745–54.
Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102.
Song Y, Liu J, Zhao K, Gao L, Zhao J. Cholesterol-induced toxicity: an integrated view of the role of cholesterol in multiple diseases. Cell Metab. 2021;33:1911–25.
Houten SM, Violante S, Ventura FV, Wanders RJ. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu Rev Physiol. 2016;78:23–44.
Forbes JM. Prolyl hydroxylase inhibitors: a breath of fresh air for diabetic kidney disease? Kidney Int. 2020;97:855–7.
Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–23.
Wang Z, Ying Z, Bosy-Westphal A, Zhang J, Schautz B, Later W, et al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. 2010;92:1369–77.
Sas KM, Kayampilly P, Byun J, Nair V, Hinder LM, Hur J, et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight. 2016;1:e86976.
Kanasaki K. The aberrant glycolysis in kidney proximal tubule: potential therapeutic target for DKD. Kidney Int. 2023;104:1056–9.
Mitrofanova A, Burke G, Merscher S, Fornoni A. New insights into renal lipid dysmetabolism in diabetic kidney disease. World J Diabetes. 2021;12:524–40.
Pérez-Martí A, Ramakrishnan S, Li J, Dugourd A, Molenaar MR, De La Motte LR, et al. Reducing lipid bilayer stress by monounsaturated fatty acids protects renal proximal tubules in diabetes. Elife. 2022;11:e74391.
Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res. 2014;55:561–72.
Lewis S, Chen L, Raghuram V, Khundmiri SJ, Chou CL, Yang CR, et al. “SLC-omics” of the kidney: solute transporters along the nephron. Am J Physiol Cell Physiol. 2021;321:C507–18.
Felig P, Marliss E, Cahill GF Jr. Plasma amino acid levels and insulin secretion in obesity. N Engl J Med. 1969;281:811–6.
Xu J, Kitada M, Koya D. NAD(+) homeostasis in diabetic kidney disease. Front Med. 2021;8:703076.
Kwiatkowska I, Hermanowicz JM, Mysliwiec M, Pawlak D. Oxidative storm induced by tryptophan metabolites: missing link between atherosclerosis and chronic kidney disease. Oxid Med Cell Longev. 2020;2020:6656033.
Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. 2018;359:1376–83.
Descamps HC, Herrmann B, Wiredu D, Thaiss CA. The path toward using microbial metabolites as therapies. EBioMedicine. 2019;44:747–54.
Cha Y, Kim T, Jeon J, Jang Y, Kim PB, Lopes C, et al. SIRT2 regulates mitochondrial dynamics and reprogramming via MEK1-ERK-DRP1 and AKT1-DRP1 axes. Cell Rep. 2021;37:110155.
Morita M, Prudent J, Basu K, Goyon V, Katsumura S, Hulea L, et al. mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol Cell. 2017;67:922–35.e5.
Hu Q, Zhang H, Gutiérrez Cortés N, Wu D, Wang P, Zhang J, et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res. 2020;126:456–70.
Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–17.
Guido C, Whitaker-Menezes D, Lin Z, Pestell RG, Howell A, Zimmers TA, et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget. 2012;3:798–810.
Qin X, Zhao Y, Gong J, Huang W, Su H, Yuan F, et al. Berberine protects glomerular podocytes via inhibiting Drp1-mediated mitochondrial fission and dysfunction. Theranostics. 2019;9:1698–713.
Cleveland KH, Brosius FC 3rd, Schnellmann RG. Regulation of mitochondrial dynamics and energetics in the diabetic renal proximal tubule by the β(2)-adrenergic receptor agonist formoterol. Am J Physiol Ren Physiol. 2020;319:F773–9.
Wang J, Yue X, Meng C, Wang Z, ** X, Cui X, et al. Acute hyperglycemia may induce renal tubular injury through mitophagy inhibition. Front Endocrinol. 2020;11:536213.
Huang D, Li T, Li X, Zhang L, Sun L, He X, et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014;8:1930–42.
Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol. 2001;166:7543–8.
Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46.
Chen L, Sha ML, Chen FT, Jiang CY, Li D, Xu CL, et al. Upregulation of KLF14 expression attenuates kidney fibrosis by inducing PPARα-mediated fatty acid oxidation. Free Radic Biol Med. 2023;195:132–44.
Chung KW, Ha S, Kim SM, Kim DH, An HJ, Lee EK, et al. PPARα/β activation alleviates age-associated renal fibrosis in Sprague Dawley rats. J Gerontol A Biol Sci Med Sci. 2020;75:452–8.
Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes. 2006;55:2502–9.
Yang W, Luo Y, Yang S, Zeng M, Zhang S, Liu J, et al. Ectopic lipid accumulation: potential role in tubular injury and inflammation in diabetic kidney disease. Clin Sci. 2018;132:2407–22.
Lin S, Wang L, Jia Y, Sun Y, Qiao P, Quan Y, et al. Lipin-1 deficiency deteriorates defect of fatty acid β-oxidation and lipid-related kidney damage in diabetic kidney disease. Transl Res. 2024;266:1–15.
Feng L, Gu C, Li Y, Huang J. High glucose promotes CD36 expression by upregulating peroxisome proliferator-activated receptor γ levels to exacerbate lipid deposition in renal tubular cells. Biomed Res Int. 2017;2017:1414070.
Hou Y, Wang Q, Han B, Chen Y, Qiao X, Wang L. CD36 promotes NLRP3 inflammasome activation via the mtROS pathway in renal tubular epithelial cells of diabetic kidneys. Cell Death Dis. 2021;12:523.
Hou Y, Wu M, Wei J, Ren Y, Du C, Wu H, et al. CD36 is involved in high glucose-induced epithelial to mesenchymal transition in renal tubular epithelial cells. Biochem Biophys Res Commun. 2015;468:281–6.
Li X, Zhang T, Geng J, Wu Z, Xu L, Liu J, et al. Advanced oxidation protein products promote lipotoxicity and tubulointerstitial fibrosis via CD36/β-catenin pathway in diabetic nephropathy. Antioxid Redox Signal. 2019;31:521–38.
Khan S, Gaivin R, Abramovich C, Boylan M, Calles J, Schelling JR. Fatty acid transport protein-2 regulates glycemic control and diabetic kidney disease progression. JCI Insight. 2020;5:e136845.
Chen J, Wu K, Lei Y, Huang M, Cheng L, Guan H, et al. Inhibition of fatty acid β-oxidation by fatty acid binding protein 4 induces ferroptosis in HK2 cells under high glucose conditions. Endocrinol Metab. 2023;38:226–44.
Shen S, Ji C, Wei K. Cellular senescence and regulated cell death of tubular epithelial cells in diabetic kidney disease. Front Endocrinol. 2022;13:924299.
Baek J, Sas K, He C, Nair V, Giblin W, Inoki A, et al. The deacylase sirtuin 5 reduces malonylation in nonmitochondrial metabolic pathways in diabetic kidney disease. J Biol Chem. 2023;299:102960.
Zhang Y, Wen P, Luo J, Ding H, Cao H, He W, et al. Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis. 2021;12:847.
An S, Yao Y, Hu H, Wu J, Li J, Li L, et al. PDHA1 hyperacetylation-mediated lactate overproduction promotes sepsis-induced acute kidney injury via Fis1 lactylation. Cell Death Dis. 2023;14:457.
Shao M, Chen D, Wang Q, Guo F, Wei F, Zhang W, et al. Canagliflozin regulates metabolic reprogramming in diabetic kidney disease by inducing fasting-like and aestivation-like metabolic patterns. Diabetologia. 2024;67:738–54.
Sharma K, Zhang G, Hansen J, Bjornstad P, Lee HJ, Menon R, et al. Endogenous adenine mediates kidney injury in diabetic models and predicts diabetic kidney disease in patients. J Clin Invest. 2023;133:e170341.
Kriz W. The inability of podocytes to proliferate: cause, consequences, and origin. Anat Rec. 2020;303:2588–96.
Reynolds PA. The mechanobiology of kidney podocytes in health and disease. Clin Sci. 2020;134:1245–53.
Yuan Q, Miao J, Yang Q, Fang L, Fang Y, Ding H, et al. Role of pyruvate kinase M2-mediated metabolic reprogramming during podocyte differentiation. Cell Death Dis. 2020;11:355.
Mohandes S, Doke T, Hu H, Mukhi D, Dhillon P, Susztak K. Molecular pathways that drive diabetic kidney disease. J Clin Invest. 2023;133:e165654.
Imasawa T, Obre E, Bellance N, Lavie J, Imasawa T, Rigothier C, et al. High glucose repatterns human podocyte energy metabolism during differentiation and diabetic nephropathy. FASEB J. 2017;31:294–307.
Li J, Sun YBY, Chen W, Fan J, Li S, Qu X, et al. Smad4 promotes diabetic nephropathy by modulating glycolysis and OXPHOS. EMBO Rep. 2020;21:e48781.
He JY, Hong Q, Chen BX, Cui SY, Liu R, Cai GY, et al. Ginsenoside Rb1 alleviates diabetic kidney podocyte injury by inhibiting aldose reductase activity. Acta Pharm Sin. 2022;43:342–53.
Ma X, Ma J, Leng T, Yuan Z, Hu T, Liu Q, et al. Advances in oxidative stress in pathogenesis of diabetic kidney disease and efficacy of TCM intervention. Ren Fail. 2023;45:2146512.
Bao HR, Chen JL, Li F, Zeng XL, Liu XJ. Relationship between PI3K/mTOR/RhoA pathway-regulated cytoskeletal rearrangements and phagocytic capacity of macrophages. Braz J Med Biol Res. 2020;53:e9207.
Luo Q, Liang W, Zhang Z, Zhu Z, Chen Z, Hu J, et al. Compromised glycolysis contributes to foot process fusion of podocytes in diabetic kidney disease: role of ornithine catabolism. Metabolism. 2022;134:155245.
Beniwal A, Jain CJ, Jain A. Lipids: a major culprit in diabetic nephropathy. Curr Diabetes Rev. 2024;20:60–9.
Schermer B, Benzing T. Lipid-protein interactions along the slit diaphragm of podocytes. J Am Soc Nephrol. 2009;20:473–8.
Suk Kang J, Son SS, Lee JH, Lee SW, Jeong AR, Lee ES, et al. Protective effects of klotho on palmitate-induced podocyte injury in diabetic nephropathy. PLoS ONE. 2021;16:e0250666.
Wang XX, Edelstein MH, Gafter U, Qiu L, Luo Y, Dobrinskikh E, et al. G protein-coupled bile acid receptor TGR5 activation inhibits kidney disease in obesity and diabetes. J Am Soc Nephrol. 2016;27:1362–78.
Wakisaka M, Nakamura K, Nakano T, Kitazono T. Roles of sodium-glucose cotransporter 2 of mesangial cells in diabetic kidney disease. J Endocr Soc. 2021;5:bvab083.
Staruschenko A, Ma R, Palygin O, Dryer SE. Ion channels and channelopathies in glomeruli. Physiol Rev. 2023;103:787–854.
Huang K, Zhao X. USP9X prevents AGEs-induced upregulation of FN and TGF-β1 through activating Nrf2-ARE pathway in rat glomerular mesangial cells. Exp Cell Res. 2020;393:112100.
Jha JC, Dai A, Garzarella J, Charlton A, Urner S, Østergaard JA, et al. Independent of renox, NOX5 promotes renal inflammation and fibrosis in diabetes by activating ROS-sensitive pathways. Diabetes. 2022;71:1282–98.
Hu F, Xue R, Wei X, Wang Z, Luo S, Lin J, et al. Egr1 knockdown combined with an ACE inhibitor ameliorates diabetic kidney disease in mice: blockade of compensatory renin increase. Diabetes Metab Syndr Obes. 2020;13:1005–13.
Opazo-Ríos L, Mas S, Marín-Royo G, Mezzano S, Gómez-Guerrero C, Moreno JA, et al. Lipotoxicity and diabetic nephropathy: novel mechanistic insights and therapeutic opportunities. Int J Mol Sci. 2020;21:2632.
Jabarpour M, Rashtchizadeh N, Ghorbani Haghjo A, Argani H, Nemati M, Dastmalchi S, et al. Protection of renal damage by HMG-CoA inhibitors: a comparative study between atorvastatin and rosuvastatin. Iran J Basic Med Sci. 2020;23:206–13.
Nagai Y, Matoba K, Takeda Y, Yako H, Akamine T, Sekiguchi K, et al. Rho-associated, coiled-coil-containing protein kinase 1 regulates development of diabetic kidney disease via modulation of fatty acid metabolism. Kidney Int. 2022;102:536–45.
Park SH, Chung S, Chung MY, Choi HK, Hwang JT, Park JH. Effects of Panax ginseng on hyperglycemia, hypertension, and hyperlipidemia: a systematic review and meta-analysis. J Ginseng Res. 2022;46:188–205.
Chen Q, Ren D, Liu L, Xu J, Wu Y, Yu H, et al. Ginsenoside compound K ameliorates development of diabetic kidney disease through inhibiting TLR4 activation induced by microbially produced imidazole propionate. Int J Mol Sci. 2022;23:12863.
Sampei S, Okada H, Tomita H, Takada C, Suzuki K, Kinoshita T, et al. Endothelial glycocalyx disorders may be associated with extended inflammation during endotoxemia in a diabetic mouse model. Front Cell Dev Biol. 2021;9:623582.
Lavoz C, Rodrigues-Diez RR, Plaza A, Carpio D, Egido J, Ruiz-Ortega M, et al. VEGFR2 blockade improves renal damage in an experimental model of type 2 diabetic nephropathy. J Clin Med. 2020;9:302.
Casalena GA, Yu L, Gil R, Rodriguez S, Sosa S, Janssen W, et al. The diabetic microenvironment causes mitochondrial oxidative stress in glomerular endothelial cells and pathological crosstalk with podocytes. Cell Commun Signal. 2020;18:105.
Oellgaard J, Gæde P, Rossing P, Persson F, Parving HH, Pedersen O. Intensified multifactorial intervention in type 2 diabetics with microalbuminuria leads to long-term renal benefits. Kidney Int. 2017;91:982–8.
Rossing K, Christensen PK, Hovind P, Tarnow L, Rossing P, Parving HH. Progression of nephropathy in type 2 diabetic patients. Kidney Int. 2004;66:1596–605.
Li Y, Sha Z, Peng H. Metabolic reprogramming in kidney diseases: evidence and therapeutic opportunities. Int J Nephrol. 2021;2021:5497346.
Golbidi S, Laher I. Exercise induced adipokine changes and the metabolic syndrome. J Diabetes Res. 2014;2014:726861.
Zacharewicz E, Hesselink MKC, Schrauwen P. Exercise counteracts lipotoxicity by improving lipid turnover and lipid droplet quality. J Intern Med. 2018;284:505–18.
Colberg SR, Sigal RJ, Yardley JE, Riddell MC, Dunstan DW, Dempsey PC, et al. Physical activity/exercise and diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2016;39:2065–79.
Kuo HY, Huang YH, Wu SW, Chang FH, Tsuei YW, Fan HC, et al. The effects of exercise habit on albuminuria and metabolic indices in patients with type 2 diabetes mellitus: a cross-sectional study. Medicina. 2022;58:577.
Monno I, Ogura Y, Xu J, Koya D, Kitada M. Exercise ameliorates diabetic kidney disease in type 2 diabetic fatty rats. Antioxidants. 2021;10:1754.
Vartak T, Godson C, Brennan E. Therapeutic potential of pro-resolving mediators in diabetic kidney disease. Adv Drug Deliv Rev. 2021;178:113965.
Michas G, Micha R, Zampelas A. Dietary fats and cardiovascular disease: putting together the pieces of a complicated puzzle. Atherosclerosis. 2014;234:320–8.
Koh A, Molinaro A, Ståhlman M, Khan MT, Schmidt C, Mannerås-Holm L, et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. 2018;175:947–61.e17.
Wu H, Zhou H, Zhang Q, Zhou Y, Fu L, Zhuang Y. Systematic review and meta-analysis: the effect and safety of peritoneal dialysis in patients with end-stage diabetic kidney disease. Ann Palliat Med. 2022;11:695–707.
Srivastava SP, Goodwin JE, Kanasaki K, Koya D. Metabolic reprogramming by N-acetyl-seryl-aspartyl-lysyl-proline protects against diabetic kidney disease. Br J Pharm. 2020;177:3691–711.
Nagai T, Kanasaki M, Srivastava SP, Nakamura Y, Ishigaki Y, Kitada M, et al. N-acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial-mesenchymal transition. Biomed Res Int. 2014;2014:696475.
de Almeida Pinheiro T, de Almeida Pinheiro T, Feltenberger JD, Andrade JMO, Neves Ferreira EC, De Farias Lelis D, et al. Effects of resveratrol and ACE inhibitor enalapril on glucose and lipid profiles in mice. Protein Pept Lett. 2017;24:854–60.
Mahmoudabady M, Kazemi N, Niazmand S, Rezaee SA, Soukhtanloo M, Hosseini M. The effect of angiotensin-converting enzyme inhibition on inflammatory and angiogenic factors in hypercholesterolemia. Pharm Rep. 2015;67:837–41.
Marcovecchio ML, Chiesa ST, Bond S, Daneman D, Dawson S, Donaghue KC, et al. ACE inhibitors and statins in adolescents with type 1 diabetes. N Engl J Med. 2017;377:1733–45.
Bakris G, Oshima M, Mahaffey KW, Agarwal R, Cannon CP, Capuano G, et al. Effects of canagliflozin in patients with baseline eGFR <30 ml/min per 1.73 m(2): subgroup analysis of the randomized CREDENCE trial. Clin J Am Soc Nephrol. 2020;15:1705–14.
Li J, Liu H, Takagi S, Nitta K, Kitada M, Srivastava SP, et al. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight. 2020;5:e129034.
Kogot-Levin A, Hinden L, Riahi Y, Israeli T, Tirosh B, Cerasi E, et al. Proximal tubule mTORC1 is a central player in the pathophysiology of diabetic nephropathy and its correction by SGLT2 inhibitors. Cell Rep. 2020;32:107954.
Lu YP, Zhang ZY, Wu HW, Fang LJ, Hu B, Tang C, et al. SGLT2 inhibitors improve kidney function and morphology by regulating renal metabolic reprogramming in mice with diabetic kidney disease. J Transl Med. 2022;20:420.
Yu WC, Huang RY, Chou TC. Oligo-fucoidan improves diabetes-induced renal fibrosis via activation of Sirt-1, GLP-1R, and Nrf2/HO-1: an in vitro and in vivo study. Nutrients. 2020;12:3068.
Yao H, Zhang A, Li D, Wu Y, Wang CZ, Wan JY, et al. Comparative effectiveness of GLP-1 receptor agonists on glycaemic control, body weight, and lipid profile for type 2 diabetes: systematic review and network meta-analysis. BMJ. 2024;384:e076410.
Aaseth J, Ellefsen S, Alehagen U, Sundfør TM, Alexander J. Diets and drugs for weight loss and health in obesity—an update. Biomed Pharmacother. 2021;140:111789.
Fang S, Cai Y, Lyu F, Zhang H, Wu C, Zeng Y, et al. Exendin-4 improves diabetic kidney disease in C57BL/6 mice independent of brown adipose tissue activation. J Diabetes Res. 2020;2020:9084567.
Tian J, Zhao L, Zhou Q, Liu W, Chen X, Lian F, et al. Efficacy of Shenzhuo formula on diabetic kidney disease: a retrospective study. J Tradit Chin Med. 2015;35:528–36.
Zhang L, Yang L, Shergis J, Zhang L, Zhang AL, Guo X, et al. Chinese herbal medicine for diabetic kidney disease: a systematic review and meta-analysis of randomised placebo-controlled trials. BMJ Open. 2019;9:e025653.
Pan Y, Liu T, Wang X, Sun J. Research progress of coumarins and their derivatives in the treatment of diabetes. J Enzym Inhib Med Chem. 2022;37:616–28.
Liao Q, Xu W, Luo Q, Wen X. Zhenqing recipe relieves diabetic nephropathy through the SIK1/SREBP-1c axis in type 2 diabetic rats. Am J Transl Res. 2021;13:13776–83.
Gao J, Liu P, Shen Z, Xu K, Wu C, Tian F, et al. Morroniside promotes PGC-1α-mediated cholesterol efflux in sodium palmitate or high glucose-induced mouse renal tubular epithelial cells. Biomed Res Int. 2021;2021:9942152.
Qin X, Jiang M, Zhao Y, Gong J, Su H, Yuan F, et al. Berberine protects against diabetic kidney disease via promoting PGC-1α-regulated mitochondrial energy homeostasis. Br J Pharm. 2020;177:3646–61.
Rong G, Weng W, Huang J, Chen Y, Yu X, Yuan R, et al. Artemether alleviates diabetic kidney disease by modulating amino acid metabolism. Biomed Res Int. 2022;2022:7339611.
Han P, Wang Y, Zhan H, Weng W, Yu X, Ge N, et al. Artemether ameliorates type 2 diabetic kidney disease by increasing mitochondrial pyruvate carrier content in db/db mice. Am J Transl Res. 2019;11:1389–402.
Mehlem A, Palombo I, Wang X, Hagberg CE, Eriksson U, Falkevall A. PGC-1α coordinates mitochondrial respiratory capacity and muscular fatty acid uptake via regulation of VEGF-B. Diabetes. 2016;65:861–73.
Funding
This work was funded by National Natural Science Foundation of China (Nos: 82370721, 82070744, 81873615, 81770723), Shandong Province Natural Science Foundation Joint Fund (No. ZR2022LSW020), and Taishan Scholars Program (Nos: tsqn201812138, ts201712090).
Author information
Authors and Affiliations
Contributions
**aoting Fan and Meilin Yang wrote the original draft. Yating Lang, Shangwei Lu, Zhijuan Kong, Ying Gao, and Ning Shen prepared the figures and tables. **aoting Fan, Meilin Yang, Yating Lang, and Dongdong Zhang revised the article. Zhimei Lv reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by: Angelo Peschiaroli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, X., Yang, M., Lang, Y. et al. Mitochondrial metabolic reprogramming in diabetic kidney disease. Cell Death Dis 15, 442 (2024). https://doi.org/10.1038/s41419-024-06833-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06833-0
- Springer Nature Limited