
Abstract
Epithelial tissue homeostasis is closely associated with the self-renewal and differentiation behaviors of epithelial stem cells (ESCs). p63, a well-known marker of ESCs, is an indispensable factor for their biological activities during epithelial development. The diversity of p63 isoforms expressed in distinct tissues allows this transcription factor to have a wide array of effects. p63 coordinates the transcription of genes involved in cell survival, stem cell self-renewal, migration, differentiation, and epithelial-to-mesenchymal transition. Through the regulation of these biological processes, p63 contributes to, not only normal epithelial development, but also epithelium-derived cancer pathogenesis. In this review, we provide an overview of the role of p63 in epithelial stemness regulation, including self-renewal, differentiation, proliferation, and senescence. We describe the differential expression of TAp63 and ΔNp63 isoforms and their distinct functional activities in normal epithelial tissues and in epithelium-derived tumors. Furthermore, we summarize the signaling cascades modulating the TAp63 and ΔNp63 isoforms as well as their downstream pathways in stemness regulation.
Similar content being viewed by others


Introduction
Structural features and expression patterns of p63
p63 is the most ancient member belonging to the p53 family together with p73 [1,2,3,4] (Fig. 1A). All members are involved in essential biological functions, including cell death [5, 6], tumor suppression [7,8,9,10], and cell differentiation [11,12,13,14]. Its encoding gene TP63 generates two main classes of isoforms by the usage of alternative promoters. TAp63 transcripts are produced from the promoter upstream of exon 1 and transcribe for protein isoforms that contain an N-terminus acidic transactivation domain (transcriptional activation domain 1, TA1), homologous to the one of p53 (Fig. 1B). The usage of an alternative promoter located within the intron downstream of exon 3 generates transcripts encoding ΔNp63 isoforms that lack the canonical N-terminal TA domain 1 (TA1*). Furthermore, the TP63 gene is expressed as, at least, three alternatively spliced C-terminal isoforms (α, β, γ) [15]. The full-length p63α proteins possess a C-terminus sterile alpha motif (SAM) domain, a protein-protein interaction domain, flanked by a second transactivation domain (TA2) and a transcription inhibitory domain (TID), which can auto-inhibit the transcriptional activity of the TA isoforms. All of the isoforms share some functional domains including the DNA-binding domain (DBD) and the oligomerization domain (OD). Altogether, the TP63 gene expresses at least six different p63 protein isoforms (TAp63α, TAp63β, TAp63γ, ΔNp63α, ΔNp63β and ΔNp63γ). Minor alpha-helix changes produce profound functional differences; accordingly, these six isoforms display distinct functions. The identification of two new C-terminus variants, p63δ and ε, has expanded the isoforms number from 6 to 10 [16, 17]. Moreover, two additional N-terminal short splicing isoforms lacking exon 4 have been found in invasive breast carcinomas (d4TAp63 and ΔNp73L) [18].

A Timeline of important discoveries in the research field of p63 in epithelial tissues. B Schematic representation of the alternative promoters generating the TAp63 and ΔNp63 isoforms along with structural features and domains of the p63 protein variants. p63 protein are shown: TA transactivating domain, DBD DNA-binding domain, OD oligomerization domain, SAM sterile alpha motif, TID trans-inhibitory domain. C The differential expression pattern of TAp63 and ΔNp63 variants (α, β, and γ) in different normal tissues (Data source, GTex portal).
The TAp63 isoforms are generally expressed at low basal levels in epithelial cells, while their expression can be induced in response to DNA damage or other cellular stresses. ΔNp63 isoforms are instead constitutively expressed in the basal compartment of the epidermis and in other epithelial tissues [19,20,21]. Unlike TAp63, ΔNp63 expression is downregulated in response to genotoxic stress. TAp63 binds canonical p53-responsive elements (RE), thus activating the transcription of several p53 target genes, and therefore acting as a tumor suppressor through the regulation of cell-cycle arrest and apoptosis [16, 22,23,24,25,26]. Conversely, the ΔNp63 isoform acts as an oncogene [27, 28]. The ΔNp63 isoforms are mainly involved in maintaining the self-renewing capacity of progenitor cells in different epithelia [29,69].
Mammary gland
The mammary gland consists of a bilayered epithelium that is composed by an inner luminal layer of secretory epithelial cells, and an outer basal compartment of myoepithelial cells, including rare stem cells [70] (Fig. 3A). During embryonic development, sustained p63 expression in multipotent progenitors facilitates the differentiation of unipotent basal cell lineage in mammary gland [71]. TP63 null mice genetically deleted for all the isoforms, display complete lack of mammary glands [20]. Similarly, humans harboring germline p63 mutations exhibit mammary hypoplasia [72]. Subsequent studies indicated that ΔNp63 is the predominant variant governing mammary glands morphogenesis. Romano and collaborators reported that selective genetic ablation of ΔNp63 recapitulates the mammary gland defective phenotype observed in the p63 global knockout mice [73]. In the adult mammary gland, ΔNp63 and TAp63 exhibit segregated expression in mammary stem cells and progenitor cells [74, 75]. ΔNp63, especially the ΔNp63α isoform, is mainly expressed in the basal cell layer that is composed of myoepithelial and stem/progenitor cells, while its expression is maintained very low in luminal cells [76, 77]. TAp63 protein is instead expressed in luminal cells [78]. The expression of ΔNp63 in mammary progenitor cells serves to maintain their undifferentiated state and quiescence and therefore to promote stem cell activity [31, 74, 79,80,81]. In addition, it has been reported that ΔNp63 can reprogram adult luminal into basal cells by promoting an intermediate multipotent-like state, thus supporting the concept of reversible plasticity in epithelial cell fate [71, 82]. ΔNp63 is expressed in the adult mammary gland during all the physiological stages, virgin, pregnant, lactation, and post-lactation involution states [75]. On the other hand, the mammary glands of p63+/− mice appear normal in the virgin, pregnant and lactating states.

A A schematic model depicting the structure of the mammary gland epithelium, composed by basal stem cells, basal myoepithelial cells, and luminal cells. B The model shows the distribution of basal stem cells, luminal cells, and neuroendocrine cells in the prostate gland epithelium. C Schematic representation of the cornea. LESCs produce daughter TA cells that migrate towards the apical layer of the central cornea, and, eventually, become terminally differentiated (arrowed). ΔNp63 is the dominant p63 isoform expressed in LESCs and its expression decreases from limbus to the central cornea. Ep epithelium, BL Bowman’s layer, St stroma.
Prostate gland
Normal prostate epithelium contains three major epithelial cell types, basal cells (including stem and TA cells), luminal cells, and neuroendocrine cells [83] (Fig. 3B). In the adult, prostate stem cells can give rise to all three lineages of prostate epithelial cells [84]. p63 has been shown to play an essential role in normal stem cell function during prostate development. At birth, mice lacking p63 manifest prostate bud agenesis and urothelial abnormalities [4, 45]. Prostaspheres derived from p63-expressing basal cells efficiently form organoids expressing the differentiation marker cytokeratin 18 (CK18) and a functional androgen receptor [85]. The prostate agenesis indicates that p63 is required for the formation of prostate stem cells residing in the basal layer of the primitive urogenital sinus epithelium. In the absence of p63, the epithelial lining of the urethra indeed shows lack of stratification and complete absence of basal cells [4]. p63-null blastocyst complementation experiments have demonstrated that p63-positive basal cells are necessary for the development of p63-negative luminal cells [86,87,88]. Within the prostate epithelium, ΔNp63 is selectively expressed in the basal cell layer, while it is absent in the luminal and neuroendocrine cells [89]. In an in vitro study of prostate cell spheroid, p63-expressing cells are located in the outer layer of the spheres and exhibit self-renewal potential, while cells located in the center display a differentiated phenotype [32]. Correspondingly, ΔNp63 isoform is the most abundant in both cortical and medullary TESCs [29, 105].
Lung
Club cells are regional progenitor cells involved in the repair of the bronchiolar epithelium in response to lung damage. They give rise to epithelial alveolar type 2 cells (AT2s) and alveolar type 1 cells (AT1s) during the repair process. In addition, the distal airway stem/progenitor cells (DASCs) expressing p63 as well as keratin-5 (K5) are located in the airway basal epithelium and are responsible for lung regeneration after lung injury [106, 107]. In addition, induced p63+/KRT5+ lung epithelial cells can give rise to alveoli, indicating the stemness potential [108, 121]. As enriched ΔNp63 expression indeed confers a basal phenotype in purified luminal cells, while Notch activation specifies a luminal cell fate [82]. Sierra Kent and colleagues demonstrated that ΔNp63α inhibits cell-cycle progression and promotes cellular quiescence of mammary epithelial cells, predominantly through Notch3 regulation, thus providing novel mechanistic insights into the maintenance of mammary cellular quiescence by the p63/Notch axis [122]. In summary, the reciprocal antagonistic interactions of p63 isoforms and Notch family members determine the proper output of stemness regulation.
The WNT/β-catenin pathway
WNT/β-catenin pathway is a highly conserved pathway involved in the maintenance of the progenitor cell population in the skin, limbus, intestine, mammary gland, and other epithelial tissues [123,124,125,126]. It’s initiated by the interaction of the WNT ligands with two cell-surface receptors Frizzled (Fzd) and an Lrp (Fig. 4A, middle panel). Then, induced Disheveled (DVL) signaling cascade inhibits the β-catenin destruction complex in the cytoplasm, where β-catenin is constantly phosphorylated by the glycogen synthase kinase-3β (GSK-3β) and, subsequently, rapidly ubiquitinated and targeted for proteasomal degradation. Finally, activated β-catenin translocates into the nucleus and regulates the expression of numerous target genes [127].
An investigation in normal mammary stem cell (MaSC) demonstrateed that ΔNp63 maintains their self-renewal ability through directly upregulating the expression of Fzd7 [31]. In turn, the WNT/β-catenin signaling could also be the upstream regulator of ΔNp63, because of the presence of a putative β-catenin-RE within its promoter region. Limb-Bud and Heart (LBH), a transcription co-factor in the WNT pathway, upregulates the transcription of ΔNp63α but downregulates TAp63α expression. By this way, LBH enhances the replicative potential as well as stemness of mammary basal ESCs and inhibits the transcription of luminal differentiation genes, such as Esr1/ERα (estrogen receptor α) [81].
The Hedgehog signaling pathway
The Hedgehog (Hh) signaling is triggered by the binding of one of the three secreted ligands, Sonic Hedgehog (Shh), Indian Hedgehog (Ihh) or Desert Hedgehog (Dhh) to the Patched1 (PTCH1) receptor. Upon binding, PTCH1 releases its inhibition on Smoothened (SMO), that then transduces the Hh signal across the cell membrane, activating the Glioma-associated oncogene homologs family (GLI1, GLI2, and GLI3) of transcription factors [128].
The Hh signaling pathway is a crucial driver of ESC self-renewal [129]. Reciprocal interactions between p63 and the Hh signaling components regulate quiescence and activation of ESCs (Fig. 4A, right panel). p63 contributes to the stemness phenotype through its ability to bind to the promoters of Hh components, assigning positive and negative regulation to Ihh by TAp63 and ΔNp63 isoforms, respectively [75]. Conversely, Ihh-induced GLI3 upregulates TAp63 expression but reduces the usage of the ΔNp63 promoter, through which the Hh signaling plays an important role in the elaboration and differentiation of mammary progenitors and deceased clonogenicity of MaSCs in vitro [75]. During keratinocyte differentiation, an active Hh signaling pathway is observed in proliferating cells, in which it triggers the expression of ΔNp63α and further activates SUFU transcription. In turn, increased SUFU inhibits GLI-targeted genes, including ΔNp63α, and then restrains proliferation and promotes differentiation of keratinocytes [130]. On the other hand, constitutively activated Hh signaling in palatal epithelium results in the persistence of p63 signaling and p63-targeted genes in medial edge epithelium, which directly enhancing the epithelial progenitor cell-associated genes [131]. In epididymal, ARL13B is responsible for epithelium integrity and tissue regeneration. In its absence, the Hh signaling is impaired and basal cell markers, such as p63 are lost [132].
Noncoding RNAs
The reciprocal microRNA (miRNA)-p63 interaction is a vital factor that modulates p63 function [133], and miRNA plays a crucial role in the modulation of stem cell senescence and proliferation [134,135,136,137] (Fig. 4B). miR-203 is a key molecule controlling the proliferative potential and inducing cell-cycle arrest of epithelial precursor cells during epithelial development. Mechanistically, it inhibits p63 activity through targeting human and mouse p63 3’-UTR, especially ΔNp63 [44, 55, 138]. Consistently, increased expression of miR-203 by oleic acid (OA), an unsaturated free fatty acid (FFA), enhances the differentiation of keratinocytes through reducing p63 expression [139]. Besides, the proliferation of human primary keratinocytes in vitro coincides with the increased miR-130b, whose overexpression will induce cell senescence through inhibition of ΔNp63 expression. However, ΔNp63α, in turn, inhibits miR-130b expression in primary keratinocytes, thus promoting their growth [140]. ΔNp63α-inhibited miR-138, miR-181a, miR-181b, and miR-130b participate in the growth-promoting of epidermal proliferating cells and keratinocyte senescence [140]. Furthermore, miR-574-3p and miR-720 function as regulator of keratinocyte differentiation by linking iASPP, an inhibitory member of ASPP (apoptosis stimulating protein of p53) family, and silencing of iASPP in keratinocytes accelerates the differentiation pathway through indirectly increasing protein level of ΔNp63 [141].
Several long noncoding RNAs (lncRNAs) have been implicated in keratinocyte biology, through either the maintenance of the undifferentiated state of the progenitor cells or the induction of epidermal differentiation [142,143,144]. In addition, lncRNAs are crucially involved in the development and progression of several human epithelial cancers [142, 145,146,155].
Cell adhesion molecules
Interestingly, p63 has also been reported to participate in the regulation of anoikis by modulating cell adhesion [156], which is essential for progenitor cell maintenance. The interaction between integrins and receptors for growth factors and cytokines creates a complex web of signaling pathways that impact ESCs niche. As depicted in Fig. 4D, deficiency of p63 results in the downregulation of genes associated with cell adhesion, such as ITGB4, which encodes β4 integrin (also known as CD29). ΔNp63α specifically upregulates β4 integrin, leading to cell detachment and anoikis in mammary epithelial cells and keratinocytes. Consistently, β4 integrin partially protects against p63 loss-induced anoikis [156]. Furthermore, the knockdown of p63 (TAp63γ and ΔNp63α) impairs the expression of cell adhesion molecules, such as human ITGA3, which encodes the integrin subunit α3 that combines with integrin β1 to form the α3β1 dimer, by which p63 is thought to facilitate the anchoring of ESCs to their niche [156, 157].
Other signaling
There are many other signaling molecules that are also involved in p63-mediated regulation of epithelial stem cell stemness (Fig. 4E). Metabolic regulation [158, 159], and reactive oxygen species (ROS) [160] are essential in all biological functions. The epidermis is subjected to significant oxidative stress due to the production of ROS during aerobic metabolism [161, 162]. REDD1 (regulated in development and DNA damage responses 1), a regulator in DNA damage response (DRR), appears to function as a regulator of cellular ROS. p63 expression, especially TAp63γ and ΔNp63α, coincides with that of REDD1 in undifferentiated human primary keratinocytes. The downregulation of anyone of this two isoforms dramatically restricts REDD1 expression and promotes the differentiation program through increased ROS levels, suggesting that the p63-REDD1-ROS axis is responsible for preserving the undifferentiated state [163]. p63 isoforms are also known as vital regulators of energy metabolism. Hamanaka and Mutlu proposed that p63 affects cellular glycolytic rate of keratinocytes by regulating the transcriptional expression of metabolic enzyme phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3), thereby resulting in a proliferation/differentiation switch [164, 165]. Smirnov and colleagues demonstrated that Zinc finger protein 185 (ZNF185) contributes to maintain epithelial integrity and epithelial differentiation potential through the specific binding of p63 to its enhancer region in keratinocytes [166]. In the mouse epidermis, the activation of protein kinase p38α phosphorylates and destabilizes ΔNp63α. Conversely, p38α deficiency stabilizes ΔNp63α and promote keratinocyte colony formation [167]. Steap4 (Six-Transmembrane Epithelial Antigen of Prostate 4)-ΔNp63 axis induced by interleukin-17 (IL-17) is positively related to the TRAF4 (TNF Receptor-Associated Factor 4)-ERK5 axis through p63-mediated TRAF4 expression, by which IL-17 favors keratinocyte proliferation [168]. Gatti et al. found that Senataxin (SETX), an RNA/DNA helicase, acts as a key molecule of skin homeostasis by regulating a series of genes involved in the early step of keratinocyte differentiation program together with ΔNp63 [169]. Additionally, the RING finger E3 ubiquitin ligase PIR2 (also known as Rnf144b) has been identified as a target of ΔNp63α, which in turn induces the degradation of ΔNp63α, impairing keratinocyte proliferation and differentiation [170]. In the mammary epithelium, ΔNp63 activates canonical bone morphogenic proteins (BMPs) signaling by inducing the transcription of BMP7, which then fosters the stem cell phenotype of mammary progenitors [171]. IκB kinase (IKK), one of the three main subunits of NF-κB complex, consists of two catalytic subunits, IKKα and IKKβ, as well as a regulatory subunit, IKKγ. TAp63 acts as the upstream regulator of IKKα through direct transactivation and also indirectly through Ets-1(E26 transformation-specific-1). In contrast, ΔNp63 only indirectly activates IKKα through GATA3 in keratinocytes during epithelial development [172].
p63 and cancer stem cell in epithelial tumor
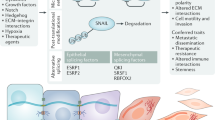
TP63 is rarely mutated in human cancers but its expression and activity are often increased [173,174,175] (Fig. 5A). The human TP63 gene exhibits increased expression in 88% squamous carcinomas and 42% of large cell carcinomas and lung adenocarcinomas [176]. Among p63 isoforms, ΔNp63 is the predominant expressed isoform in various epithelial tumors and its alterations positively correlate with increased metastatic potential and therapy resistance [174, 177, 178]. Emerging evidence suggests that several tumors are maintained by tumor-initiating cells (TICs), also referred as cancer stem cells (CSCs), possessing stem-like properties [179, 180]. CSCs are considered as the cellular “seeds” of tumor recurrence, metastasis and therapy resistance [181,182,183,184]. Like in normal tissues, self-renewal and differentiation of rare CSCs are essential and responsible for the growth of the tumor mass [185, 186]. Growing evidence suggests that abnormal signaling within pathways that regulate the stemness properties of normal stem cells may contribute to foster survival of CSCs. Most importantly, increasing evidence is pointing at the regulation of CSCs by p63 in various epithelial cancers [87, 174].

A Distinct expression levels of the p63 protein in different tumor tissues (The Human Protein Atlas, https://www.proteinatlas.org/ENSG00000073282-TP63/pathology). B The ΔNp63 isoform plays a crucial role in regulating various signaling pathways implicated in CSC stemness regulation, particularly in breast and SCC cancers. C p63 regulates ALDH1A1 in the context of prostate cancer, in which p63 affects CSC stemness. D TAp63 modulates the expression of specific Hippo pathway components to regulate the stemness of CSCs in breast cancer.
ΔNp63 is enriched in breast cancer CSCs and promotes their colony formation ability [31] (Fig. 5B). Memmi and colleagues reported that ΔNp63 is preferentially expressed in CSCs from transgenic mice with mammary gland targeted ErbB2 (MMTV-ErB2) tumors and is required to maintain their self-renewal potential. Mechanistically, ΔNp63 directly binds to the gene regulatory regions of Shh, Gli2, and Ptch1 genes, regulating their expression and ultimately contributing to pathway activation [187]. Additionally, p63 inactivation in ErbB2 progenitors reduces the transcription of genes for encoding components of the Shh signaling, to which mainly ΔNp63 isoform contributes, thus attenuating mammary CSC self-renewal [187]. Similar to the normal counterparts, the tumor-initiating activity of CSCs of basal breast cancer is governed by ΔNp63-dependent transcription of Fzd7 [31]. A clinical study showed that ΔNp63 was enriched in a cell subpopulation from triple negative breast cancer patients. Furthermore, ΔNp63α positively correlates with Notch1 expression by directly binding to its promoter, and increases the fraction of CSC-like cells in a luminal breast cancer cell line [188]. The interaction between ΔNp63 and BMPs, especially BMP7, is able to commit human breast cancer cells to a stem-like phenotype [171]. The ΔNp63 isoform stimulates the transcription of the gene that encodes the cell-surface antigen CD44, a marker associated with cancer stem cells in breast, colorectal, and squamous cell carcinomas [189, 190]. Albeit at levels lower than in normal prostate, p63 expression is essential for regulating stem cell functions of neoplastic prostate epithelial cells. Cytoplastic aberrance of p63 is associated with high ALDH1A1(Aldehyde Dehydrogenase 1 Family, Member A1) expression, thus leading to increased cell proliferation rate of prostate CSCs [191] (Fig. 5C). Su and colleagues demonstrated that TAp63 is the main regulator of the transition from mammary cells to TICs (Fig. 5D). In the absence of TAp63, mammary epithelial cells (MECs) unleash stemness potential by expressing MaSCs signatures as well as by activating the Hippo pathway. Therefore, TAp63 null mice develop aggressive metastasis [192].
In the skin, ΔNp63α acts as an oncogene, capable of inhibiting oncogene-induced senescence and promoting development of squamous cell carcinoma (SCC) (Fig. 5B). ΔNp63α cooperates with oncogenic RAS to promote proliferation and to maintain the KRT15-positive stem cell population in order to drive tumorigenesis in vivo [193]. The expression of Lsh, a chromatin-remodeling protein is regulated by ΔNp63α during senescence bypass and tumor initiation, suggesting that p63 plays a role in epigenetic regulation of stemness [193]. Enhancer zest homolog 2 (EZH2) and SET domain bifurcated 1 (SETDB1) are two well-known oncogenic methyltransferases that are highly expressed in stem-like SSC sub-populations. SETDB1 cooperates with ΔNp63α to regulate the SCC stem cell phenotype. In addition, EZH2-activated RUNX3 acts as an inhibitor of both SETD1 and ΔNp63α [194]. ΔNp63 is often overexpressed in SCCs, in which it acts as a master regulator of CSCs maintenance via its functional connection with signal transducer and activator of transcription 3 (STAT3), assigning STAT3 induces ΔNp63 expression through binding to its promoter [195,196,197]. The gene encoding the chromatin-modifying protein that ACTL6A (Actin-like protein 6 A) is coamplified with p63 locus in HNSCC and induces a CSC phenotype characterized by undifferentiated regenerative proliferation. They also found ACTL6A/p63 complex is a direct transcription suppressor of Hippo regulator WWC1 and thus promotes YAP-mediated tumorigenesis [198]. Additionally, transglutaminase 2 (TG2) is an effector required for CSC survival in epidermal SCC. The interaction between TG2 and α6/β4-integrin is able to induce YAP-dependent ΔNp63α stabilization and therefore leads to enhanced tumor stem cell properties [199]. The Ribosomal S6 protein kinase 4 (RSK4) is one of the direct downstream targets of ΔNp63α in CSC maintaining in esophageal squamous cell carcinoma. As found, ΔNp63α directly transactivates RSK4 expression, thereby inhibiting radiosensitivity through GSK-3β/β-catenin axis [200]. Additionally, ΔNp63 regulates the self-renewal and maintenance of LESCs, which are indispensable for lung adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC) initiation as well as progression through the regulation of a common landscape of enhancer-associated cell identity genes, such as BCL9L (B-cell CLL/lymphoma 9-like) [201].
Altered expression of p63 in tumors correlates with aberrant expression of p53 or with the presence of TP53 mutants, indicating that the p63-p53 axis is relevant to tumorigenesis. Several p53 mutant proteins can interact with p63 through the OD, resulting in the inhibition of the transcriptional activity of the TAp63 variants and, subsequently, of their tumor suppressive behavior [2, 202]. Other p53 mutations occurring in the DBD render this domain very unstable, thus resulting in p63α binding via a co-aggregation mechanism mediated by the TIDs of the α-isoforms [203]. Inactivation of p63 by p53 mutants has been associated with the ability of mutant p53 proteins to promote tumor growth, chemoresistance, and metastasis [204]. Mechanistically, during tumor progression, tumor growth factor-β (TGF-β), acting in concert with oncogenic RAS, induces the formation of a mutant-p53/p63/SMADs ternary protein complex, in which the SMADs are essential platforms for the complex to assembly [202]. As a result of this interplay, the anti-metastatic properties of TAp63 are inhibited, allowing cancer cells to acquire pro-invasive and migratory capabilities. Mutant p53 proteins was also found to recruit p63 to gene promoters. In this context, p63 serves as a molecular chaperone through which p53 mutants aberrantly reprogram the cancer cell transcriptome and facilitate tumor invasion [205]. Zhang and colleagues reported that mutant p53 is capable of antagonizing p63-mediated tumor suppression in an in vivo model of T lymphoblastic lymphomas (T-ALL) [206]. In particular, mutant p53 contributes to the pathogenesis of T-ALL by collaborating with NOTCH1 to hijack p63-mediated transcription. Other studies have highlighted the ability of some p53 mutants to potentiate ΔNp63 function via protein-protein interaction [207].
Growing evidence has implicated p63 as a critical factor in driving the metastatic cascade (Fig. 5B). ΔNp63 acts as a vital regulator of quasi-mesenchymal CSCs which drive the metastasis events, and is required for the post-extravasation proliferation of CSCs in human breast cancer [208]. In the early stage, ΔNp63α is responsible for local invasion through collaborating with the membrane-type 1 (MT1)-matrix metalloproteinase (MMP) in basal-like BC, assigning to ΔNp63α the role in inhibiting the transcription of MT1-MMP [209]. One of the main traits of CSCs is the epithelial-mesenchymal transition process. Activation of oncogenic RAS or the TGF-β signaling in breast cancer cells stimulates ΔNp63 transcriptional activity and then induces the transcription of the dual-specificity phosphatase 6 (DUSP6) gene, whose expression is mainly restricted to stem cells [210]. As a result, DUSP6 empowers ΔNp63 and promotes metastatic behaviors in breast cancer cells [211]. In prostate adenocarcinomas, ΔNp63 is associated with the onset of basal-like cancer stem cell population but not with metastasis, as none of the tumors expressing p63 showed bone metastasis [212].
Conclusions
p63 is a p53 family protein responsible for morphogenesis and postnatal regeneration of epithelial tissues [213]. TAp63 and ΔNp63 isoforms, expressed in a variety of epithelial tissues and in epithelium-derived tumors, affect the transcriptions of genes involved in different mechanisms of stemness regulation. In this review, we outline the contribution of TAp63 and ΔNp63 isoforms to stemness in epithelial tissues. We also report about their involvement in the regulation of multiple signaling pathways, in both physiological and pathological conditions. In general, by sharing the ability of p53 to induce cell-cycle arrest and apoptosis, TAp63 often acts as a tumor suppressor [23, 214]. In contrast, ΔNp63 mainly works as an oncogene through orchestrating several tumor-promoting pathways, thus being suggested as a marker of CSCs [215, 216]. Therefore, the balance between TAp63 and ΔNp63 appears to modulate cell fate decisions. In either normal ESCs or CSCs, our work clearly shows that p63 participates in multiple pathways related to the regulation of self-renewal and functional differentiation. Collectively, p63 isoforms play an indispensable role in regulating normal ESCs and CSCs stemness for epithelium regeneration and tumorigenesis, respectively. Further investigations on p63 upstream signaling and affected targets would allow to acquire thorough insights into the network centered on this pivotal transcription factor, finally contributing to the achievement of a deeper knowledge of stemness mechanisms, aimed at the development of new therapeutic strategies.
References
De Laurenzi V, Melino G. Evolution of functions within the p53/p63/p73 family. Ann N Y Acad Sci. 2000;926:90–100.
Candi E, Agostini M, Melino G, Bernassola F. How the TP53 family proteins TP63 and TP73 contribute to tumorigenesis: regulators and effectors. Hum Mutat. 2014;35:702–14.
Bernassola F, Salomoni P, Oberst A, Di Como CJ, Pagano M, Melino G, et al. Ubiquitin-dependent degradation of p73 is inhibited by PML. J Exp Med. 2004;199:1545–57.
Pignon JC, Grisanzio C, Geng Y, Song J, Shivdasani RA, Signoretti S. p63-expressing cells are the stem cells of develo** prostate, bladder, and colorectal epithelia. Proc Natl Acad Sci USA 2013;110:8105–10.
Vitale I, Pietrocola F, Guilbaud E, Aaronson SA, Abrams JM, Adam D, et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ. 2023;30:1097–154.
Priami C, Montariello D, De Michele G, Ruscitto F, Polazzi A, Ronzoni S, et al. Aberrant activation of p53/p66Shc-mInsc axis increases asymmetric divisions and attenuates proliferation of aged mammary stem cells. Cell Death Differ. 2022;29:2429–44.
Thomas AF, Kelly GL, Strasser A. Of the many cellular responses activated by TP53, which ones are critical for tumour suppression? Cell Death Differ. 2022;29:961–71.
Kennedy MC, Lowe SW. Mutant p53: it’s not all one and the same. Cell Death Differ. 2022;29:983–7.
Hoyos D, Greenbaum B, Levine AJ. The genotypes and phenotypes of missense mutations in the proline domain of the p53 protein. Cell Death Differ. 2022;29:938–45.
de Andrade KC, Lee EE, Tookmanian EM, Kesserwan CA, Manfredi JJ, Hatton JN, et al. The TP53 Database: transition from the International Agency for Research on Cancer to the US National Cancer Institute. Cell Death Differ. 2022;29:1071–3.
Levine AJ. Exploring the future of research in the Tp53 field. Cell Death Differ. 2022;29:893–4.
El-Saafin F, Bergamasco MI, Chen Y, May RE, Esakky P, Hediyeh-Zadeh S, et al. Loss of TAF8 causes TFIID dysfunction and p53-mediated apoptotic neuronal cell death. Cell Death Differ. 2022;29:1013–27.
Lindstrom MS, Bartek J, Maya-Mendoza A. p53 at the crossroad of DNA replication and ribosome biogenesis stress pathways. Cell Death Differ. 2022;29:972–82.
Engeland K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022;29:946–60.
Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13:962–72.
Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–16.
Mangiulli M, Valletti A, Caratozzolo MF, Tullo A, Sbisa E, Pesole G, et al. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res. 2009;37:6092–104.
de Biase D, Morandi L, Degli Esposti R, Ligorio C, Pession A, Foschini MP, et al. p63 short isoforms are found in invasive carcinomas only and not in benign breast conditions. Virchows Arch. 2010;456:395–401.
Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–13.
Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–8.
Koster MI, Dai D, Marinari B, Sano Y, Costanzo A, Karin M, et al. p63 induces key target genes required for epidermal morphogenesis. Proc Natl Acad Sci USA 2007;104:3255–60.
Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–4.
Guo X, Keyes WM, Papazoglu C, Zuber J, Li W, Lowe SW, et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol. 2009;11:1451–7.
Suh EK, Yang A, Kettenbach A, Bamberger C, Michaelis AH, Zhu Z, et al. p63 protects the female germ line during meiotic arrest. Nature. 2006;444:624–8.
Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–73.
Candi E, Melino G, Toth A, Dotsch V. Mechanisms of quality control differ in male and female germ cells. Cell Death Differ. 2021;28:2300–2.
Hibi K, Trink B, Patturajan M, Westra WH, Caballero OL, Hill DE, et al. AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci USA 2000;97:5462–7.
Moore JE, McMullen CB, Mahon G, Adamis AP. The corneal epithelial stem cell. DNA Cell Biol. 2002;21:443–51.
Senoo M, Pinto F, Crum CP, McKeon F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007;129:523–36.
**n L, Lukacs RU, Lawson DA, Cheng D, Witte ON. Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells. 2007;25:2760–9.
Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, et al. DeltaNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol. 2014;16:1004–15.
Candi E, Rufini A, Terrinoni A, Giamboi-Miraglia A, Lena AM, Mantovani R, et al. DeltaNp63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proc Natl Acad Sci USA 2007;104:11999–2004.
Su X, Chakravarti D, Flores ER. p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer. 2013;13:136–43.
Pecorari R, Bernassola F, Melino G, Candi E. Distinct interactors define the p63 transcriptional signature in epithelial development or cancer. Biochem J. 2022;479:1375–92.
King KE, Weinberg WC. p63: defining roles in morphogenesis, homeostasis, and neoplasia of the epidermis. Mol Carcinog. 2007;46:716–24.
Koster MI, Roop DR. Transgenic mouse models provide new insights into the role of p63 in epidermal development. Cell Cycle. 2004;3:411–3.
Tai K, Cockburn K, Greco V. Flexibility sustains epithelial tissue homeostasis. Curr Opin Cell Biol. 2019;60:84–91.
Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science. 2014;344:1242281.
Harris-Tryon TA, Grice EA. Microbiota and maintenance of skin barrier function. Science. 2022;376:940–5.
Yuan X, Chen J, Grauer JA, Xu Q, Van Brunt LA, Helms JA. The junctional epithelium is maintained by a stem cell population. J Dent Res. 2021;100:209–16.
Forster N, Saladi SV, van Bragt M, Sfondouris ME, Jones FE, Li Z, et al. Basal cell signaling by p63 controls luminal progenitor function and lactation via NRG1. Dev Cell. 2014;28:147–60.
McKeon F. p63 and the epithelial stem cell: more than status quo? Genes Dev. 2004;18:465–9.
Ratajczak MZ, Bujko K, Mack A, Kucia M, Ratajczak J. Cancer from the perspective of stem cells and misappropriated tissue regeneration mechanisms. Leukemia. 2018;32:2519–26.
Yi R, Poy MN, Stoffel M, Fuchs E. A skin microRNA promotes differentiation by repressing ‘stemness’. Nature. 2008;452:225–9.
Melino G, Memmi EM, Pelicci PG, Bernassola F. Maintaining epithelial stemness with p63. Sci Signal. 2015;8:re9.
Koster MI, Huntzinger KA, Roop DR. Epidermal differentiation: transgenic/knockout mouse models reveal genes involved in stem cell fate decisions and commitment to differentiation. J Investig Dermatol Symp Proc. 2002;7:41–45.
Su X, Cho MS, Gi YJ, Ayanga BA, Sherr CJ, Flores ER. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009;28:1904–15.
Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 2004;18:126–31.
Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci USA 2001;98:3156–61.
Kai-Hong J, Jun X, Kai-Meng H, Ying W, Hou-Qi L. P63 expression pattern during rat epidermis morphogenesis and the role of p63 as a marker for epidermal stem cells. J Cutan Pathol. 2007;34:154–9.
Nguyen BC, Lefort K, Mandinova A, Antonini D, Devgan V, Della Gatta G, et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006;20:1028–42.
Koster MI, Roop DR. The role of p63 in development and differentiation of the epidermis. J Dermatol Sci. 2004;34:3–9.
Crum CP, McKeon FD. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu Rev Pathol. 2010;5:349–71.
Voroteliak EA, Chermnykh ES, Tkachenko SB, Vasil’ev AV, Terskikh VV. [Expression and function of p63 gene in epithelial cells]. Izv Akad Nauk Ser Biol. 2007;4:389–93.
Lena AM, Shalom-Feuerstein R, Rivetti di Val Cervo P, Aberdam D, Knight RA, Melino G, et al. miR-203 represses ‘stemness’ by repressing DeltaNp63. Cell Death Differ. 2008;15:1187–95.
Koster MI, Kim S, Roop DR. P63 deficiency: a failure of lineage commitment or stem cell maintenance? J Investig Dermatol Symp Proc. 2005;10:118–23.
Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009;5:64–75.
Beaudry VG, Attardi LD. SKP-ing TAp63: stem cell depletion, senescence, and premature aging. Cell Stem Cell. 2009;5:1–2.
Mikkola ML. p63 in skin appendage development. Cell Cycle. 2007;6:285–90.
Koster MI, Roop DR. p63 and epithelial appendage development. Differentiation. 2004;72:364–70.
Sun X, Joost S, Kasper M. Plasticity of epithelial cells during skin wound healing. Cold Spring Harb Perspect Biol. 2023;15:a041232.
Gebel J, Luh LM, Coutandin D, Osterburg C, Lohr F, Schafer B, et al. Mechanism of TAp73 inhibition by DeltaNp63 and structural basis of p63/p73 hetero-tetramerization. Cell Death Differ. 2016;23:1930–40.
Liu F, Zhou H, Du W, Huang X, Zheng X, Zhang C, et al. Hair follicle stem cells combined with human allogeneic acellular amniotic membrane for repair of full thickness skin defects in nude mice. J Tissue Eng Regen Med. 2020;14:723–35.
Drewa T, Joachimiak R, Kaznica A, Sarafian V, Pokrywczynska M. Hair stem cells for bladder regeneration in rats: preliminary results. Transpl Proc. 2009;41:4345–51.
Shin JW, Choi HR, Nam KM, Lee HS, Kim SA, Joe HJ, et al. The co-expression pattern of p63 and HDAC1: a potential way to disclose stem cells in interfollicular epidermis. Int J Mol Sci. 2017;18:1360.
Fiuraskova M, Brychtova S, Kolar Z, Kucerova R, Bienova M. Expression of beta-catenin, p63 and CD34 in hair follicles during the course of androgenetic alopecia. Arch Dermatol Res. 2005;297:143–6.
Van Tri M, Polonsky J, Merienne C, Sevenet T. Soularubinone, a new antileukemic quassionoid from Soulamea tomentosa. J Nat Prod. 1981;44:279–84.
Nelson AM, Katseff AS, Ratliff TS, Garza LA. Interleukin 6 and STAT3 regulate p63 isoform expression in keratinocytes during regeneration. Exp Dermatol. 2016;25:155–7.
Romano RA, Smalley K, Liu S, Sinha S. Abnormal hair follicle development and altered cell fate of follicular keratinocytes in transgenic mice expressing DeltaNp63alpha. Development. 2010;137:1431–9.
Yang X, Wang H, Jiao B. Mammary gland stem cells and their application in breast cancer. Oncotarget. 2017;8:10675–91.
Wuidart A, Sifrim A, Fioramonti M, Matsumura S, Brisebarre A, Brown D, et al. Early lineage segregation of multipotent embryonic mammary gland progenitors. Nat Cell Biol. 2018;20:666–76.
van Bokhoven H, Jung M, Smits AP, van Beersum S, Ruschendorf F, van Steensel M, et al. Limb mammary syndrome: a new genetic disorder with mammary hypoplasia, ectrodactyly, and other Hand/Foot anomalies maps to human chromosome 3q27. Am J Hum Genet. 1999;64:538–46.
Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139:772–82.
Coates PJ, Nenutil R, Holcakova J, Nekulova M, Podhorec J, Svoboda M, et al. p63 isoforms in triple-negative breast cancer: DeltaNp63 associates with the basal phenotype whereas TAp63 associates with androgen receptor, lack of BRCA mutation, PTEN and improved survival. Virchows Arch. 2018;472:351–9.
Li N, Singh S, Cherukuri P, Li H, Yuan Z, Ellisen LW, et al. Reciprocal intraepithelial interactions between TP63 and hedgehog signaling regulate quiescence and activation of progenitor elaboration by mammary stem cells. Stem Cells. 2008;26:1253–64.
Yallowitz AR, Alexandrova EM, Talos F, Xu S, Marchenko ND, Moll UM. p63 is a prosurvival factor in the adult mammary gland during post-lactational involution, affecting PI-MECs and ErbB2 tumorigenesis. Cell Death Differ. 2014;21:645–54.
Barbareschi M, Pecciarini L, Cangi MG, Macri E, Rizzo A, Viale G, et al. p63, a p53 homologue, is a selective nuclear marker of myoepithelial cells of the human breast. Am J Surg Pathol. 2001;25:1054–60.
Nylander K, Vojtesek B, Nenutil R, Lindgren B, Roos G, Zhanxiang W, et al. Differential expression of p63 isoforms in normal tissues and neoplastic cells. J Pathol. 2002;198:417–27.
Kudo KI, Tsuyama N, Nagata K, Imaoka T, Iizuka D, Sugai-Takahashi M, et al. DeltaNp63alpha transcriptionally represses p53 target genes involved in the radiation-induced DNA damage response: DeltaNp63alpha may cause genomic instability in epithelial stem cells. Radiat Oncol. 2022;17:183.
DiRenzo J, Signoretti S, Nakamura N, Rivera-Gonzalez R, Sellers W, Loda M, et al. Growth factor requirements and basal phenotype of an immortalized mammary epithelial cell line. Cancer Res. 2002;62:89–98.
Lindley LE, Curtis KM, Sanchez-Mejias A, Rieger ME, Robbins DJ, Briegel KJ. The WNT-controlled transcriptional regulator LBH is required for mammary stem cell expansion and maintenance of the basal lineage. Development. 2015;142:893–904.
Yalcin-Ozuysal O, Fiche M, Guitierrez M, Wagner KU, Raffoul W, Brisken C. Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates. Cell Death Differ. 2010;17:1600–12.
Wang Y, Hayward S, Cao M, Thayer K, Cunha G. Cell differentiation lineage in the prostate. Differentiation. 2001;68:270–9.
Signoretti S, Waltregny D, Dilks J, Isaac B, Lin D, Garraway L, et al. p63 is a prostate basal cell marker and is required for prostate development. Am J Pathol. 2000;157:1769–75.
Huang Y, Hamana T, Liu J, Wang C, An L, You P, et al. Prostate sphere-forming stem cells are derived from the P63-expressing basal compartment. J Biol Chem. 2015;290:17745–52.
Signoretti S, Loda M. Defining cell lineages in the prostate epithelium. Cell Cycle. 2006;5:138–41.
Lee DK, Liu Y, Liao L, Wang F, Xu J. The prostate basal cell (BC) heterogeneity and the p63-positive BC differentiation spectrum in mice. Int J Biol Sci. 2014;10:1007–17.
Signoretti S, Pires MM, Lindauer M, Horner JW, Grisanzio C, Dhar S, et al. p63 regulates commitment to the prostate cell lineage. Proc Natl Acad Sci USA 2005;102:11355–60.
Grisanzio C, Signoretti S. p63 in prostate biology and pathology. J Cell Biochem. 2008;103:1354–68.
Rai S, Alsaidan OA, Yang H, Cai H, Wang L. Heparan sulfate inhibits transforming growth factor beta signaling and functions in cis and in trans to regulate prostate stem/progenitor cell activities. Glycobiology. 2020;30:381–95.
Amatu JB, Baudouin C, Trinh L, Labbe A, Buffault J. [Corneal epithelial biomechanics: resistance to stress and role in healing and remodeling]. J Fr Ophtalmol. 2023;46:287–99.
Tang Q, Luo C, Lu B, Fu Q, Yin H, Qin Z, et al. Thermosensitive chitosan-based hydrogels releasing stromal cell derived factor-1 alpha recruit MSC for corneal epithelium regeneration. Acta Biomater. 2017;61:101–13.
Sridhar MS. Anatomy of cornea and ocular surface. Indian J Ophthalmol. 2018;66:190–4.
Arpitha P, Prajna NV, Srinivasan M, Muthukkaruppan V. High expression of p63 combined with a large N/C ratio defines a subset of human limbal epithelial cells: implications on epithelial stem cells. Invest Ophthalmol Vis Sci. 2005;46:3631–6.
Shalom-Feuerstein R, Lena AM, Zhou H, De La Forest Divonne S, Van Bokhoven H, Candi E, et al. DeltaNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ. 2011;18:887–96.
Novelli F, Ganini C, Melino G, Nucci C, Han Y, Shi Y, et al. p63 in corneal and epidermal differentiation. Biochem Biophys Res Commun. 2022;610:15–22.
Ekpo P, Inthasin N, Matamnan S, Wongprompitak P, Wattanapanitch M, Boonwong C, et al. Characterization of limbal explant sites: optimization of stem cell outgrowth in vitro culture. PLoS ONE. 2020;15:e0233075.
Li Y, Inoue T, Takamatsu F, Kobayashi T, Shiraishi A, Maeda N, et al. Differences between niche cells and limbal stromal cells in maintenance of corneal limbal stem cells. Invest Ophthalmol Vis Sci. 2014;55:1453–62.
Shaharuddin B, Harvey I, Ahmad S, Ali S, Meeson A. Characterisation of human limbal side population cells isolated using an optimised protocol from an immortalised epithelial cell line and primary limbal cultures. Stem Cell Rev Rep. 2014;10:240–50.
Jeong WY, Yoo HY, Kim CW. beta-cellulin promotes the proliferation of corneal epithelial stem cells through the phosphorylation of erk1/2. Biochem Biophys Res Commun. 2018;496:359–66.
Di Iorio E, Barbaro V, Ruzza A, Ponzin D, Pellegrini G, De Luca M. Isoforms of DeltaNp63 and the migration of ocular limbal cells in human corneal regeneration. Proc Natl Acad Sci USA 2005;102:9523–8.
Wang DY, Cheng CC, Kao MH, Hsueh YJ, Ma DH, Chen JK. Regulation of limbal keratinocyte proliferation and differentiation by TAp63 and DeltaNp63 transcription factors. Invest Ophthalmol Vis Sci. 2005;46:3102–8.
Gordon J, Wilson VA, Blair NF, Sheridan J, Farley A, Wilson L, et al. Functional evidence for a single endodermal origin for the thymic epithelium. Nat Immunol. 2004;5:546–53.
Stefanski HE, **ng Y, Nicholls J, Jonart L, Goren E, Taylor PA, et al. P63 targeted deletion under the FOXN1 promoter disrupts pre-and post-natal thymus development, function and maintenance as well as induces severe hair loss. PLoS ONE. 2022;17:e0261770.
Di Como CJ, Urist MJ, Babayan I, Drobnjak M, Hedvat CV, Teruya-Feldstein J, et al. p63 expression profiles in human normal and tumor tissues. Clin Cancer Res. 2002;8:494–501.
Shi Y, Dong M, Zhou Y, Li W, Gao Y, Han L, et al. Distal airway stem cells ameliorate bleomycin-induced pulmonary fibrosis in mice. Stem Cell Res Ther. 2019;10:161.
Zhou Y, Wang Y, Li D, Zhang T, Ma Y, Zuo W. Stable long-term culture of human distal airway stem cells for transplantation. Stem Cells Int. 2021;2021:9974635.
Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell. 2011;147:525–38.
**ao Z, Jiang Q, Willette-Brown J, ** S, Zhu F, Burkett S, et al. The pivotal role of IKKalpha in the development of spontaneous lung squamous cell carcinomas. Cancer Cell. 2013;23:527–40.
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–36.
Fernanda de Mello Costa M, Weiner AI, Vaughan AE. Basal-like progenitor cells: a review of dysplastic alveolar regeneration and remodeling in lung repair. Stem Cell Rep. 2020;15:1015–25.
Peng W, Chang M, Wu Y, Zhu W, Tong L, Zhang G, et al. Lyophilized powder of mesenchymal stem cell supernatant attenuates acute lung injury through the IL-6-p-STAT3-p63-JAG2 pathway. Stem Cell Res Ther. 2021;12:216.
Arason AJ, Jonsdottir HR, Halldorsson S, Benediktsdottir BE, Bergthorsson JT, Ingthorsson S, et al. deltaNp63 has a role in maintaining epithelial integrity in airway epithelium. PLoS ONE. 2014;9:e88683.
Tanaka Y, Yamaguchi M, Hirai S, Sumi T, Tada M, Saito A, et al. Characterization of distal airway stem-like cells expressing N-terminally truncated p63 and thyroid transcription factor-1 in the human lung. Exp Cell Res. 2018;372:141–9.
Weiner AI, Zhao G, Zayas HM, Holcomb NP, Adams-Tzivelekidis S, Wong J, et al. DeltaNp63 drives dysplastic alveolar remodeling and restricts epithelial plasticity upon severe lung injury. Cell Rep. 2022;41:111805.
Fukumoto J, Sidramagowda Patil S, Krishnamurthy S, Saji S, John I, Narala VR, et al. Altered expression of p63 isoforms and expansion of p63- and club cell secretory protein-positive epithelial cells in the lung as novel features of aging. Am J Physiol Cell Physiol. 2019;316:C492–508.
Bouras T, Pal B, Vaillant F, Harburg G, Asselin-Labat ML, Oakes SR, et al. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell. 2008;3:429–41.
Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–6.
Okuyama R, Ogawa E, Nagoshi H, Yabuki M, Kurihara A, Terui T, et al. p53 homologue, p51/p63, maintains the immaturity of keratinocyte stem cells by inhibiting Notch1 activity. Oncogene. 2007;26:4478–88.
Koh LF, Ng BK, Bertrand J, Thierry F. Transcriptional control of late differentiation in human keratinocytes by TAp63 and Notch. Exp Dermatol. 2015;24:754–60.
Tadeu AM, Horsley V. Notch signaling represses p63 expression in the develo** surface ectoderm. Development. 2013;140:3777–86.
Kent S, Hutchinson J, Balboni A, Decastro A, Cherukuri P, Direnzo J. DeltaNp63alpha promotes cellular quiescence via induction and activation of Notch3. Cell Cycle. 2011;10:3111–8.
Teuliere J, Faraldo MM, Deugnier MA, Shtutman M, Ben-Ze’ev A, Thiery JP, et al. Targeted activation of beta-catenin signaling in basal mammary epithelial cells affects mammary development and leads to hyperplasia. Development. 2005;132:267–77.
Zhang X, Dong N, Hu X. Wnt/beta-catenin signaling inhibitors. Curr Top Med Chem. 2023;23:880–96.
Holland JD, Klaus A, Garratt AN, Birchmeier W. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol. 2013;25:254–64.
Zhang Y, Wang X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J Hematol Oncol. 2020;13:165.
Rim EY, Clevers H, Nusse R. The Wnt pathway: from signaling mechanisms to synthetic modulators. Annu Rev Biochem. 2022;91:571–98.
Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416–29.
Sari IN, Phi LTH, Jun N, Wijaya YT, Lee S, Kwon HY. Hedgehog signaling in cancer: a prospective therapeutic target for eradicating cancer stem cells. Cells. 2018;7:208.
Chari NS, Romano RA, Koster MI, Jaks V, Roop D, Flores ER, et al. Interaction between the TP63 and SHH pathways is an important determinant of epidermal homeostasis. Cell Death Differ. 2013;20:1080–8.
Li J, Yuan Y, He J, Feng J, Han X, **g J, et al. Constitutive activation of hedgehog signaling adversely affects epithelial cell fate during palatal fusion. Dev Biol. 2018;441:191–203.
Girardet L, Cyr DG, Belleannee C. Arl13b controls basal cell stemness properties and Hedgehog signaling in the mouse epididymis. Cell Mol Life Sci. 2022;79:556.
Candi E, Amelio I, Agostini M, Melino G. MicroRNAs and p63 in epithelial stemness. Cell Death Differ. 2015;22:12–21.
Suh N. MicroRNA controls of cellular senescence. BMB Rep. 2018;51:493–9.
Sun L, Zhu W, Zhao P, Zhang J, Lu Y, Zhu Y, et al. Down-regulated exosomal microRNA-221 - 3p derived from senescent mesenchymal stem cells impairs heart repair. Front Cell Dev Biol. 2020;8:263.
Ortiz GGR, Mohammadi Y, Nazari A, Ataeinaeini M, Kazemi P, Yasamineh S, et al. A state-of-the-art review on the MicroRNAs roles in hematopoietic stem cell aging and longevity. Cell Commun Signal. 2023;21:85.
Potter ML, Hill WD, Isales CM, Hamrick MW, Fulzele S. MicroRNAs are critical regulators of senescence and aging in mesenchymal stem cells. Bone. 2021;142:115679.
Chen HL, Chiang PC, Lo CH, Lo YH, Hsu DK, Chen HY, et al. Galectin-7 regulates keratinocyte proliferation and differentiation through JNK-miR-203-p63 signaling. J Invest Dermatol. 2016;136:182–91.
Kim YJ, Lee SB, Lee HB. Oleic acid enhances keratinocytes differentiation via the upregulation of miR-203 in human epidermal keratinocytes. J Cosmet Dermatol. 2019;18:383–9.
Rivetti di Val Cervo P, Lena AM, Nicoloso M, Rossi S, Mancini M, Zhou H, et al. p63-microRNA feedback in keratinocyte senescence. Proc Natl Acad Sci USA 2012;109:1133–8.
Chikh A, Matin RN, Senatore V, Hufbauer M, Lavery D, Raimondi C, et al. iASPP/p63 autoregulatory feedback loop is required for the homeostasis of stratified epithelia. EMBO J. 2011;30:4261–73.
Panatta E, Lena AM, Mancini M, Smirnov A, Marini A, Delli Ponti R, et al. Long non-coding RNA uc.291 controls epithelial differentiation by interfering with the ACTL6A/BAF complex. EMBO Rep. 2020;21:e46734.
Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. 2013;493:231–5.
Mancini M, Cappello A, Pecorari R, Lena AM, Montanaro M, Fania L, et al. Involvement of transcribed lncRNA uc.291 and SWI/SNF complex in cutaneous squamous cell carcinoma. Discov Oncol. 2021;12:14.
Ma Y, Di Y, Li Q, Zhan Q, He X, Liu S, et al. LncRNAs as epigenetic regulators of epithelial to mesenchymal transition in pancreatic cancer. Discov Oncol. 2022;13:61.
Lin Z, Feng F, Liang J, Zeng H, Li J. lncRNA RP11-10A14.5: a potential prognosis biomarker for LUAD through regulation on proliferation and metastasis. Discov Oncol. 2022;13:32.
Chen Z, Bian C, Huang J, Li X, Chen L, **e X, et al. Tumor-derived exosomal HOTAIRM1 regulates SPON2 in CAFs to promote progression of lung adenocarcinoma. Discov Oncol. 2022;13:92.
Wei W, Xu T, Zhang Y, Huang Y, Wang X. Upregulation of long noncoding RNA linc02544 and its association with overall survival rate and the influence on cell proliferation and migration in lung squamous cell carcinoma. Discov Oncol. 2022;13:41.
Wang J, Li L, Jiang X, Wang B, Hu X, Liu W, et al. Silencing of long non-coding RNA TUC338 inhibits the malignant phenotype of nasopharyngeal cancer cells via modulating the miR-1226-3p/FGF2 axis. Discov Oncol. 2022;13:102.
Tanis SEJ, Koksal ES, van Buggenum J, Mulder KW. BLNCR is a long non-coding RNA adjacent to integrin beta-1 that is rapidly lost during epidermal progenitor cell differentiation. Sci Rep. 2019;9:31.
Fierro C, Gatti V, La Banca V, De Domenico S, Scalera S, Corleone G, et al. The long non-coding RNA NEAT1 is a DeltaNp63 target gene modulating epidermal differentiation. Nat Commun. 2023;14:3795.
Fessing MY, Mardaryev AN, Gdula MR, Sharov AA, Sharova TY, Rapisarda V, et al. p63 regulates Satb1 to control tissue-specific chromatin remodeling during development of the epidermis. J Cell Biol. 2011;194:825–39.
Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, et al. Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell. 2016;19:491–501.
Liu B, Liu YF, Du YR, Mardaryev AN, Yang W, Chen H, et al. Cbx4 regulates the proliferation of thymic epithelial cells and thymus function. Development. 2013;140:780–8.
Mardaryev AN, Liu B, Rapisarda V, Poterlowicz K, Malashchuk I, Rudolf J, et al. Cbx4 maintains the epithelial lineage identity and cell proliferation in the develo** stratified epithelium. J Cell Biol. 2016;212:77–89.
Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, et al. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol. 2006;8:551–61.
Kurata S, Okuyama T, Osada M, Watanabe T, Tomimori Y, Sato S, et al. p51/p63 controls subunit alpha3 of the major epidermis integrin anchoring the stem cells to the niche. J Biol Chem. 2004;279:50069–77.
Favaloro B, Tamburro A, Angelucci S, Luca AD, Melino S, di Ilio C, et al. Molecular cloning, expression and site-directed mutagenesis of glutathione S-transferase from Ochrobactrum anthropi. Biochem J. 1998;335:573–9.
Yan Y, He M, Zhao L, Wu H, Zhao Y, Han L, et al. A novel HIF-2alpha targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPR(ER) axis. Cell Death Differ. 2022;29:1769–89.
Baumeister R, Murphy CT, Heimbucher T. Metabolic adaptation to hypoxia: do worms and cancer cells share common metabolic responses to hypoxic stress? Cell Death Differ. 2021;28:1434–6.
Chen J, Liu Y, Zhao Z, Qiu J. Oxidative stress in the skin: impact and related protection. Int J Cosmet Sci. 2021;43:495–509.
Liu HM, Cheng MY, Xun MH, Zhao ZW, Zhang Y, Tang W, et al. Possible mechanisms of oxidative stress-induced skin cellular senescence, inflammation, and cancer and the therapeutic potential of plant polyphenols. Int J Mol Sci. 2023;24:3755.
Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell. 2002;10:995–1005.
Amelio I, Melino G, Candi E. p63 adjusts sugar taste of epidermal layers. J Invest Dermatol. 2017;137:1204–6.
Hamanaka RB, Mutlu GM. PFKFB3, a direct target of p63, is required for proliferation and inhibits differentiation in epidermal keratinocytes. J Invest Dermatol. 2017;137:1267–76.
Smirnov A, Lena AM, Cappello A, Panatta E, Anemona L, Bischetti S, et al. ZNF185 is a p63 target gene critical for epidermal differentiation and squamous cell carcinoma development. Oncogene. 2019;38:1625–38.
Choo MK, Kraft S, Missero C, Park JM. The protein kinase p38alpha destabilizes p63 to limit epidermal stem cell frequency and tumorigenic potential. Sci Signal. 2018;11:eaau0727.
Wu L, Chen X, Zhao J, Martin B, Zepp JA, Ko JS, et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J Exp Med. 2015;212:1571–87.
Gatti V, Fierro C, Compagnone M, La Banca V, Mauriello A, Montanaro M, et al. DeltaNp63-Senataxin circuit controls keratinocyte differentiation by promoting the transcriptional termination of epidermal genes. Proc Natl Acad Sci USA 2022;119:e2104718119.
Conforti F, Yang AL, Piro MC, Mellone M, Terrinoni A, Candi E, et al. PIR2/Rnf144B regulates epithelial homeostasis by mediating degradation of p21WAF1 and p63. Oncogene. 2013;32:4758–65.
Balboni AL, Hutchinson JA, DeCastro AJ, Cherukuri P, Liby K, Sporn MB, et al. DeltaNp63alpha-mediated activation of bone morphogenetic protein signaling governs stem cell activity and plasticity in normal and malignant mammary epithelial cells. Cancer Res. 2013;73:1020–30.
Candi E, Terrinoni A, Rufini A, Chikh A, Lena AM, Suzuki Y, et al. p63 is upstream of IKK alpha in epidermal development. J Cell Sci. 2006;119:4617–22.
Orzol P, Holcakova J, Nekulova M, Nenutil R, Vojtesek B, Coates PJ. The diverse oncogenic and tumour suppressor roles of p63 and p73 in cancer: a review by cancer site. Histol Histopathol. 2015;30:503–21.
Fisher ML, Balinth S, Mills AA. DeltaNp63alpha in cancer: importance and therapeutic opportunities. Trends Cell Biol. 2022;33:280–92.
Gatti V, Bongiorno-Borbone L, Fierro C, Annicchiarico-Petruzzelli M, Melino G, Peschiaroli A. p63 at the crossroads between stemness and metastasis in breast cancer. Int J Mol Sci. 2019;20:2683.
Massion PP, Taflan PM, Rahman SM, Yildiz P, Shyr Y, Carbone DP, et al. Role of p63 amplification and overexpression in lung cancer development. Chest. 2004;125:102S.
Botchkarev VA, Flores ER. p53/p63/p73 in the epidermis in health and disease. Cold Spring Harb Perspect Med. 2014;4:a015248.
Gonfloni S, Caputo V, Iannizzotto V. P63 in health and cancer. Int J Dev Biol. 2015;59:87–93.
Verma P, Shukla N, Kumari S, Ansari MS, Gautam NK, Patel GK. Cancer stem cell in prostate cancer progression, metastasis and therapy resistance. Biochim Biophys Acta Rev Cancer. 2023;1878:188887.
Qureshi-Baig K, Ullmann P, Haan S, Letellier E. Tumor-initiating cells: a criTICal review of isolation approaches and new challenges in targeting strategies. Mol Cancer. 2017;16:40.
Chaudhary A, Raza SS, Haque R. Transcriptional factors targeting in cancer stem cells for tumor modulation. Semin Cancer Biol. 2023;88:123–37.
Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–43.
Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–28.
Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–96.
McMullen JRW, Soto U. Newly identified breast luminal progenitor and gestational stem cell populations likely give rise to HER2-overexpressing and basal-like breast cancers. Discov Oncol. 2022;13:38.
Nagao K, Kato C, Ikemoto Y, Motojima T, Fujii K, Umezawa A, et al. PTCH1-null induced pluripotent stem cells exclusively differentiate into immature ectodermal cells with large areas of medulloblastoma-like tissue. Discov Oncol. 2022;13:36.
Memmi EM, Sanarico AG, Giacobbe A, Peschiaroli A, Frezza V, Cicalese A, et al. p63 sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci USA 2015;112:3499–504.
Du Z, Li J, Wang L, Bian C, Wang Q, Liao L, et al. Overexpression of DeltaNp63alpha induces a stem cell phenotype in MCF7 breast carcinoma cell line through the Notch pathway. Cancer Sci. 2010;101:2417–24.
Boldrup L, Coates PJ, Gu X, Nylander K. DeltaNp63 isoforms regulate CD44 and keratins 4, 6, 14 and 19 in squamous cell carcinoma of head and neck. J Pathol. 2007;213:384–91.
Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA 2007;104:973–8.
Ferronika P, Triningsih FX, Ghozali A, Moeljono A, Rahmayanti S, Shadrina AN, et al. p63 cytoplasmic aberrance is associated with high prostate cancer stem cell expression. Asian Pac J Cancer Prev. 2012;13:1943–8.
Su X, Napoli M, Abbas HA, Venkatanarayan A, Bui NHB, Coarfa C, et al. TAp63 suppresses mammary tumorigenesis through regulation of the Hippo pathway. Oncogene. 2017;36:2377–93.
Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011;8:164–76.
Balinth S, Fisher ML, Hwangbo Y, Wu C, Ballon C, Sun X, et al. EZH2 regulates a SETDB1/DeltaNp63alpha axis via RUNX3 to drive a cancer stem cell phenotype in squamous cell carcinoma. Oncogene. 2022;41:4130–44.
Wei S, Li J, Tang M, Zhang K, Gao X, Fang L, et al. STAT3 and p63 in the regulation of cancer stemness. Front Genet. 2022;13:909251.
Ripamonti F, Albano L, Rossini A, Borrelli S, Fabris S, Mantovani R, et al. EGFR through STAT3 modulates DeltaN63alpha expression to sustain tumor-initiating cell proliferation in squamous cell carcinomas. J Cell Physiol. 2013;228:871–8.
Galoczova M, Coates P, Vojtesek B. STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett. 2018;23:12.
Saladi SV, Ross K, Karaayvaz M, Tata PR, Mou H, Rajagopal J, et al. ACTL6A is co-amplified with p63 in squamous cell carcinoma to drive YAP activation, regenerative proliferation, and poor prognosis. Cancer Cell. 2017;31:35–49.
Fisher ML, Kerr C, Adhikary G, Grun D, Xu W, Keillor JW, et al. Transglutaminase interaction with alpha6/beta4-integrin stimulates YAP1-dependent DeltaNp63alpha stabilization and leads to enhanced cancer stem cell survival and tumor formation. Cancer Res. 2016;76:7265–76.
Li MY, Fan LN, Han DH, Yu Z, Ma J, Liu YX, et al. Ribosomal S6 protein kinase 4 promotes radioresistance in esophageal squamous cell carcinoma. J Clin Invest. 2020;130:4301–19.
Napoli M, Wu SJ, Gore BL, Abbas HA, Lee K, Checker R, et al. DeltaNp63 regulates a common landscape of enhancer associated genes in non-small cell lung cancer. Nat Commun. 2022;13:614.
Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98.
Kehrloesser S, Osterburg C, Tuppi M, Schafer B, Vousden KH, Dotsch V. Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family. Cell Death Differ. 2016;23:1952–60.
Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–86.
Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, et al. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget. 2011;2:1203–17.
Zhang J, Sun W, Kong X, Zhang Y, Yang HJ, Ren C, et al. Mutant p53 antagonizes p63/p73-mediated tumor suppression via Notch1. Proc Natl Acad Sci USA 2019;116:24259–67.
Gong L, Pan X, Lim CB, de Polo A, Little JB, Yuan ZM. A functional interplay between Delta133p53 and DeltaNp63 in promoting glycolytic metabolism to fuel cancer cell proliferation. Oncogene. 2018;37:2150–64.
Lambert AW, Fiore C, Chutake Y, Verhaar ER, Strasser PC, Chen MW, et al. DeltaNp63/p73 drive metastatic colonization by controlling a regenerative epithelial stem cell program in quasi-mesenchymal cancer stem cells. Dev Cell. 2022;57:2714–30.e2718.
Lodillinsky C, Infante E, Guichard A, Chaligne R, Fuhrmann L, Cyrta J, et al. p63/MT1-MMP axis is required for in situ to invasive transition in basal-like breast cancer. Oncogene. 2016;35:344–57.
Ganini C, Amelio I, Bertolo R, Bove P, Buonomo OC, Candi E, et al. Global map** of cancers: The Cancer Genome Atlas and beyond. Mol Oncol. 2021;15:2823–40.
Vasilaki E, Morikawa M, Koinuma D, Mizutani A, Hirano Y, Ehata S, et al. Ras and TGF-beta signaling enhance cancer progression by promoting the DeltaNp63 transcriptional program. Sci Signal. 2016;9:ra84.
Galoczova M, Nenutil R, Pokorna Z, Vojtesek B, Coates PJ. TAp63 and DeltaNp63 (p40) in prostate adenocarcinomas: DeltaNp63 associates with a basal-like cancer stem cell population but not with metastasis. Virchows Arch. 2021;478:627–36.
Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006;20:3185–97.
Park E, Kim H, Kim JM, Primack B, Vidal-Cardenas S, Xu Y, et al. FANCD2 activates transcription of TAp63 and suppresses tumorigenesis. Mol Cell. 2013;50:908–18.
Xu Y, Yang X, **ong Q, Han J, Zhu Q. The dual role of p63 in cancer. Front Oncol. 2023;13:1116061.
Fisher ML, Balinth S, Mills AA. DeltaNp63alpha in cancer: importance and therapeutic opportunities. Trends Cell Biol. 2023;33:280–92.
Acknowledgements
The authors thank Richard Knight for helpful and constructive criticisms. All the Figures except Fig. 1 were prepared using the BioRender online website (https://www.biorender.com).
Funding
This work has been supported by the MUR-PNRR M4C2I1.3 PE6 project PE00000019 Heal Italia (CUP: E83C22004670001) to FB, GM, EC, MA, MF, MP. The Research leading to these results has received funding from AIRC under IG 2019 – 23232 – P.I. Bernassola Francesca, under IG 2022 - ID. 27366 project – P.I. Melino Gennaro, under IG 2019 - ID. 22206 – P.I. Candi Eleonora. Work has been also partially supported by Fondazione Luigi Maria Monti IDI-IRCCS (R.C. to EC) and by Regione Lazio through LazioInnova Progetto Gruppo di Ricerca n 85-2017-14986; n 33 & 55-2021-T0002E0001.
Author information
Authors and Affiliations
Consortia
Contributions
EC, YS, FB, and GM conceived the project; YL prepared the first draft; YL, SG, TW, JF, PL, CS, EC, YS, GM, and FB wrote the manuscript; YL prepared figures. All of the Authors have approved this submitted version.
Corresponding authors
Ethics declarations
Competing interests
GM and YS are members of the Editorial Board of Oncogene. The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Giovannini, S., Wang, T. et al. p63: a crucial player in epithelial stemness regulation. Oncogene 42, 3371–3384 (2023). https://doi.org/10.1038/s41388-023-02859-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-023-02859-4
- Springer Nature Limited