
Abstract
Disrupted redox homeostasis contributes to renal ischemia–reperfusion (IR) injury. Abundant natural products can activate nuclear factor erythroid-2-related factor 2 (Nrf2), thereby providing therapeutic benefits. Methyl eugenol (ME), an analog of the phenolic compound eugenol, has the ability to induce Nrf2 activity. In this study, we investigated the protective effects of ME against renal oxidative damage in vivo and in vitro. An IR-induced acute kidney injury (AKI) model was established in mice. ME (20 mg·kg−1·d−1, i.p.) was administered to mice on 5 consecutive days before IR surgery. We showed that ME administration significantly attenuated renal destruction, improved the survival rate, reduced excessive oxidative stress and inhibited mitochondrial lesions in AKI mice. We further demonstrated that ME administration significantly enhanced Nrf2 activity and increased the expression of downstream antioxidative molecules. Similar results were observed in vitro in hypoxia/reoxygenation (HR)-exposed proximal tubule epithelial cells following pretreatment with ME (40 μmol·L−1). In both renal oxidative damage models, ME induced Nrf2 nuclear retention in tubular cells. Using specific inhibitors (CC and DIF-3) and molecular docking, we demonstrated that ME bound to the binding pocket of AMPK with high affinity and activated the AMPK/GSK3β axis, which in turn blocked the Nrf2 nuclear export signal. In addition, ME alleviated the development of renal fibrosis induced by nonfatal IR, which is frequently encountered in the clinic. In conclusion, we demonstrate that ME modulates the AMPK/GSK3β axis to regulate the cytoplasmic–nuclear translocation of Nrf2, resulting in Nrf2 nuclear retention and thereby enhancing antioxidant target gene transcription that protects the kidney from oxidative damage.
Similar content being viewed by others

Introduction
Acute kidney injury (AKI) is a common sudden but serious clinical event characterized by the rapid loss of renal function and potential subsequent chronic fibrosis that is associated with high morbidity and mortality [1]. Ischemia–reperfusion (IR) injury is one of the leading causes of AKI in the clinic [2, 3]. Currently, no proven pharmacological therapies are available to prevent or attenuate AKI [4], and there is an urgent need to identify effective drugs and thus improve clinical outcomes.
IR disrupts the cellular redox balance and triggers excessive reactive oxygen species (ROS) production in the kidney, which impairs mitochondria and triggers inflammation, apoptosis and necrosis [5]. Therefore, ROS are among the potential critical targets for the management of IR-induced AKI. The transcription factor nuclear factor erythroid factor 2-related factor 2 (Nrf2) maintains cell homeostasis and plays a crucial role in many inflammation- and oxidative stress-related conditions by regulating the expression of antioxidant genes [6,7,8,9]. Under physiological conditions, Nrf2 binds to its negative regulatory factor kelch-like epichlorohydrin-associated protein 1 (Keap1) in the cytoplasm and is degraded through the ubiquitin–proteasome pathway. In response to oxidative stress, the interaction between Nrf2 and Keap1 is blocked, and Nrf2 translocates into the nucleus, where it binds to conserved antioxidant response elements (AREs) and activates downstream cytoprotective antioxidants, such as heme oxygenase-1 (HO-1) [10], NAD(P)H quinone dehydrogenase 1 (NQO1) [11] and superoxide dismutase (SOD) [12]. Nrf2 activation exerts protective effects against brain, heart, and liver IR injury via the inhibition of oxidative stress, which reduces the inflammatory response and cell apoptosis [13,14,15]. We previously showed that enhancing Nrf2 activity by reducing its ubiquitylation effectively attenuates renal postreperfusion injury [16], further confirming that the Nrf2 pathway is a prospective therapeutic target to ameliorate organ damage by reducing oxidative stress. However, in addition to modulating the ubiquitin-mediated degradation of Nrf2, other strategies for activating the Nrf2 pathway deserve further investigation.
Methyl eugenol (4-allyl-1,2-dimethoxybenzene, ME), an analog of the phenolic compound eugenol, is a natural component of many aromatic plants, such as Myristica fragrans, Ocimum basilicum, and Pimenta officinalis, that has been used extensively by humans as a flavoring agent in daily life [17, 18]. ME has been shown to exert protective effects in a variety of disease models due to its unique anticancer [19, 20], anti-inflammatory [21, 22], antibacterial [23], antinociceptive [24, 25], and anxiolytic [26] activities. Recently, we reported that ME exerted a powerful protective effect against acute liver injury in mice by reducing the inflammatory response and apoptosis both in vivo and in vitro [27]. Furthermore, studies have shown that ME can induce Nrf2 activation, acting as a master regulator of cellular resistance to oxidative insults [28, 29]. However, the specific protective effects of ME against oxidative damage and the underlying mechanisms of Nrf2 activation remain unknown and thus inspire further investigation.
Materials and methods
Animals
Male C57BL/6J mice (6–8 weeks old) were purchased from Bei**g Vital River Laboratory Animal Technology Co., Ltd. (Bei**g, China). All animals were maintained under a constant 12 h light/dark cycle, 25 °C and 55% relative humidity, with free access to food and water. All animal experiments were performed under the guidelines of the Chinese Council on Animal Care and approved by the Institutional Animal Care and Use Committee of Tongji Medical College, Huazhong University of Science and Technology.
Drug preparation and administration
For animal experiments, ME was freshly prepared in 0.5% sodium carboxymethylcellulose aqueous solution, and either drug or an equal volume of vehicle was administered intraperitoneally once a day on 5 consecutive days. The IR operation was performed 2 h after the final injection. ML385 was dissolved in DMSO and injected intraperitoneally (30 mg/kg) 1 h before the operation [30], and the final concentration of DMSO was less than 2.5%. For in vitro experiments, ME was dissolved in DMSO, diluted to the desired final concentration in culture medium and applied to HK-2 cells. CC (4 μmol/L), CDDO (100 nmol/L) or DIF (10 μmol/L) was dissolved in DMSO and added to cells before exposure to hypoxia. The DMSO concentration was kept at less than 0.1% in all the groups.
Experimental grou**
After acclimating to the housing environment for 5 days, the mice were randomly assigned to different groups. Fatal renal IR injury groups: sham, sham+ML385, IR, IR + ME (at different concentrations) and IR + ME + ML385; nonfatal model (25 min of ischemia) groups: sham, IR, and IR + ME.
Mouse model of fatal acute renal IR injury
Mice were subjected to ischemia/reperfusion as described in our previous publication with some modifications [16]. Briefly, mice were anesthetized, and the right kidney was removed. Then, a nontraumatic vascular clip was used to occlude the left renal pedicle for 35 min. The clip was then removed, and mice were sacrificed 24 h later to harvest blood and the remaining kidney for analysis. The same operation was performed on mice in the sham group without occlusion of the renal artery.
Mouse model of nonfatal renal IR injury
To explore the potential longer-term therapeutic effect of ME, a modified, moderate 25-min renal IR model was adopted [31]. Mice were prepared in the same way as described above, and the left renal pedicle was occluded for 25 min. Then, blood and the left kidney were harvested at the indicated reperfusion times (Days 1, 3, 7, 15, and 30) for further experiments.
Reagents
Details of the reagents used are presented in Supplementary Table 1.
UPLC–MS/MS quantitative analysis
Prior to surgery, the ME concentration in the plasma and renal tissue 2 h after the last injection was analyzed by UPLC–MS/MS as previously described [32, 33]. Chromatographic separation and mass spectrometry experiments were performed on a SCIEX LC-30A HPLC system coupled to an AB SCIEX QTRAP® 6500 (Foster City, USA).
Creatinine and blood urea nitrogen analysis
Creatinine (Cr) and blood urea nitrogen (BUN) levels were measured in the collected serum to assess renal function using an automated biochemical analyzer BS-200 (Mindray, Shenzhen, China).
Hematoxylin and eosin (H&E) staining
Fixed kidney tissues were embedded in paraffin and sliced into 5-μm-thick sections, which were stained with H&E in accordance with the manufacturer’s instructions. Tubular damage was scored as described previously [34] by a pathologist in a blinded fashion.
TUNEL assay
Apoptotic cells in renal tissues were identified by terminal transferase dUTP nick end labeling (TUNEL) staining using an in situ cell death detection kit (11684817910; Roche, Basel, Switzerland) as described previously [27]. The average number of TUNEL-positive cells was calculated in ten randomly selected fields per animal.
Immunohistochemical staining
Immunohistochemical staining of cleaved caspase-3 and Nrf2 was performed as described previously [35, 36]. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Abcam, ab205718, 1:2000) was used, and the other antibodies used are detailed in Supplementary Table 3. The stained sections were digitally imaged and analyzed using ImageJ software (NIH, MD, USA).
Oil Red O staining
Once harvested, the kidneys were rapidly frozen in liquid nitrogen and cut into 8-μm-thick sections, which were then incubated in 60% isopropanol for 5 min and stained with Oil Red O working solution for 20 min. Mayer hematoxylin counterstaining was performed for 2 min at room temperature. Sections were washed three times with water and then mounted.
Masson’s trichrome staining
To assess fibrotic lesions in the kidneys at different time periods after nonfatal renal IR injury, collagen deposition was examined by Masson’s trichrome staining. Briefly, kidney sections were stained with Masson trichrome staining reagents, and collagen was stained blue according to the standard protocol.
8-OHdG immunofluorescence staining
To evaluate oxidative damage in renal tissues, immunofluorescence staining of the characteristic marker of oxidative stress-associated DNA adducts, 8-hydroxydeoxyguanosine (8-OHdG), was performed as previously described [37]. Briefly, prepared kidney tissue sections were incubated with 8-OHdG antibody at 4 °C overnight. Alexa Fluor 488-conjugated donkey anti-mouse IgG (Life Technologies, A21202, 1:400) was used as the secondary antibody. Then, the sections were counterstained with DAPI, and a fluorescence microscope (ECLIPSE C1, NIKON, Tokyo, Japan) was used to acquire images.
MDA and SOD measurements
Malondialdehyde (MDA, a marker of oxidative stress) production and SOD (a primary antioxidant enzyme) activity in the kidneys or HK-2 cells were measured by corresponding assay kits (Supplementary Table 1) according to the manufacturer’s instructions.
Mitochondrial morphology assay
To observe alterations in kidney mitochondrial morphology in different groups of mice, transmission electron microscopy (TEM) was applied as previously described [38]. Additionally, the mitochondrial morphology in HK-2 cells was characterized after staining with MitoTracker Red CMXRos (200 nmol/L, Beyotime) as described previously [36] and observation under confocal microscopy.
Cell culture and RNA interference
The human renal proximal tubule epithelial cell line HK-2 was purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China). Cells were cultured as described previously [34], and after the cultures reached 70%–80% confluency, the cells were serum-starved for 12 h and randomly divided into the following groups: normal controls, HR, HR + ME, HR + CDDO, HR + ME + ML385 and HR + ME + CC/DIF-3. Small interfering RNA targeting Nrf2 (si-Nrf2) and scrambled siRNA (si-NC) were provided (sequences listed in Supplementary Table 2) by Hanbio (Shanghai, China). HK-2 cells in 6-well plates at 40%–50% confluence were transfected with 100 pmol si-Nrf2 or si-NC with an equal volume of Lipofectamine 2000 (Invitrogen, USA) following previously reported instructions [37]. After 48 h, routine Western blotting was performed to evaluate the efficiency of the siRNAs, and other experiments were conducted.
Hypoxia/reoxygenation model
Hypoxia/reoxygenation (HR) experiments were performed in the H35 Hypoxystation (Don Whitley Scientific, West Yorkshire, UK) as previously described [16]. Briefly, for hypoxic induction, HK-2 cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2, 1% O2 and 94% N2 for 24 h and then reoxygenated under normal conditions for 2 h. As the nonischemic control, HK-2 cells were exposed to normoxic conditions and complete medium supplemented with glucose and 10% FBS. Cells were treated with ME or an equal volume of DMSO prior to HR. Then, the cells were harvested for further experiments.
Cell viability assay
The cytotoxicity of ME in HK-2 cells was determined by a CCK-8 kit according to the manufacturer’s instructions. Cell viability in the treatment groups was normalized to that in the control group, for which the mean was set to one.
BrdU incorporation assay
To assess the effect of ME on HK-2 cell proliferation, a bromodeoxyuridine (BrdU) cell proliferation assay kit was utilized (MultiSciences). Briefly, cells were seeded onto 6-well plates with 2 mL culture medium/well, treated as indicated, and then incubated with 4 μL BrdU solution for 4 h. Then, the cells were harvested, fixed, stained with the corresponding antibody and subjected to flow cytometry analysis (BD Biosciences, USA). Data were analyzed with FlowJo v.10.5.3 (FlowJo LLC).
Apoptosis assay
Cell apoptosis was detected using an Annexin V Apoptosis Detection Kit (Beyotime) as previously described by our group [27]. A FACSCalibur flow cytometer (BD Biosciences, USA) was used to assess the percentage of apoptotic cells.
MMP measurement
The mitochondrial membrane potential (MMP) of HK-2 cells was detected by a JC-1 fluorescent probe, which shifts from red (JC-1 aggregate) to green (JC-1 monomer) upon mitochondrial depolarization. After incubation with JC-1 solution for 30 min at 37 °C, the cells were harvested, and data were acquired on a FACSCalibur flow cytometer (BD Biosciences, USA). The MMP was evaluated based on the JC-1 aggregate/JC-1 monomer fluorescence ratio [39].
Immunofluorescence (IF)
Briefly, prepared kidney tissue sections or cells were immunolabeled with primary antibodies (Supplementary Table 3) overnight at 4 °C followed by incubation with DyLight 488 goat anti-rabbit IgG (Abbkine, A23220, 1:200) or CoraLite 594-conjugated goat anti-mouse IgG (Proteintech, SA00013-1, 1:200) for 1 h at 37 °C. Nuclei were counterstained with DAPI for 15 min at room temperature. Digital images were captured using a fluorescence microscope or confocal laser scanning microscope (Nikon TiE, Tokyo, Japan).
Intracellular ROS and mtROS measurements
ROS production in HK-2 cells was measured using the Reactive Oxygen Species Assay Kit (S0033S, Beyotime), and a FACSCalibur system was used for quantitative analysis. Mitochondrial ROS (mtROS) were measured using the MitoSOX™ Red mitochondrial superoxide indicator (5 µmol/L, Invitrogen, CA, USA) according to the manufacturer’s instructions.
Western blot
Total protein from kidney homogenates or HK-2 cells was extracted using Cell Lysis Buffer (P0013J, Beyotime), and cytoplasmic and nuclear proteins were extracted with a Nuclear and Cytoplasmic Protein Extraction Kit (P0027, Beyotime). Western blotting was performed as previously described [16]. The primary antibodies used are detailed in Supplementary Table 3. All results were analyzed using ImageJ software.
Molecular docking
Molecular docking was performed as described previously [40,41,42]. In brief, the crystal structure of AMPK was downloaded from the Protein Data Bank (PDB, http://www.rcsb.org/; PDB ID: 5UFU). The 2D structure of ME was downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov/). Then, PyMOL and AutoDockTools were used to perform component-target molecular docking, and the results were processed with Discovery Studio 2019 (DS) software.
Statistical analysis
All data are expressed as the mean ± SEM. All experiments were independently repeated at least three times. The significance of differences between two groups was determined by the two-tailed unpaired Student’s t test with GraphPad Prism 8.0.1 (GraphPad Software, La Jolla, CA, USA), one-way ANOVA was used to analyze the significance of differences across more than two groups, and survival rates were evaluated by a two-sided log-rank test. For all tests, the threshold of statistical significance was set to P < 0.05. The exact value of n (number of animals or independent replicates) is indicated in the figure legends.
Results
ME alleviated renal tissue injury and prolonged survival in mice with IR injury
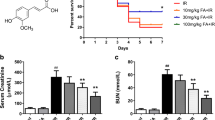
To investigate the protective effects of ME against AKI, mice were divided into 3 groups based on ME pretreatment and IR injury (Fig. 1a, b). Different doses of ME (0, 5, 10, 20 and 40 mg/kg) were administered prior to IR injury. In this experiment, IR induced obvious renal tissue injury characterized by increased plasma levels of Cr and BUN, which were decreased by ME treatment (Fig. 1c, d). The comparison of ME at 40 versus 20 mg/kg revealed no significant enhancement of the protective effect (Cr: 83.03 ± 4.46 μmol/L vs. 76.40 ± 2.33 μmol/L, P > 0.05, and BUN: 31.77 ± 1.14 mmol/L vs. 28.97 ± 0.90 mmol/L, P > 0.05), and mice treated with 20 mg/kg ME had higher drug concentrations in plasma (11.02 ng/mL vs. 1.1 ng/mL) and renal tissue homogenates (5.63 × 10−3 ng/mg vs. 1.36 × 10−3 ng/mg), as indicated by UPLC–MS/MS analysis (Supplementary Fig. S1). Therefore, a dose of 20 mg/kg was employed in the subsequent in vivo experiments to minimize possible toxic side effects. During a 2-week follow-up, the survival rate of the mice receiving ME treatment was notably higher than that of the mice without ME treatment (Fig. 1e). Renal histologic injury was then examined using H&E staining. Compared to the sham group, the IR group showed significant proximal tubular injury characterized by cast formation, vacuolization, and extensive tubular necrosis; ME treatment significantly reduced these effects of IR (Fig. 1f–g). In addition, as a sensitive marker of renal tubular damage, the expression of kidney injury molecule-1 (KIM-1) was significantly inhibited by ME treatment, as indicated by immunostaining and Western blotting (Fig. 1h–k). Collectively, ME pretreatment strikingly ameliorated AKI and reduced the mortality of mice with IR injury.

a Detailed experimental design in vivo. Black arrows indicate the injection of vehicle and green arrows indicate the treatment of ME. b Graphic presentation shows animal model of IR injury. The levels of c serum creatinine (Cr) and d urea nitrogen (BUN) in mice in different groups assessed 24 h after IR (n = 5). e The survival rate of mice in the three groups (n = 6). Representative f hematoxylin and eosin (H&E) staining (left) and g quantitative analysis of tubular injury (right, n = 5). In contrast to the normal kidney histology from sham group, IR induced significant tubular epithelial necrosis (black arrow) with some detached necrotic epithelial cells in the tubular lumen (yellow arrow), while ME treatment showed less tubular damage. h Immunostaining for KIM-1 of kidney sections (left) and i quantitative analysis (right, n = 5). j Western blotting for KIM-1 protein in the kidneys and k quantitative analysis of KIM-1 protein expression 24 h after IR (n = 4). Data represented by mean ± SEM. ##P < 0.01 vs the Sham group and **P < 0.01 vs the IR group; ns indicates no significance.
ME reduced IR-induced oxidative stress
To evaluate the changes in oxidative stress-related damage in kidney tissues following IR injury, we assessed the nuclear aggregation of 8-OHdG (a marker of free radical formation) by immunofluorescence staining and quantified MDA content and SOD activity. As shown in Fig. 2a–d, IR dramatically increased 8-OHdG formation, enhanced the renal level of MDA, and decreased the activity of SOD. Importantly, ME treatment significantly reduced these effects, providing convincing evidence that ME ameliorated IR-induced oxidative stress. Given that 8-OHdG is induced by excessive ROS [43] and that mitochondria are the primary source of intracellular ROS, which cause mitochondrial damage [44], the effects of ME on mitochondrial changes were investigated. Evidence of mitochondrial destruction, such as mitochondrial swelling, fragmentation, and disruption of cristae, was observed in the kidneys of mice in the IR group, as determined by TEM (Fig. 2e). Furthermore, IR decreased the expression of translocase of outer mitochondrial membrane 20 (TOM20) and the essential mitochondrial DNA (mtDNA) packaging protein transcription factor A, mitochondrial (TFAM), both of which are downregulated under oxidative stress [36]. In marked contrast, ME treatment significantly reduced mitochondrial fragmentation and restored the expression of TOM20 and TFAM in the kidneys (Fig. 2e–h). These findings provided further confirmation that ME reduces oxidative stress in the kidney tissue of mice with IR injury.

a Representative sections from 8-OHdG staining. b The nuclear 8-OHdG intensity was calculated (n = 5). c The MDA content and d SOD activity in the renal cells of the three groups were detected by commercial kits (n = 5). e Representative TEM images of mitochondria in the renal tubules of mice. The red arrows indicate the damage of mitochondria. f Western blotting for TOM20 and TFAM proteins in kidney tissues and g, h quantitative analysis of protein expression (n = 4). Data represented by mean ± SEM. #P < 0.05, ##P < 0.01 vs the Sham group; **P < 0.01 vs the IR group.
ME attenuated HR-induced apoptosis in HK-2 cells
To determine the effects of ME in vitro, HK-2 cells were divided into 3 groups, and an HR model was developed (Fig. 3a). Initially, CCK-8 cell cytotoxicity assays were conducted to test the dose-dependent effect of ME on cell viability at concentrations from 0 μmol/L to 100 μmol/L. When the concentration of ME was equal to or greater than 80 μmol/L, the viability of HK-2 cells was significantly reduced (Fig. 3b). Thus, for the remaining in vitro cellular studies, ME concentrations of less than or equal to 40 μmol/L were utilized. As shown in Fig. 3c–f, HR significantly decreased cell viability and induced apoptosis, whereas ME treatment enhanced cell viability and inhibited apoptosis in a dose-dependent manner; 40 μmol/L ME was demonstrated to be the optimal drug concentration. Additionally, the Western blot results showed that ME significantly inhibited proapoptotic caspase-3 cleavage and Bax expression and increased antiapoptotic Bcl-2 levels in HR-exposed HK-2 cells (Fig. 3g–j, ME = 40 μmol/L, P < 0.01). These findings demonstrated that ME has cytoprotective effects and inhibits HR-induced cell apoptosis in vitro.

a Experimental design in vitro. Black arrows indicate the treatment of vehicle and green arrows indicate the treatment of ME. b Effect of various concentrations of ME on cytotoxicity in cultured HK-2 cells (n = 6). c Proliferation of HK-2 cells following HR insult analyzed using BrdU cell proliferation assay kit, and d quantitative analysis of the BrdU incorporation (n = 3). e Apoptosis of HK-2 cells in different groups was detected using flow cytometry and f quantitative analysis (n = 5). g Western blot for apoptosis-associated protein Bax, Bcl-2, Caspase-3, and cleaved caspase-3 (C-caspase3) proteins in HK-2 cells and h–j quantitative analysis of protein expression (n = 3). Data represented by mean ± SEM. #P < 0.05, ##P < 0.01 vs the NC group; *P < 0.05, **P < 0.01 vs the HR group and ns indicates no significance vs the NC group.
ME alleviated HR-induced oxidative damage in HK-2 cells
Consistent with the in vivo results, HR induced a marked increase in ROS, indicative of oxidative stress, and the associated indicators were dramatically reversed by ME treatment (Fig. 4a–d). Moreover, mtROS levels were elevated in HR-exposed HK-2 cells, but ME treatment significantly ameliorated this increase (Fig. 4e, f). Mitochondrial function in HK-2 cells was detected following HR in the presence and absence of ME. As shown in Fig. 4g, h, compared with the normal control (NC) HK-2 cells, HR-exposed HK-2 cells showed an obvious loss of MMP that was mitigated by ME treatment, as revealed by JC-1 assays (P < 0.01). MitoTracker stains mitochondria in live cells, and stain accumulation is dependent upon membrane potential. The results showed that HR disrupted mitochondrial morphology, as visualized by a mitochondrial fluorescent probe (MitoTracker Red CMXRos), and as expected based on the normalization of the MMP, ME treatment prevented mitochondrial disruption (Fig. 4i). Similar to the in vivo results, the in vitro results showed decreased protein levels of TOM20 and TFAM, as determined by IF and Western blotting, in HK-2 cells exposed to HR compared to control cells, whereas these effects were reversed by ME treatment (Fig. 4j–m). Corroborating the previously presented in vivo results, these in vitro results further illustrated that ME alleviates oxidative stress in HR-exposed HK-2 cells.

a, b Intracellular ROS in HK-2 cells of the three groups were detected using a FACSCalibur system (n = 5). c MDA and d SOD in the HK-2 cells were detected by commercial kits (n = 5). e Representative micrographs showing MitoSOX in the HK-2 cells and f quantitative analysis of mtROS levels (n = 4). g, h Flow cytometry was used to analyze JC-1 staining (n = 3). Representative micrographs showing i MitoTracker staining and j double-IF staining of TOM20 (red) and TFAM (green) in HK-2 cells. k Western blotting for TOM20 and TFAM proteins in HK-2 cells in the 3 groups and l, m quantitative analysis of protein expression (n = 4). Data represented by mean ± SEM. #P < 0.05, ##P < 0.01 vs the NC group; **P < 0.01 vs the HR group.
The protective effect of ME against renal IR injury was dependent on Nrf2 nuclear retention
The Nrf2 pathway is known to be pivotal in the regulation of antioxidant enzyme expression [37, 45, 46]. Using Western blotting, we verified that Nrf2 expression was upregulated in both the in vivo and in vitro models and that ME treatment significantly increased Nrf2 expression (Fig. 5a–d). To determine whether the protective effects of ME correlate with Nrf2 activation in vivo, we utilized the Nrf2 inhibitor ML385, which specifically binds to the DNA-binding domain of Nrf2 and reduces the expression of downstream target genes and Nrf2 itself [47], as previously described [48] (Fig. S2a). The inhibition of Nrf2 was validated by Western blot, and the results were consistent with those previously reported by other investigators [48, 49]; moreover, the Nrf2 downstream genes HO-1 and NQO1 were markedly downregulated (Fig. 5e–h). Compared to the mice treated with ME only, those treated with ME and ML385 showed significantly increased serum Cr and BUN levels, but renal function was not significantly altered in the sham+ML385 group compared with the sham group (Fig. 5i, j). Histologic injury, cell apoptosis, and nuclear 8-OHdG content in the kidneys following various treatments were then investigated. As shown in Fig. 5k, l, as expected, IR injury caused significant histologic injury, cell apoptosis, and oxidative stress, which were significantly decreased by ME treatment; notably, the inhibition of Nrf2 reversed these protective effects, and no significant differences were observed in the sham + ML385 group compared with the sham group.

Representative Western blot of Nrf2 total protein level in different groups a in vivo and b in vitro and c, d quantitative data are shown (n = 4). e Western blot of renal Nrf2, HO-1, and NQO1 protein expression after IR injury in different groups as indicated and f–h quantitative analyses (n = 5). Plasma levels of i Cr and j BUN in different groups as indicated (n = 5). k Representative micrographs show H&E staining (black arrows indicate tubular epithelial necrosis necrotic, yellow arrows indicate tubular lumen), TUNEL staining, immunostaining for cleaved caspase-3 and 8-OHdG staining of kidney sections in different groups and l quantitative analysis of tubular injury, TUNEL+ apoptotic cells, cleaved caspase-3+ area and 8-OHdG intensity (n = 5). m Western blot for cytoplasmic and nuclear protein level of Nrf2 in kidney tissues in different groups as indicated and n quantitative analysis (n = 3). o Representative micrographs show immunostaining of Nrf2 in the kidneys (arrowheads indicate Nrf2 positive cells) and p in HK-2 cells in different groups as indicated. Data represented by mean ± SEM. #P < 0.05, ##P < 0.01 vs the Sham group; *P < 0.05, **P < 0.01 vs the IR group; &P < 0.05, &&P < 0.01 vs the ME group.
To further corroborate these findings, we next determined the cytoplasmic and nuclear distribution of Nrf2 following IR injury. As shown in Fig. 5m, n, the nuclear level of Nrf2 protein was increased in the kidneys of the ME-treated group after 24 h of reperfusion. The level of Nrf2 in the cytoplasm was not altered, as confirmed by immunostaining assays. In the sham group, Nrf2 protein levels were low, and Nrf2 was predominantly localized in the renal cell cytoplasm. After IR, Nrf2 levels were slightly elevated, and a portion of Nrf2 was found to have translocated to the nucleus of renal tubular cells; this effect was significantly aggravated by ME treatment of mice after IR injury (Fig. 5o). Similar findings were also observed in HK-2 cells in vitro (Fig. 5p). Furthermore, CDDO (100 nmol/L), a specific Nrf2 activator, was applied in vitro as previously described [37]. As shown in Fig. S2b, c, CDDO and ME significantly elevated Nrf2 levels and decreased HK-2 cell apoptosis compared to HR alone, and the protective effect of ME was diminished by ML385 (10 μmol/L), consistent with the in vivo results. The renoprotective effects of ME were further confirmed with siRNA targeting Nrf2 in HR-exposed HK-2 cells (Fig. S2d–g). All the data indicated that ME regulates the cytoplasmic–nuclear translocation of Nrf2 and facilitates Nrf2 nuclear retention and that the protective effects of ME are dependent on the Nrf2 signaling pathway.
The Nrf2 nuclear export signal was blocked by the AMPK/GSK3β axis activated by ME
Because ME increased total Nrf2 levels and promoted its nuclear retention, we speculated that there might be upstream regulators that coordinately mediate these effects. The nuclear export of Nrf2 was previously shown to be primarily regulated by a GSK3β/Fyn-dependent mechanism, in which Fyn phosphorylates nuclear Nrf2 at Tyr568 and promotes Nrf2 export from the nucleus [50]. As a stress sensor and potential regulator of GSK3β activity, AMPK is activated under conditions of energy shortage and inflammation, can inhibit Nrf2 nuclear export, and can activate Nrf2 [51], thereby leading to the transactivation of downstream target genes to alleviate oxidative stress [52, 53] (Fig. 6a). In addition, the protective role of the AMPK/GSK3β-Nrf2 axis has been reported in lipopolysaccharide-induced acute lung injury [54]. Taken together, our results and those of other researchers indicate that AMPK/GSK3β might be a promising therapeutic target of ME.

a The possible mechanism diagram by which ME works. b Experimental design. Cells were treated with ME and specific inhibitors. Green arrows indicate the pretreatment of ME and red arrow indicates the expose of CC or DIF-3. c Western blot for protein levels of p-AMPK, AMPK, p-GSK3β, GSK3β in HR-exposed HK-2 cells and d, e quantitative analysis of protein expression (n = 4). f Phosphorylation of AMPK, GSK3β and nuclear protein level of Nrf2 in HR-exposed HK-2 cells treated with ME, CC or DIF-3, either alone or in combination and g–i quantitative analysis (n = 3). j Protein levels of p-AMPK, AMPK, p-GSK3β, GSK3β, and Keap1 in kidney tissues in different groups as indicated and k–m quantitative analysis of protein expression (n = 4). Representative images of n Oil Red O staining used to evaluate lipid accumulation in the kidney and o LC3/LAMP1 double-IF staining performed to evaluate autolysosomes. p Proposed binding mode of ME with AMPK (PDB ID: 5UFU). The overall view of ME in the binding pocket and q 2D presentation of interaction between ME and AMPK. r Graphic presentation shows the free binding energy of ME and metformin to AMPK. Data represented by mean ± SEM. #P < 0.05, ##P < 0.01 vs the NC, HR or Sham group; *P < 0.05, **P < 0.01 vs the IR, HR or ME group; &&P < 0.01 vs the ME group; ns indicates no significance vs the Sham or ME group.
As shown in Fig. 6b, the specific AMPK inhibitor CC (4 μmol/L) and an inhibitor of GSK3β phosphorylation, DIF-3 (10 μmol/L), were employed to block the phosphorylation of AMPK and GSK3β to examine the links between AMPK/GSK3β pathway activation and nuclear Nrf2 retention. First, we examined the levels of p-AMPK and p-GSK3β with and without ME pretreatment. The results indicated that the increases in p-AMPK and p-GSK3β were amplified by ME compared to HR alone (Fig. 6c–e). Moreover, treatment with both CC and DIF-3 reversed the ME-induced phosphorylation of GSK3β and resulted in decreased nuclear Nrf2 levels, while AMPK phosphorylation was not evidently altered by DIF-3 treatment (Fig. 6f–i). These results indicated that the AMPK/GSK3β-Nrf2 axis is concordantly activated in HR-exposed HK-2 cells. Consistent with the in vitro results, ME significantly increased the phosphorylation of AMPK and GSK3β in the kidney tissue of IR-induced mice, while the expression of Keap1 was not significantly altered (Fig. 6j–m). Because AMPK is upstream of several signaling pathways and has been reported to be a positive regulator of the fatty acid oxidation and autophagy pathways [55, 56], which also play important roles in renal IR injury, Oil Red O staining of lipids was performed to evaluate lipid accumulation, and immunofluorescence staining of LC3 and LAMP1 was conducted to detect autophagosomes and lysosomes, respectively. Consistent with a previous study [57], IR induced significant lipid droplet accumulation even though p-AMPK was adaptively increased, and autophagic flux increased, as indicated by the colocalization of LC3 and LAMP1. However, ME pretreatment decreased positive staining for lipid droplets after IR injury and promoted the formation of autolysosomes, which further verified that ME pretreatment activates AMPK, thereby protecting the kidney from IR injury (Fig. 6n–o). To investigate the molecular interaction between ME and AMPK, molecular docking simulation was performed. The docking results of ME and AMPK (PDB ID: 5UFU) are shown in Fig. 6p–r. ME formed hydrogen bonds with AMPK at ASP157 and ASN144, and van der Waals and other bonds were also formed. The free binding energy of ME with AMPK was −5.7 kcal/mol, which was lower than that of the AMPK activator metformin (−5.0 kcal/mol [58]), suggesting a high binding affinity of ME for AMPK (Fig. 6r) that further supports our above findings. These results demonstrated that the ME-induced nuclear retention of Nrf2, which is mediated through the activation of the AMPK/GSK3β signaling pathway, blocks Nrf2 nuclear export.
ME attenuated the progression of IR-induced renal fibrosis
Clinically, fatal renal ischemia–reperfusion injury is relatively rare, while nonfatal AKI is quite often a risk factor for subsequent renal fibrosis [59]. Therefore, we established a mouse model of moderate renal IR injury (25 min of ischemia rather than 35 min, which induces fatal injury) and evaluated the effects of ME on kidney tubulointerstitial fibrosis at 4 weeks post injury (Fig. 7a). As anticipated, all mice survived the 4 weeks of observation (Fig. 7b). ME significantly alleviated renal injury in the moderate renal IR injury model, as indicated by marked attenuation of the increases in Cr and BUN, and both values gradually returned to normal in all three groups within 30 days (Fig. 7c, d). In addition, the Western blot results revealed that α-SMA expression was significantly upregulated at 30 days following IR injury, while ME treatment remarkably suppressed this upregulation (Fig. 7e, f). At the indicated time points after renal IR injury (Fig. 7g, h), the gradual progression over time of renal fibrosis was significantly reduced by ME pretreatment. Taken together, the results indicated that ME hinders the progression of renal fibrosis over the 30 days following IR-induced AKI.

a Experimental design of nonfatal renal IR injury. Black arrows indicate the injection of vehicle and green arrows indicate the treatment of ME. b Thirty-day survival rate of mice exposed to moderate renal IR (n = 6). c, d Plasma levels of Cr and BUN of the mice in different groups at the indicated time points after renal IRI (Days 1, 3, 7, 15, and 30, n = 3, respectively). e Western blot for protein level of α-SMA in kidneys of mice assessed at 30 days after IR injury with or without ME treatment and f quantitative analysis of protein expression (n = 4). g Representative Masson's Trichrome stain of kidneys in the three groups of mice at the time indicated (Days 1, 7, 15, and 30, respectively) and h Quantitative analysis of renal fibrotic lesions in different groups as indicated (n = 5). Data represented by mean ± SEM. #P < 0.05, ##P < 0.01 vs the Sham group; *P < 0.05, **P < 0.01 vs the IR group. ns indicates no significance.
Discussion
This study examined the effect of ME on oxidative stress using an in vivo IR-induced AKI model. The generated evidence confirmed that ME pretreatment decreased the IR-induced increases in plasma Cr and BUN levels, alleviated tubular injury, and prolonged mouse survival. Consistent with the knowledge that IR injury results in nuclear 8-OHdG accumulation, which causes mitochondrial dysfunction [60], our results concurred that while IR evoked severe oxidative injury and abnormities in mitochondrial structure and function, ME pretreatment markedly reversed this damage. These investigations were repeated in vitro. HR exposure impaired cell viability, induced apoptosis, increased intracellular ROS production and contributed to significant mitochondrial injury in HK-2 cells; critically important, ME treatment markedly attenuated these detrimental effects. Collectively, ME mitigated oxidative stress-induced damage both in vivo and in vitro following renal IR injury.
As a critical transcription factor that regulates the antioxidant response, Nrf2 is usually activated and modulated by hypoxia, and its transcriptional activity is determined by its intranuclear accumulation [54, 61]. In this study, we showed that ME treatment, as well as treatment with the Nrf2 activator CDDO, significantly elevated Nrf2 levels, which were accompanied by the increased expression of HO-1 and NQO1. By using the specific Nrf2 inhibitor ML385 and si-Nrf2, we found that the renoprotective effect was dependent on the induction of Nrf2 by ME. Furthermore, we discovered that ME treatment markedly increased Nrf2 nuclear accumulation, whereas no significant changes in cytoplasmic Nrf2 levels were observed. Even though further investigation of the distribution and function of Nrf2 is required, the data indicate that ME-induced Nrf2 nuclear retention underlies the renoprotective effect of ME against IR injury.
To ascertain how ME facilitates Nrf2 nuclear retention, the AMPK/GSK3β axis, which regulates the cytoplasmic–nuclear translocation of Nrf2, was examined. Consistent with previous reports [61, 62], the levels of Nrf2 and phosphorylated MAPK were slightly elevated after IR. With ME pretreatment, the phosphorylation of AMPK and GSK3β significantly increased in both HR-exposed HK-2 cells and IR-induced kidney tissues; Oil Red O staining and LC3/LAMP1 staining further verified these results. In addition, by individually inhibiting AMPK and GSK3β, we confirmed that AMPK phosphorylation acts upstream of GSK3β to inhibit Nrf2 nuclear exclusion. To predict the interaction between ME and AMPK (PDB ID: 5UFU), a molecular docking simulation was performed, and ME was found to have high affinity for AMPK. Collectively, the data show that ME has an effective binding capacity for AMPK, and the ME-induced nuclear retention of Nrf2 is mediated by activation of the AMPK/GSK3β-Nrf2 signaling pathway.
Optimal candidates for the treatment of AKI must not only minimize acute injury but also prevent or mitigate the subsequent fibrosis that can progress to chronic kidney disease (CKD) [63]. It is recognized that mild to moderate renal IR injury, rather than fatal renal IR injury, is encountered more frequently in the clinical setting. Therefore, a moderate renal IR injury model was developed to investigate the effects of ME on kidney function and the development of fibrosis in the weeks following AKI. Interestingly, in addition to ameliorating the immediate effects of AKI, ME promoted the recovery of kidney function and reduced markers of subsequent fibrosis, suggesting that ME could have broader therapeutic potential for the clinical treatment of renal acute/chronic injury.
One limitation of this study was that Nrf2 nuclear retention could also be affected by diverse nuclear import signals. Previous studies have reported that the phosphorylation of Ser40 in the N-terminal Neh2 domain of Nrf2 facilitates Keap1/Nrf2 dissociation and Nrf2 nuclear translocation [64, 65]. In the present study, we investigated ME regulation of Nrf2 activity through the AMPK/GSK3β axis in HR-exposed HK-2 cells. However, the level of Nrf2 phosphorylation has not yet been investigated, and it remains unknown whether ME interacts directly with Nrf2 or targets Keap1, the negative regulator of Nrf2, and affects its activity.
In summary, as illustrated in Fig. 8, we demonstrated that ME modulates the AMPK/GSK3β axis and regulates the cytoplasmic–nuclear translocation of Nrf2, which results in Nrf2 nuclear retention and therefore enhances antioxidant target gene transcription to protect the kidney against oxidative damage. This study provides novel insight and in-depth evidence for the application of ME to prevent oxidative stress-associated diseases, especially renal IR injury.

ME binds to the binding pocket of AMPK with high affinity and activated the AMPK/GSK3β axis, which results in Nrf2 nuclear retention and therefore enhances antioxidant target gene transcription to protect the kidney against oxidative damage.
References
Levey AS, James MT. Acute kidney injury. Ann Intern Med. 2017;167:Itc66–itc80.
Funk JA, Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Ren Physiol. 2012;302:F853–864.
Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–210.
Michelsen J, Cordtz J, Liboriussen L, Behzadi MT, Ibsen M, Damholt MB, et al. Prevention of rhabdomyolysis-induced acute kidney injury - A DASAIM/DSIT clinical practice guideline. Acta Anaesthesiol Scand. 2019;63:576–86.
Yu H, ** F, Liu D, Shu G, Wang X, Qi J, et al. ROS-responsive nano-drug delivery system combining mitochondria-targeting ceria nanoparticles with atorvastatin for acute kidney injury. Theranostics. 2020;10:2342–57.
Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26.
Diao C, Chen Z, Qiu T, Liu H, Yang Y, Liu X, et al. Inhibition of PRMT5 attenuates oxidative stress-induced pyroptosis via activation of the Nrf2/HO-1 signal pathway in a mouse model of renal ischemia-reperfusion injury. Oxid Med Cell Longev. 2019;2019:2345658.
Yu D, Liu X, Zhang G, Ming Z, Wang T. Isoliquiritigenin inhibits cigarette smoke-induced COPD by attenuating inflammation and oxidative stress via the regulation of the Nrf2 and NF-κB signaling pathways. Front Pharmacol. 2018;9:1001.
Tu W, Wang H, Li S, Liu Q, Sha H. The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 2019;10:637–51.
Liu H, Lin Z, Ma Y. Suppression of Fpr2 expression protects against endotoxin-induced acute lung injury by interacting with Nrf2-regulated TAK1 activation. Biomed Pharmacother. 2020;125:109943.
Liu Q, Lv H, Wen Z, Ci X, Peng L. Isoliquiritigenin activates nuclear factor erythroid-2 related factor 2 to suppress the NOD-like receptor protein 3 inflammasome and inhibits the NF-κB pathway in macrophages and in acute lung injury. Front Immunol. 2017;8:1518.
Ling L, Alattar A, Tan Z, Shah FA, Ali T, Alshaman R, et al. A potent antioxidant endogenous neurohormone melatonin, rescued MCAO by attenuating oxidative stress-associated neuroinflammation. Front Pharmacol. 2020;11:1220.
Tang C, Hong J, Hu C, Huang C, Gao J, Huang J, et al. Palmatine protects against cerebral ischemia/reperfusion injury by activation of the AMPK/Nrf2 pathway. Oxid Med Cell Longev. 2021;2021:6660193.
Duan J, Guan Y, Mu F, Guo C, Zhang E, Yin Y, et al. Protective effect of butin against ischemia/reperfusion-induced myocardial injury in diabetic mice: involvement of the AMPK/GSK-3β/Nrf2 signaling pathway. Sci Rep. 2017;7:41491.
Wu Y, Qiu G, Zhang H, Zhu L, Cheng G, Wang Y, et al. Dexmedetomidine alleviates hepatic ischaemia-reperfusion injury via the PI3K/AKT/Nrf2-NLRP3 pathway. J Cell Mol Med. 2021;25:9983–94.
Zhang J, Zhang J, Ni H, Wang Y, Katwal G, Zhao Y, et al. Downregulation of XBP1 protects kidney against ischemia-reperfusion injury via suppressing HRD1-mediated NRF2 ubiquitylation. Cell Death Discov. 2021;7:44.
Tan KH, Nishida R. Methyl eugenol: its occurrence, distribution, and role in nature, especially in relation to insect behavior and pollination. J Insect Sci. 2012;12:56.
Smith RL, Adams TB, Doull J, Feron VJ, Goodman JI, Marnett LJ, et al. Safety assessment of allylalkoxybenzene derivatives used as flavouring substances – methyl eugenol and estragole. Food Chem Toxicol. 2002;40:851–70.
Yin L, Sun ZH, Ren Q, Su X, Zhang DL. Methyl eugenol induces potent anticancer effects in RB355 human retinoblastoma cells by inducing autophagy, cell cycle arrest and inhibition of PI3K/mTOR/Akt signalling pathway. J Buon. 2018;23:1174–8.
Groh IAM, Esselen M. Methyleugenol and selected oxidative metabolites affect DNA-damage signalling pathways and induce apoptosis in human colon tumour HT29 cells. Food Chem Toxicol. 2017;108:267–75.
Saleh H, El-Shorbagy HM. Mechanism underlying methyl eugenol attenuation of intestinal ischemia/reperfusion injury. Appl Physiol Nutr Metab. 2017;42:1097–105.
Choi YK, Cho GS, Hwang S, Kim BW, Lim JH, Lee JC, et al. Methyleugenol reduces cerebral ischemic injury by suppression of oxidative injury and inflammation. Free Radic Res. 2010;44:925–35.
Nabila B, Piras A, Fouzia B, Falconieri D, Kheira G, Fedoul FF, et al. Chemical composition and antibacterial activity of the essential oil of Laurus nobilis leaves. Nat Prod Res. 2022;36:989–93.
Wang ZJ, Tabakoff B, Levinson SR, Heinbockel T. Inhibition of Nav1.7 channels by methyl eugenol as a mechanism underlying its antinociceptive and anesthetic actions. Acta Pharmacol Sin. 2015;36:791–9.
Yano S, Suzuki Y, Yuzurihara M, Kase Y, Takeda S, Watanabe S, et al. Antinociceptive effect of methyleugenol on formalin-induced hyperalgesia in mice. Eur J Pharmacol. 2006;553:1–3.
Liu YM, Fan HR, Deng SN, Zhu TL, Yan YH, Ge WH, et al. Methyleugenol potentiates central amygdala GABAergic inhibition and reduces anxiety. J Pharmacol Exp Ther. 2019;368:1–10.
Wang M, Zhang J, Zhang J, Sun K, Li Q, Kuang B, et al. Methyl eugenol attenuates liver ischemia reperfusion injury via activating PI3K/Akt signaling. Int Immunopharmacol. 2021;99:108023.
Zhou J, Ma X, Cui Y, Song Y, Yao L, Liu Y, et al. Methyleugenol protects against t-BHP-triggered oxidative injury by induction of Nrf2 dependent on AMPK/GSK3beta and ERK activation. J Pharmacol Sci. 2017;135:55–63.
Alminderej F, Bakari S, Almundarij TI, Snoussi M, Aouadi K, Kadri A. Antioxidant activities of a new chemotype of Piper cubeba L. fruit essential oil (Methyleugenol/Eugenol): in silico molecular docking and ADMET studies. Plants. 2020;9:1534.
Wang R, Wang D, Wang H, Wang T, Weng Y, Zhang Y, et al. Therapeutic targeting of Nrf2 signaling by maggot extracts ameliorates inflammation-associated intestinal fibrosis in chronic DSS-induced colitis. Front Immunol. 2021;12:670159.
Ascon M, Ascon DB, Liu M, Cheadle C, Sarkar C, Racusen L, et al. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 2009;75:526–35.
Ao L, Fang S, Zhang K, Gao Y, Cui J, Jia W, et al. Sequence-dependent synergistic effect of aumolertinib-pemetrexed combined therapy on EGFR-mutant non-small-cell lung carcinoma with pre-clinical and clinical evidence. J Exp Clin Cancer Res. 2022;41:163.
Yao L, Cheng S, Yang J, **ang F, Zhou Z, Zhang Q, et al. Metabolomics reveals the intervention effect of Zhuang medicine Longzuantongbi granules on a collagen-induced arthritis rat model by using UPLC-MS/MS. J Ethnopharmacol. 2022;294:115325.
Li Y, Xu B, Yang J, Wang L, Tan X, Hu X, et al. Liraglutide protects against lethal renal ischemia-reperfusion injury by inhibiting high-mobility group box 1 nuclear-cytoplasmic translocation and release. Pharmacol Res. 2021;173:105867.
Tian R, Wang P, Huang L, Li C, Lu Z, Lu Z, et al. Sanqi oral solution ameliorates renal ischemia/reperfusion injury via reducing apoptosis and enhancing autophagy: involvement of ERK/mTOR pathways. Front Pharmacol. 2020;11:537147.
Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. 2021;11:1845–63.
Nezu M, Souma T, Yu L, Suzuki T, Saigusa D, Ito S, et al. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017;91:387–401.
Han X, Zhao S, Song H, Xu T, Fang Q, Hu G, et al. Kaempferol alleviates LD-mitochondrial damage by promoting autophagy: implications in Parkinson’s disease. Redox Biol. 2021;41:101911.
Gong LJ, Wang XY, Gu WY, Wu X. Pinocembrin ameliorates intermittent hypoxia-induced neuroinflammation through BNIP3-dependent mitophagy in a murine model of sleep apnea. J Neuroinflammation. 2020;17:337.
Zhang J, Zhao T, Wang C, Meng Q, Huo X, Wang C, et al. Catalpol-induced AMPK activation alleviates cisplatin-induced nephrotoxicity through the mitochondrial-dependent pathway without compromising its anticancer properties. Oxid Med Cell Longev. 2021;2021:7467156.
Deng Y, Ren H, Ye X, **a L, Liu M, Liu Y, et al. Integrated phytochemical analysis based on UPLC-Q-TOF-MS/MS, network pharmacology, and experiment verification to explore the potential mechanism of Platycodon grandiflorum for chronic bronchitis. Front Pharmacol. 2020;11:564131.
Liang Y, Liang B, Chen W, Wu XR, Liu-Huo WS, Zhao LZ. Potential mechanism of dingji fumai decoction against atrial fibrillation based on network pharmacology, molecular docking, and experimental verification integration strategy. Front Cardiovasc Med. 2021;8:712398.
Jung E, Kang WS, Jo K, Kim J. Ethyl pyruvate prevents renal damage induced by methylglyoxal-derived advanced glycation end products. J Diabetes Res. 2019;2019:4058280.
Li H, **ao Y, Tang L, Zhong F, Huang G, Xu JM, et al. Adipocyte fatty acid-binding protein promotes palmitate-induced mitochondrial dysfunction and apoptosis in macrophages. Front Immunol. 2018;9:81.
Sajadimajd S, Khazaei M. Oxidative stress and cancer: the role of Nrf2. Curr Cancer Drug Targets. 2018;18:538–57.
Liu M, Grigoryev DN, Crow MT, Haas M, Yamamoto M, Reddy SP, et al. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009;76:277–85.
Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem Biol. 2016;11:3214–25.
Feng LX, Zhao F, Liu Q, Peng JC, Duan XJ, Yan P, et al. Role of Nrf2 in lipopolysaccharide-induced acute kidney injury: protection by human umbilical cord blood mononuclear cells. Oxid Med Cell Longev. 2020;2020:6123459.
Deng S, Essandoh K, Wang X, Li Y, Huang W, Chen J, et al. Tsg101 positively regulates P62-Keap1-Nrf2 pathway to protect hearts against oxidative damage. Redox Biol. 2020;32:101453.
Jain AK, Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282:16502–10.
Wu YX, Wang YY, Gao ZQ, Chen D, Liu G, Wan BB, et al. Ethyl ferulate protects against lipopolysaccharide-induced acute lung injury by activating AMPK/Nrf2 signaling pathway. Acta Pharmacol Sin. 2021;42:2069–81.
Liu XM, Peyton KJ, Shebib AR, Wang H, Korthuis RJ, Durante W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am J Physiol Heart Circ Physiol. 2011;300:H84–93.
Matzinger M, Fischhuber K, Pölöske D, Mechtler K, Heiss EH. AMPK leads to phosphorylation of the transcription factor Nrf2, tuning transactivation of selected target genes. Redox Biol. 2020;29:101393.
Lv H, Liu Q, Wen Z, Feng H, Deng X, Ci X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3beta-Nrf2 signal axis. Redox Biol. 2017;12:311–24.
Li X, Zhuge Z, Carvalho L, Braga VA, Lucena RB, Li S, et al. Inorganic nitrate and nitrite ameliorate kidney fibrosis by restoring lipid metabolism via dual regulation of AMP-activated protein kinase and the AKT-PGC1α pathway. Redox Biol. 2022;51:102266.
Zhu M, Shen W, Li J, Jia N, **ong Y, Miao J, et al. AMPK activator O304 protects against kidney aging through promoting energy metabolism and autophagy. Front Pharmacol. 2022;13:836496.
Ma H, Guo X, Cui S, Wu Y, Zhang Y, Shen X, et al. Dephosphorylation of AMP-activated protein kinase exacerbates ischemia/reperfusion-induced acute kidney injury via mitochondrial dysfunction. Kidney Int. 2022;101:315–30.
Li J, Gui Y, Ren J, Liu X, Feng Y, Zeng Z, et al. Metformin protects against cisplatin-induced tubular cell apoptosis and acute kidney injury via AMPKα-regulated autophagy induction. Sci Rep. 2016;6:23975.
Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20:223–8.
Hu Q, Ren J, Li G, Wu J, Wu X, Wang G, et al. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018;9:403.
Li Z, Zhu J, Wan Z, Li G, Chen L, Guo Y. Theaflavin ameliorates renal ischemia/reperfusion injury by activating the Nrf2 signalling pathway in vivo and in vitro. Biomed Pharmacother. 2020;134:111097.
Pan JS, Huang L, Belousova T, Lu L, Yang Y, Reddel R, et al. Stanniocalcin-1 inhibits renal ischemia/reperfusion injury via an AMP-activated protein kinase-dependent pathway. J Am Soc Nephrol. 2015;26:364–78.
Li F, Bahnson EM, Wilder J, Siletzky R, Hagaman J, Nickekeit V, et al. Oral high dose vitamin B12 decreases renal superoxide and post-ischemia/reperfusion injury in mice. Redox Biol. 2020;32:101504.
Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–82.
Zhuang Y, Wu H, Wang X, He J, He S, Yin Y. Resveratrol attenuates oxidative stress-induced intestinal barrier injury through PI3K/Akt-mediated Nrf2 signaling pathway. Oxid Med Cell Longev. 2019;2019:7591840.
Acknowledgements
The present study was supported by grants from the National Natural Science Foundation of China (grant nos. 82170772, 81873623 and 81570678), the Major State Basic Research Development Program of China (grant no. 2013CB530803), the Clinical Research Physician Program of Tongji Medical College, HUST and the Non-Profit Central Research Institute Fund of Chinese Academy of Medical Sciences (grant no. 2018PT32018). The authors would like to express their gratitude to EditSprings (https://www.editsprings.cn/) for expert linguistic services.
Author information
Authors and Affiliations
Contributions
BCK and ZHW performed the experiments, analyzed the data, and wrote the draft. SHH, MQW, and KLS performed the experiments and assisted in data interpretation. JZ, JSZ, and HQN helped with the acquisition of samples and data collection. NQG designed the study and edited the manuscript. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary Information
Rights and permissions
About this article

Cite this article
Kuang, Bc., Wang, Zh., Hou, Sh. et al. Methyl eugenol protects the kidney from oxidative damage in mice by blocking the Nrf2 nuclear export signal through activation of the AMPK/GSK3β axis. Acta Pharmacol Sin 44, 367–380 (2023). https://doi.org/10.1038/s41401-022-00942-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00942-2
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Targeting cardiovascular risk factors with eugenol: an anti-inflammatory perspective
Inflammopharmacology (2023)