
Abstract
Glioblastoma is the most common primary malignant brain tumour. Survival is poor and improved treatment options are urgently needed. Although immunotherapies have emerged as effective treatments for a number of cancers, translation of these through to brain tumours is a distinct challenge, particularly due to the blood–brain barrier and the unique immune tumour microenvironment afforded by CNS-specific cells. This review discusses the immune system within the CNS, mechanisms of immune escape employed by glioblastoma, and the immunological effects of conventional glioblastoma treatments. Novel therapies for glioblastoma that harness the immune system and their current clinical progress are outlined, including cancer vaccines, T-cell therapies and immune checkpoint modulators.
Similar content being viewed by others


Introduction
Glioblastoma is the most common malignant primary brain tumour in adults.1 Survival is poor, with a median survival of 14.6 months with standard treatment of surgical debulking followed by external-beam radiotherapy with concurrent temozolomide (TMZ) chemotherapy and adjuvant TMZ chemotherapy.2 In the last decade, clinical trials investigating targeted therapies have failed to demonstrate any significant improvement in survival, and innovative new treatments are urgently needed.3 Recently, focus has shifted towards novel strategies that modulate the immune response towards the tumour and the surrounding tumour microenvironment.
Recognition and eradication of malignant cells via immune surveillance of tumour-associated antigens (TAAs) is a key function of the immune system.4 TAAs typically represent peptides which are present within tumour cells but usually absent in the surrounding normal tissue. In glioblastoma, these antigens most commonly fall into three main classes; (i) aberrantly expressed non-mutated self-antigens, (ii) mutated self-antigens and (iii) unique antigens or neo-antigens - novel peptide sequences which are the result of somatic mutations in the cancer genome.5 Tumours manipulate the immune system to avoid detection of TAAs and to facilitate their own growth and survival.6
Immunotherapy aims to harness the immune system against tumours, with breakthroughs observed in a number of cancers, most notably malignant melanoma and haematological malignancies. However, translating these approaches into therapies for primary brain tumours represents a distinct challenge due the unique tumour microenvironment and distinctive immune system within the CNS.
The CNS was traditionally considered immune privileged due to (i) the presence of the blood–brain barrier (BBB) that restricts access to immune cells, (ii) an absence of conventional lymphatic drainage restricting the trafficking of antigens to lymph nodes,7 (iii) a scarcity of specialised antigen-presenting cells,8 and iv) downregulation of the major histocompatibility complex (MHC) expression in normal brain parenchyma, limiting antigen presentation.9 In recent years, this dogma has been eroded with substantial evidence now demonstrating that these interlinked factors tightly regulate a fully functional, innate and adaptive immune system within the CNS (Table 1).
In this review, we describe the features of the immune system in the CNS, discuss the immune evasion strategies of glioblastoma, the molecular properties of its microenvironment, and the current clinical evidence supporting a role for immunomodulation in the treatment of glioblastoma.
Glioblastoma tumour microenvironment and mechanisms of immune escape
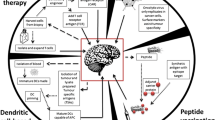
Glioblastoma arises from glial cells, with surrounding brain parenchyma comprising CNS-specific cells including astrocytes, neurons and microglia, and a distinctive extracellular matrix (ECM) composition.10 Glioblastoma induces a TME characterised by immunosuppressive cytokines secreted by tumour cells, microglia and tumour-associated macrophages (TAMs). These factors, notably interleukin-6, interleukin-10, transforming growth factor-beta (TGF-β), and prostaglandin-E collectively inhibit both the innate and adaptive immune systems with suppression of NK activity and T-cell activation and proliferation, induction of T-cell apoptosis, downregulation of MHC expression, and skew of TAMs towards an M2 (immunosuppressive) phenotype.11 The TME is also characterised by tissue hypoxia provided by irregular vascularity and high-tumour oxygen consumption. Tissue hypoxia activates the immunosuppressive STAT3 pathway, leading to hypoxia-inducible factor-1 alpha (HIF-1α) synthesis, activation of regulatory T cells (T-regs) and production of vascular endothelial growth factor (VEGF),11 which can inhibit the maturation and function of dendritic cells. These mechanisms of immune escape are discussed below in more detail and summarised in Fig. 1.

Immune gateways (left). In addition to the resident microglia, there are three distinct macrophage populations within the CNS present at so-called ‘immune gateways’ that act as ports of entry for activated T cells into the CNS. Perivascular macrophages, derived from the embryonic yolk sac, are located around parenchymal vessels (top). The other two populations, derived from bone marrow, are located in the meningeal spaces (middle) and the choroid plexus (bottom) (adapted from ref. 156). Immune evasion in glioblastoma (right): the immunosuppressive tumour microenvironment (TME) of glioblastoma is the result of complex interactions between tumour cells, microglia, tumour-associated macrophages (TAMs), components of the extracellular matrix and tumour infiltrating lymphocytes (TILs), which are predominantly regulatory in phenotype (T-regs). Hypoxia promotes angiogenesis of abnormal blood vessels, further driving tumour growth (adapted from ref. 10)
Extracellular matrix composition
ECM proteins commonly found in abundance in peripheral tissues including collagens, laminins and fibronectin are typically only associated with vascular basement membranes within the CNS. Instead, predominant ECM proteins in the glioma TME include glycoproteins, hyaluronic acid and heparan sulphate proteoglycans (HSPGs), which may be concentrated in cancer stem cell niches.12 HSPGs, in particular, are upregulated in glioblastoma13 and cause retention of heparin-binding angiogenic growth factors such as fibroblast growth factor (FGF) and VEGF, the local release of which promotes tumour angiogenesis and progression.14 Furthermore, glioma vasculature can upregulate the vessel-associated macromolecules periostin and tenascin C (TNC), which can promote tumour survival.15 These macromolecules can also promote immune evasion with TNC shown to block T-cell movement across glioma-associated blood vessels, preventing their migration into brain parenchyma.16 Periostin, when secreted by glioma stem cells, is able to promote recruitment of tumour promoting macrophages from peripheral circulation.17
Macrophages and microglia
Tumour-associated macrophages (TAMs), along with the resident CNS microglia, can constitute up to 30% of the tumour mass.10 Transcriptome analysis of TAMs has found that they may possess markers consistent with both M1 (classically activated or immunopermissive) and M2 (alternatively activated or immunosuppressive) phenotypes, incongruous with the traditional M1/M2 dichotomy.18 TAM populations can be described both functionally and spatially. For example, CNS-resident microglia exist within the TME alongside distinct populations of bone marrow-derived macrophages (outlined in Table 1),19 and recent research has suggested that within the TME, bone marrow-derived macrophages may localise preferentially to the perivascular niche, while resident microglia localise to peritumoural regions.20 Accumulation of TAMs expressing CD163 (haemoglobin scavenger receptor) and CD204 (macrophage scavenger receptor), considered ‘M2′ phenotype markers, increases as tumour grade increases.21 In glioblastoma, higher CD163 TAM expression correlates with poorer outcomes.11 Cancer stem cells in glioblastoma are able to recruit TAMs by overexpression of the macrophage/microglia cytokine colony-stimulating factor-1 (CSF-1).22 This induces a pro-tumourigenic microenvironment via release of immunosuppressive factors such as IL-10 and overexpression of the indoleamine 2,3-dioxygenase (IDO) enzyme.17,23 M2-TAMs also lack expression of key T-cell co-stimulation molecules24 and drive both tumour angiogenesis and resistance to anti-VEGF agents. Angiogenesis is driven through production of pro-angiogenic molecules including VEGF and FGF, with resistance to anti-angiogenics due to upregulation of alternative angiogenic pathways and stimulation of pericytes to proliferate in the perivascular niche. Pericytes are perivascular cells responsible for modulating blood flow, vessel permeability and remodelling, and their proliferation stabilises new vessels.25,26 TAMs can also enhance the invasiveness of glioma stem cells (GSCs) via the TGF-β1 signalling pathway.27 Further evidence, incongruous with the M1/M2 dichotomy, is that blockade of CSF-1R on the macrophage surface does not deplete glioblastoma TAMs in vivo, but leads to reprogramming of TAMs away from immunosuppressive phenotypes.28 Other emerging evidence includes the discovery of a link between tissue hypoxia and macrophage polarisation; with M1 macrophages present within normoxic tumour regions and M2 macrophages present in areas of hypoxia.29 These findings support the view that macrophage polarisation is not simply location dependent but instead dependent on distinct signals present in the local microenvironment.30 TAMs retain both plasticity and the ability to undergo reprogramming;31 characteristics which are potentially exploitable for therapeutic benefit.32
Downregulation of MHC
An effective T-cell immune response requires antigen presentation and subsequent recognition, which is dependent on the co-expression of the human MHC proteins. Comparative analyses of gene expression profiles suggest that invading glioblastoma cells are able to escape immune recognition by downregulating expression of MHC molecules.33 The role of antigen presentation within the CNS is thought to fall primarily to the resident microglia, with supporting evidence in vivo demonstrating that microglia are able to cross-present tumour antigens to CD8+ T cells via MHC Class I.34 However, the presence of immunosuppressive cytokines such as IL-10 and TGF-ß within the glioblastoma TME cause microglia to lose MHC expression.11,35 Pericytes may also play a role in antigen presentation within the CNS, and pericyte MHC Class II expression is shown to increase in response to inflammatory cytokine release.36 In the TME, pericytes in contact with glioblastoma cells possess immunosuppressive functions evidenced by changes including a reduction in MHC expression and T-cell co-stimulatory signals.37 Low levels of MHC Class I molecules are also found on glioblastoma cancer stem cells, rendering them resistant to T-cell-mediated killing, and thus contributing towards tumour initiation, progression and resistance to therapy.38
T lymphocytes
In patients with glioblastoma, increased T-cell infiltration is found in both tumour (tumour-infiltrating lymphocyte; TIL) and brain parenchyma.39 Studies of human tumour samples have shown that this influx is counteracted with a number of events that evade the immune response including further downregulating MHC to prevent antigen presentation,40 increasing expression of the inhibitory protein programmed death ligand-1 (PD-L1),41 and increased recruitment of immunosuppressive regulatory T cells (T-regs) which express co-inhibitory molecules including cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed death receptor 1 (PD-1).42,43 Fourfold more T-regs are found in human glioblastoma samples than benign pituitary adenomas and meningiomas, with CTLA-4 expression on these T-regs threefold that on T-regs in peripheral blood.42 PD-L1 expression on circulating monocytes is significantly higher in patients with glioblastoma compared to healthy controls, while expression in tumour infiltrating monocytes is, on average, twice that of circulating monocytes from the same patient.41
Immunological effects of standard therapy
Standard therapy for patients diagnosed with glioblastoma consists of maximal safe debulking surgery, followed by radiotherapy and temozolomide chemotherapy (RT-TMZ).2 In addition, high doses of glucocorticoids are frequently administered to reduce tumour-associated and radiotherapy-induced cerebral oedema. These therapies may potentially both augment or diminish immune responses.
Radiotherapy, chemotherapy and glucocorticoid therapy are independently immunosuppressive. Approximately 70% of patients experience clinically significant reductions in their circulating CD4+ lymphocyte counts following standard treatment, persisting for up to a year and associated with early tumour progression.44 TMZ-induced lymphopenia is considered a significant limitation to clinical translation of immunotherapies.44 Radiation induces M2 macrophages and activation of TGF-β.45,46,47 RT-TMZ has been shown to increase PD-L1 expression in preclinical glioblastoma models, and acquired resistance to radiotherapy has been overcome by PD-L1 blockade in cancer models.48,49 Further, as well as depleting systemic lymphocyte counts, RT-TMZ also tilts the balance between regulatory and effector peripheral blood lymphocytes towards an immunosuppressive state.50,147,148
Tumour heterogeneity is well characterised in glioblastoma, and may contribute towards immune cell heterogeneity within tumours.149,150 Glioblastoma heterogeneity is at least partly maintained by extracellular vesicles (EVs) that contain molecules including DNA, RNA and proteins. The shedding and uptake of these vesicles from tumour cells form an additional communication network capable of modulating the TME. Recent evidence has also shown that PD-L1 is expressed on glioblastoma EVs.151,152 Further, vascular heterogeneity exists, with vessel co-option (glioblastoma migration along existing blood vessels) occurring alongside angiogenesis, increasing the complexity of heterogeneity within glioblastoma.153 Further research into these factors, including utility of techniques such as single-cell RNA sequencing to characterise both tumour, and immune cell heterogeneity154,155 may facilitate a future shift towards more personalised treatment.
In summary, the CNS has an active immune system and glioblastoma employs a number of strategies to evade immune-mediated death. To design therapies that effectively harness the immune system against glioblastoma, a deeper understanding of the complex interactions between tumours and the tightly regulated immune system in the CNS is needed. Future therapies will need to address multi-dimensional challenges including identifying truly tumour-specific antigens to target, increasing tumour immunogenicity, targeting the immune microenvironment and controlling toxicity of therapy.
References
Louis, D. N. et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820 (2016).
Stupp, R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009).
Chen R., Cohen A. L., Colman H. Targeted therapeutics in patients with high-grade gliomas: past, present, and future. Curr. Treat. Options Oncol. 17, 42 (2016).
Corthay, A. Does the immune system naturally protect against cancer? Front. Immunol. 5, 197 (2014).
Coulie, P. G., Van den Eynde, B. J., Van Der Bruggen, P. & Boon, T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14, 135 (2014).
Lakshmi Narendra, B., Eshvendar Reddy, K., Shantikumar, S. & Ramakrishna, S. Immune system: a double-edged sword in cancer. Inflamm. Res. 62, 823–834 (2013).
Engelhardt, B. et al. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 132, 317–338 (2016).
Heimberger, A. B. & Sampson, J. H. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro. Oncol. 13, 3–13 (2011).
Chandramohan, V., Mitchell, D. A., Johnson, L. A., Sampson, J. H. & Bigner, D. D. Antibody, T-cell and dendritic cell immunotherapy for malignant brain tumors. Future Oncol. 9, 977–990 (2013).
Quail, D. F. & Joyce, J. A. The microenvironmental landscape of brain tumors. Cancer Cell. 31, 326–341 (2017).
Razavi, S. M. et al. Immune evasion strategies of glioblastoma. Front. Surg. 3, 11 (2016).
Reinhard, J., Brösicke, N., Theocharidis, U. & Faissner, A. The extracellular matrix niche microenvironment of neural and cancer stem cells in the brain. Int. J. Biochem. Cell Biol. 81, 174–183 (2016).
Wade, A. et al. Proteoglycans and their roles in brain cancer. FEBS J. 280, 2399–2417 (2013).
Kundu, S. et al. Heparanase promotes glioma progression and is inversely correlated with patient survival. Mol. Cancer Res. 14, 1243–1253 (2016).
Oskarsson, T., Batlle, E. & Massagué, J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell 14, 306–321 (2014).
Huang, J.-Y. et al. Extracellular matrix of glioblastoma inhibits polarization and transmigration of T cells: the role of tenascin-C in immune suppression. J. Immunol. 185, 1450–1459 (2010).
Zhou, W. et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 17, 170–182 (2015).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Bowman, R. L. & Joyce, J. A. Therapeutic targeting of tumor-associated macrophages and microglia in glioblastoma. Immunotherapy 6, 663–666 (2014).
Chen, Z. et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 77, 2266–2278 (2017).
Komohara, Y., Ohnishi, K., Kuratsu, J. & Takeya, M. Possible involvement of the M2 anti‐inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 216, 15–24 (2008).
De, I. et al. CSF1 overexpression promotes high-grade glioma formation without impacting the polarization status of glioma-associated microglia and macrophages. Cancer Res. 76, 2552–2560 (2016).
Komohara, Y., **ushi, M. & Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 105, 1–8 (2014).
Hussain, S. F. et al. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro. Oncol. 8, 261–279 (2006).
Brandenburg, S. et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 131, 365–378 (2016).
Bergers, G. & Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592 (2008).
X.-z, Ye et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 189, 444–453 (2012).
Pyonteck, S. M. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264 (2013).
Geissmann, F. et al. Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661 (2010).
Mantovani, A., Biswas, S. K., Galdiero, M. R., Sica, A. & Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 229, 176–185 (2013).
Venteicher, A. S. et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 (2017).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399 (2017).
Perng, P. & Lim, M. Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Front. Oncol. 5, 153 (2015).
Jarry, U. et al. Efficiently stimulated adult microglia cross‐prime naive CD8+T cells injected in the brain. Eur. J. Immunol. 43, 1173–1184 (2013).
Roy, L.-O., Poirier, M.-B. & Fortin, D. Transforming growth factor-beta and its implication in the malignancy of gliomas. Target Oncol. 10, 1–14 (2015).
Pieper, C., Marek, J. J., Unterberg, M., Schwerdtle, T. & Galla, H.-J. Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Res. 1550, 1–8 (2014).
Valdor, R. et al. Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget 8, 68614 (2017).
Di Tomaso, T. et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. 16, 800–813 (2010).
Ampie, L., Woolf, E. C. & Dardis, C. Immunotherapeutic advancements for glioblastoma. Front. Oncol. 5, 12 (2015).
Leone, P. et al. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J. Natl Cancer Inst. 105, 1172–1187 (2013).
Bloch, O. et al. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 19, 3165–3175 (2013).
Jacobs, J. F. et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro. Oncol. 11, 394–402 (2009).
Reardon, D. A. et al. Immunotherapy advances for glioblastoma. Neuro. Oncol. 16, 1441–1458 (2014).
Grossman, S. A. et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res. 17, 5473–5480 (2011).
Tsai, C. S. et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 68, 499–507 (2007).
Chiang, C. S. et al. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front. Oncol. 2, 89 (2012).
Jobling, M. F. et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 166, 839–848 (2006).
Derer, A. et al. Chemoradiation increases PD-L1 expression in certain melanoma and glioblastoma cells. Front. Immunol. 7, 610 (2016).
Dovedi, S. J. et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 74, 5458–5468 (2014).
Fadul, C. E. et al. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro. Oncol. 13, 393–400 (2011).
Schaue, D., **e, M. W., Ratikan, J. A. & McBride, W. H. Regulatory T cells in radiotherapeutic responses. Front. Oncol. 2, 90 (2012).
Olnes, M. J. et al. Effects of systemically administered hydrocortisone on the human immunome. Sci. Rep. 6, 23002 (2016).
Benedetti, S. et al. Dexamethasone inhibits the anti-tumor effect of interleukin 4 on rat experimental gliomas. Gene Ther. 10, 188–192 (2003).
Wong, E. T., Lok, E., Gautam, S. & Swanson, K. D. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br. J. Cancer 113, 232–241 (2015).
Barker, H. E., Paget, J. T., Khan, A. A. & Harrington, K. J. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425 (2015).
Formenti, S. C. & Demaria, S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J. Natl Cancer Inst. 105, 256–265 (2013).
Demaria, S. & Formenti, S. C. Sensors of ionizing radiation effects on the immunological microenvironment of cancer. Int. J. Radiat. Biol. 83, 819–825 (2007).
McBride, W. H. et al. A sense of danger from radiation. Radiat. Res. 162, 1–19 (2004).
Garnett, C. T. et al. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 64, 7985–7994 (2004).
Chakraborty, M. et al. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J. Immunol. 170, 6338–6347 (2003).
Reits, E. A. et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 203, 1259–1271 (2006).
Vereecque, R. et al. γ-ray irradiation induces B7.1 expression in myeloid leukaemic cells. Br. J. Haematol. 108, 825–831 (2000).
Weiss, T., Weller, M. & Roth, P. Immunological effects of chemotherapy and radiotherapy against brain tumors. Expert Rev. Anticancer Ther. 16, 1087–1094 (2016).
Sahebjam, S., Sharabi, A., Lim, M., Kesarwani, P. & Chinnaiyan, P. Immunotherapy and radiation in glioblastoma. J. Neurooncol. 134, 531–539 (2017).
Fernandez-Palomo, C. et al. Bystander effects in tumor-free and tumor-bearing rat brains following irradiation by synchrotron X-rays. Int. J. Radiat. Biol. 89, 445–453 (2013).
Preusser, M., Lim, M., Hafler, D. A., Reardon, D. A. & Sampson, J. H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 11, 504–514 (2015).
Stepanenko, A. A. et al. Temozolomide promotes genomic and phenotypic changes in glioblastoma cells. Cancer Cell. Int. 16, 36 (2016).
Yarchoan, M., Hopkins, A. & Jaffee, E. M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med 377, 2500–2501 (2017).
Van Allen, E. M. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 348, 124–128 (2015).
Rosenberg, J. E. et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387, 1909–1920 (2016).
Bouffet, E. et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 34, 2206–2211 (2016).
Kim, T. G. et al. Immunological factors relating to the antitumor effect of temozolomide chemoimmunotherapy in a murine glioma model. Clin. Vaccine Immunol. 17, 143–153 (2010).
Vaios, E. J., Nahed, B. V., Muzikansky, A., Fathi, A. T. & Dietrich, J. Bone marrow response as a potential biomarker of outcomes in glioblastoma patients. J. Neurosurg. 127, 132–138 (2017).
Fadul, C. E. et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J. Immunother. 34, 382–389 (2011).
Mathios, D. et al. Anti–PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 8, 370ra180 (2016).
Stupp, R. et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 318, 2306–2316 (2017).
Hottinger, A. F., Pacheco, P. & Stupp, R. Tumor treating fields: a novel treatment modality and its use in brain tumors. Neuro. Oncol. 18, 1338–1349 (2016).
Lee, S. & Margolin, K. Cytokines in cancer immunotherapy. Cancers 3, 3856–3893 (2011).
Rand, R. W. et al. Intratumoral administration of recombinant circularly permuted interleukin-4-Pseudomonas exotoxin in patients with high-grade glioma. Clin. Cancer Res. 6, 2157–2165 (2000).
Zaidi, M. R. & Merlino, G. The two faces of interferon-γ in cancer. Clin. Cancer Res. 17, 6118–6124 (2011).
Zhu, V. F., Yang, J., LeBrun, D. G. & Li, M. Understanding the role of cytokines in glioblastoma multiforme pathogenesis. Cancer Lett. 316, 139–150 (2012).
Jackson, C., Ruzevick, J., Phallen, J., Belcaid, Z. & Lim, M. Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment. Clin. Dev. Immunol. 1, 1–20 (2011).
Reardon, D. A. et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro. Oncol. 19, iii21–iii21 (2017).
Weller, M. et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 18, 1373–1385 (2017).
Wilmotte, R. et al. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport 16, 1081–1085 (2005).
Berghoff, A. S. et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro. Oncol. 17, 1064–1075 (2015).
Wei, B. et al. The upregulation of programmed death 1 on peripheral blood T cells of glioma is correlated with disease progression. Tumour Biol. 35, 2923–2929 (2014).
Xue, S., Song, G. & Yu, J. The prognostic significance of PD-L1 expression in patients with glioma: a meta-analysis. Sci. Rep. 7, 4231 (2017).
Omuro, A. et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro. Oncol. 20, 674–686 (2018).
Reardon, D. A. et al. Phase 2 study to evaluate safety and efficacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: an update. J. Clin. Oncol. 35, 2042 (2017).
Reardon, D. A. et al. ATIM-35. Results of the phase IB keynote-028 multi-cohort trial of pembrolizumab monotherapy in patients with recurrent PD-L1-positive glioblastoma multiforme (GBM). Neuro. Oncol. 18, vi25–vi26 (2016).
Haanen, J. et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 28, iv119–iv142 (2017).
Zhu, X. et al. Severe cerebral edema following nivolumab treatment for pediatric glioblastoma: case report. J. Neurosurg. Pediatr. 19, 249–253 (2017).
Melief, C. J., van Hall, T., Arens, R., Ossendorp, F. & van der Burg, S. H. Therapeutic cancer vaccines. J. Clin. Invest. 125, 3401–3412 (2015).
Winograd, E. K., Ciesielski, M. J. & Fenstermaker, R. A. Novel vaccines for glioblastoma: clinical update and perspective. Immunotherapy 8, 1293–1308 (2016).
Bloch, O. et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro. Oncol. 16, 274–279 (2014).
Ishikawa, E. et al. Phase I/IIa trial of fractionated radiotherapy, temozolomide, and autologous formalin-fixed tumor vaccine for newly diagnosed glioblastoma. J. Neurosurg. 121, 543–553 (2014).
Terasaki, M. et al. Phase I trial of a personalized peptide vaccine for patients positive for human leukocyte antigen–A24 with recurrent or progressive glioblastoma multiforme. J. Clin. Oncol. 29, 337–344 (2011).
Lim, W. A. & June, C. H. The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017).
Schuster, S. J. et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 377, 2545–2554 (2017).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Brudno, J. N. & Kochenderfer, J. N. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321–3330 (2016).
Garber, K. Driving T-cell immunotherapy to solid tumors. Nat. Biotechnol. 36, 215–219 (2018).
Thaci, B. et al. Significance of interleukin-13 receptor alpha 2–targeted glioblastoma therapy. Neuro. Oncol. 16, 1304–1312 (2014).
Wikstrand, C. J., Reist, C. J., Archer, G. E., Zalutsky, M. R. & Bigner, D. D. The class III variant of the epidermal growth factor receptor (EGFRvIII): characterization and utilization as an immunotherapeutic target. J. Neurovirol. 4, 148–158 (1998).
Zhang, C. et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J. Natl Cancer Inst. 108, djv375 (2016).
Yan, M. et al. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastas. Rev. 34, 157–164 (2015).
Yan, M., Parker, B. A., Schwab, R. & Kurzrock, R. HER2 aberrations in cancer: Implications for therapy. Cancer Treat. Rev. 40, 770–780 (2014).
Ahmed, N. et al. Her2-specific chimeric antigen receptor–modified virus-specific t cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 3, 1094–1101 (2017).
O’rourke, D. M. et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984 (2017).
Brown, C. E. et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569 (2016).
Bielamowicz, K., Khawja, S. & Ahmed, N. Adoptive cell therapies for glioblastoma. Front. Oncol. 3, 275 (2013).
Schuessler, A. et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 74, 3466–3476 (2014).
Mitchell, D. A. et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro. Oncol. 10, 10–18 (2008).
Jackson, H. J., Rafiq, S. & Brentjens, R. J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 13, 370–383 (2016).
Han, J. et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 5, 11483 (2015).
Genssler, S. et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 5, e1119354 (2016).
Kaufman, H. L., Kohlhapp, F. J. & Zloza, A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 14, 642 (2015).
Wang, P. et al. Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun. 8, 1395 (2017).
Martuza, R. L., Malick, A., Markert, J. M., Ruffner, K. L. & Coen, D. M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 252, 854–856 (1991).
Post, D. E. et al. Targeted cancer gene therapy using a hypoxia inducible factor dependent oncolytic adenovirus armed with interleukin-4. Cancer Res. 67, 6872–6881 (2007).
Leber, M. F. et al. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol. Ther. 19, 1097–1106 (2011).
Geletneky, K. et al. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer 12, 99 (2012).
Wollmann, G., Ozduman, K. & van den Pol, A. N. Oncolytic virus therapy of glioblastoma multiforme – concepts and candidates. Cancer J. 18, 69–81 (2012).
Uchida, H. et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol. Ther. 21, 561–569 (2013).
Saha, D. et al. Combinatorial effects of VEGFR kinase inhibitor axitinib and oncolytic virotherapy in mouse and human glioblastoma stem-like cell models. Clin. Cancer Res. 24, 3409–3422 (2018).
Weller, M. et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 18, e315–e329 (2017).
Gilbert, M. R. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 699–708 (2014).
Chinot, O. L. et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 709–722 (2014).
Oyama, T. et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-κB activation in hemopoietic progenitor cells. J. Immunol. 160, 1224–1232 (1998).
Ohm, J. E. & Carbone, D. P. VEGF as a mediator of tumor-associated immunodeficiency. Immunol. Res. 23, 263–272 (2001).
Jain, R. K. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 26, 605–622 (2014).
Allen, E. et al. Combined antiangiogenic and anti–PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 9, eaak9679 (2017).
Fukumura, D., Kloepper, J., Amoozgar, Z., Duda, D. G. & Jain, R. K. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 15, 325–340 (2018).
Hodi, F. S. et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2, 632–642 (2014).
Awan, M. et al. Extra-CNS metastasis from glioblastoma: a rare clinical entity. Expert Rev. Anticancer Ther. 15, 545–552 (2015).
Huang, A. C. et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017).
Barua, N. U., Gill, S. S. & Love, S. Convection-enhanced drug delivery to the brain: therapeutic potential and neuropathological considerations. Brain Pathol. 24, 117–127 (2014).
Qiao, J. et al. Intratumoral oncolytic adenoviral treatment modulates the glioma microenvironment and facilitates systemic tumor-antigen-specific T cell therapy. Oncoimmunology 4, e1022302 (2015).
Vogelbaum, M. A. et al. First-in-human evaluation of the Cleveland Multiport Catheter for convection-enhanced delivery of topotecan in recurrent high-grade glioma: results of pilot trial 1. J. Neurosurg. https://doi.org/10.3171/2017.10.JNS171845 (2018)
Sears, C. L. & Pardoll, D. M. The intestinal microbiome influences checkpoint blockade. Nat. Med. 24, 254 (2018).
Nelson, M. H., Diven, M. A., Huff, L. W. & Paulos, C. M. Harnessing the microbiome to enhance cancer immunotherapy. J. Immunol. Res. 2015, 368736 (2015).
Cogdill, A. P., Andrews, M. C. & Wargo, J. A. Hallmarks of response to immune checkpoint blockade. Br. J. Cancer 117, 1 (2017).
Pitt, J. M. et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and-extrinsic factors. Immunity 44, 1255–1269 (2016).
Evans, S. S., Repasky, E. A. & Fisher, D. T. Fever and the thermal regulation of immunity: the immune system feels the heat. Nat. Rev. Immunol. 15, 335–349 (2015).
Sheridan, C. IDO inhibitors move center stage in immuno-oncology. Nat. Biotechnol. 33, 321–322 (2015).
Chang, C.-H. & Pearce, E. L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 17, 364 (2016).
Aldape, K., Zadeh, G., Mansouri, S., Reifenberger, G. & von Deimling, A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 129, 829–848 (2015).
Doucette, T. et al. Immune heterogeneity of glioblastoma subtypes: extrapolation from the cancer genome atlas. Cancer Immunol. Res. 1, 112–122 (2013).
Ricklefs, F. et al. Extracellular vesicles from high-grade glioma exchange diverse pro-oncogenic signals that maintain intratumoral heterogeneity. Cancer Res. 76, 2876–2881 (2016).
Ricklefs, F. L. et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 4, eaar2766 (2018).
Wang, N., Jain, R. K. & Batchelor, T. T. New directions in anti-angiogenic therapy for glioblastoma. Neurotherapeutics 14, 321–332 (2017).
Papalexi, E. & Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 18, 35 (2018).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014).
Korn, T. & Kallies, A. T cell responses in the central nervous system. Nat. Rev. Immunol. 17, 179–194 (2017).
Pachter, J. S., De Vries, H. E. & Fabry, Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J. Neuropathol. Exp. Neurol. 62, 593–604 (2003).
Daneman, R. & Prat, A. The blood–brain barrier. Perspect. Biol. 7, a020412 (2015).
Dombrowski, Y. et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 20, 674 (2017).
Laman, J. D. & Weller, R. O. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J. Neuroimmune Pharmacol. 8, 840–856 (2013).
Jessen, N. A., Munk, A. S. F., Lundgaard, I. & Nedergaard, M. The glymphatic system: a beginner’s guide. Neurochem. Res. 40, 2583–2599 (2015).
Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337 (2015).
Herz, J., Filiano, A. J., Smith, A., Yogev, N. & Kipnis, J. Myeloid cells in the central nervous system. Immunity 46, 943–956 (2017).
Nimmerjahn, A., Kirchhoff, F. & Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 (2005).
Acknowledgements
N.B. is funded by Cancer Research UK. T.C. is funded by the University College London Hospitals Charity. P.M. is supported by the University College Hospital/University College London Biomedical Research Centre and the National Brain Appeal.
Authors contributions
N.B., T.C., D.O. and P.M. wrote, reviewed and edited the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
P.M. has received honoraria from Bristol-Myers Squibb. The remaining authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brown, N.F., Carter, T.J., Ottaviani, D. et al. Harnessing the immune system in glioblastoma. Br J Cancer 119, 1171–1181 (2018). https://doi.org/10.1038/s41416-018-0258-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-018-0258-8
- Springer Nature Limited
This article is cited by
-
Understanding the immunosuppressive microenvironment of glioma: mechanistic insights and clinical perspectives
Journal of Hematology & Oncology (2024)
-
Glioblastoma stem cell metabolism and immunity
Cancer and Metastasis Reviews (2024)
-
Global stability and parameter analysis reinforce therapeutic targets of PD-L1-PD-1 and MDSCs for glioblastoma
Journal of Mathematical Biology (2024)
-
Upregulation of the canonical signaling pathway of interferon-gamma is associated with glioblastoma progression
Molecular Biology Reports (2024)
-
Emerging hallmark of gliomas microenvironment in evading immunity: a basic concept
The Egyptian Journal of Neurology, Psychiatry and Neurosurgery (2023)