
Abstract
Cancer stem cells (CSCs), also known as tumor-initiating cells (TICs), contribute to tumorigenesis, resistance to chemoradiotherapy and recurrence in human cancers, suggesting targeting CSCs may represent a potential therapeutic strategy. In the current study, we found family with sequence similarity 83, member A (FAM83A) is significantly overexpressed and associated with poorer overall survival and disease-free survival in pancreatic cancer. Overexpression of FAM83A markedly promoted, whereas inhibition of FAM83A decreased, CSC-like traits and chemoresistance both in vitro and in an in vivo mouse model of pancreatic cancer. Furthermore, overexpression of FAM83A activated the well-characterized CSC-associated pathways transforming growth factor-β (TGF-β) signaling and Wnt/β-catenin signaling. Importantly, the FAM83A locus was amplified in a number of human cancers and silencing FAM83A in associated cancer cell lines inhibited activation of the WNT/β-catenin and TGF-β signaling pathways and reduced tumorigenicity. Taken together, these results indicate that FAM83A has a vital oncogenic role to promote pancreatic cancer progression and may represent a potential clinical target.
Similar content being viewed by others

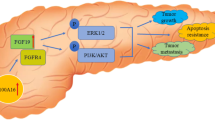

Introduction
Pancreatic cancer is the seventh leading cause of cancer-related mortality.1, 2 Despite advances in modern medical technology, pancreatic cancer has benefited from marginal improvements in survival outcomes; the 5-year overall survival rate of patients with pancreatic cancer is only 6% and the median survival time is <9 months.3, 4 Failure of conventional chemotherapy, including both intrinsic and acquired chemoresistant behavior, is a major factor that significantly decreases the clinical efficacy of chemotherapy for pancreatic cancer.5, 6 The response rates to common chemotherapeutic drugs, such as gemcitabine, erlotinib and 5-fluorouracil (5-FU), in pancreatic cancer have been reported to be lower than 25%.5, 7, 8 Therefore, better understanding the molecular mechanisms that underlie drug resistance in pancreatic cancer could lead to the development novel therapeutic strategies for this highly lethal malignancy.
The intrinsic resistance of cancer stem cells (CSCs), also known as tumor-initiating cells (TICs), to conventional therapy is currently regarded as a potential therapeutic target.9 For instance, it has recently been reported that the high rates and patterns of therapeutic failure observed in ovarian cancer are closely associated with stable accumulation of drug-resistant CSCs.10 Li et al.11 found that the percentage of the CD44+CD24–/low CSC sub-population, which exhibits intrinsic resistance to chemotherapy, was significantly increased in patients with breast cancer treated with chemotherapeutic drugs such as docetaxel, doxorubicin or cyclophosphamide. Similarly, CD133+ pancreatic CSCs have been demonstrated to be exclusively tumorigenic and highly resistant to chemotherapy and radiation therapy, and the CD133+ CXCR4+ sub-population of pancreatic CSCs is critical for tumor metastasis,12, 13, 14 suggesting that CSCs have important roles in pancreatic cancer progression. Therefore, targeting pancreatic CSCs could potentially increase chemosensitivity and thus improve the response to treatment.
Family with sequence similarity 83, member A (FAM83A), also known as BJ-TSA-9, is located on chromosome 8q24 and was originally identified as a potential tumor-specific gene by a bioinformatics approach.15 Furthermore, FAM83A is overexpressed in multiple human tumors, including lung, breast, testis and bladder cancer,16, 17, Primers and oligonucleotides Cloning primer human FAM83A-ORF, forward: 5′-AGCCGGTCAAGGCACCTGGG-3′ and reverse 5′-TCAGAAGTGAGGGGAGGCCTGCAGGAAGGGCCTCCAGGTT-3′; real-time PCR primer: FAM83A, forward: 5′-CCCATCTCAGTCACTGGCATT-3′ and reverse: 5′-CCGCCAACATCTCCTTGTTC-3′; ABCG2 forward: 5'-TGGTGTTTCCTTGTGACACTG-3′ and reverse: 5′-TGAGCCTTTGGTTAAGACCG-3′; BMI1 forward: 5′-TCGTTGTTCGATGCATTTCT-3′ and reverse: 5′-CTTTCATTGTCTTTTCCGCC-3′; SOX2 forward: 5′-GCTTAGCCTCGTCGATGAAC-3′ and reverse: 5′-AACCCCAAGATGCACAACTC-3′; OCT4 forward: 5′-GGTTCTCGATACTGGTTCGC-3′ and reverse: 5′-GTGGAGGAAGCTGACAACAA-3; NANOG forward: 5′-ATGGAGGAGGGAAGAGGAGA-3′ and reverse: 5′-GATTTGTGGGCCTGAAGAAA-3′; GAPDH forward: 5′-AATGAAGGGGTCATTGATGG-3′ and reverse: 5′-AAGGTGAAGGTCGGAGTCAA-3′. FAM83A primer used for genomic copy number detection: forward: 5′-CGCCACTGTGTACTTCCAGA-3′ and reverse: 5′-TCCACATCCGTGAACACATC-3′, FAM83A RNAi#1: 5′-GCACAACAACATCAGAGACCT-3′; FAM83A RNAi#2: 5′-GACTGGAGATTTGTCCTGTCT-3′. The human FAM83A gene was PCR-amplified from cDNA and cloned into pMSCV retroviral vector (Clontech, Mountain View, CA, USA). ShRNAs targeting FAM83A were cloned into the pSuper-retroviral vector. Transfection of plasmids was performed using the Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. Stable cell lines expressing FAM83A and FAM83A shRNA(s) were generated via retroviral infection as previously described,45 and were selected with 0.5 μg/ml puromycin for 10 days. Immunohistochemistry analysis was performed on the 103 paraffin-embedded pancreatic adenocarcinoma tissues, using anti-FAM83A antibody (Sigma). In brief, paraffin-embedded specimens were cut into 4-μm sections and baked at 65 °C for 30 min. The sections were deparaffinized with xylenes and rehydrated. Sections were submerged into EDTA antigenic retrieval buffer and microwaved for antigenic retrieval. The sections were treated with 3% hydrogen peroxide in methanol to quench the endogenous peroxidase activity, followed by incubation with 1% bovine serum albumin to block the nonspecific binding. Rabbit anti- FAM83A (1:500; Sigma) was incubated with the sections overnight at 4 °C. For negative controls, the IgG antibody or normal goat serum was co-incubation at 4 °C overnight preceding the immunohistochemical staining procedure. After washing, the tissue sections were treated with biotinylated anti-rabbit secondary antibody (Zymed, San Francisco, CA, USA), followed by further incubation with streptavidin-horseradish peroxidase complex (Zymed). The tissue sections were immersed in 3-amino-9-ethyl carbazole and counterstained with 10% Mayer's hematoxylin, dehydrated, and mounted in Crystal Mount. The degree of immunostaining were reviewed and scored separately by two independent pathologists blindly. The scores were determined by combining the proportion of positively stained tumor or normal pancreatic epithelial cells and the intensity of staining. Cell proportions were scored as follows: 0, no positive cells; 1, <10% positive cells; 2, 10–35% positive cells; 3, 35–75% positive cells; 4, >75% positive cells. Staining intensity was graded according to the following standard: 1, no staining; 2, weak staining (light yellow); 3, moderate staining (yellow brown); 4, strong staining (brown). The staining index (SI) was calculated as the product of the staining intensity score and the proportion of positive cells. Using this method of assessment, we evaluated protein expression in benign pancreatic epithelia and malignant lesions by determining the SI, with possible scores of 0, 2, 3, 4, 6, 8, 9, 12 and 16. Then the median value, which SI=8, was chosen as the cut off value. Therefore, samples with a SI⩾8 were determined as high expression and samples with a SI<8 were determined as low expression. Five hundred cells were seeded in six-well ultralow cluster plates (Corning, NY, USA) for 10 days. Spheres were cultured in Dulbecco’s modified Eagle’s medium/F12 serum-free medium (Invitrogen, Grand Island, NY, USA) supplemented with 2% B27 (Invitrogen, Grand Island, NY, USA), 20 ng/ml of EGF, and 20 ng/ml of bFGF (PeproTech, Offenbach, Germany), 0.4% bovine serum albumin (Sigma) and 5 μg/ml insulin. Gemcitabine (Gemzar, Lilly SA, Alcobendas, Spain) and 5-FU (Sigma; 03738) were dissolved in phosphate-buffered saline with concentration of 50 μM. β-Catenin/TCF inhibitor (FH535)(S7484), TGF-β inhibitor (S2704) were purchased from Selleck (Houston, TX, USA). The male/female BALB/c nude mice (6–7 weeks of age, 18–20 g) were randomly divided into 15 groups (n=6 per group). The indicated cells were inoculated with Matrigel subcutaneously into the inguinal folds of nude mice. Tumor volume was determined using external caliper and calculated using the equation (L × W2)/2. The mice were killed 31 days after inoculation, tumors were excised and subjected to pathologic examination. In the experiment testing, the chemoresistance effect of FAM83A, the BALB/c nude mice were implanted subcutaneously with the indicated cells (1 × 106) in order to rapidly induce exponentially growing tumors. When tumors reached a volume of approximate 100 mm3, animals were randomly assigned to five groups (n=6 per group), followed by intraperitoneal injection of Gemcitabine (80 mg/kg) twice a week. On day 43, animals were killed, and tumors were excised, weighed and subjected to pathological examination. All experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and conformed to our institutional ethical guidelines for animal experiments. Cells were dissociated with trypsin and re-suspended at 1 × 106 cells/ml in Dulbecco’s modified Eagle’s medium containing 2% fetal bovine serum and then pre-incubated at 37 °C for 30 min with or without 100 μM verapamil (Sigma-Aldrich, Deisenhofen, Germany) to inhibit ABC transporters. The cells were subsequently incubated for 90 min at 37 °C with 5 μg/ml Hoechst 33342 (Sigma-Aldrich). Finally, the cells were incubated on ice for 10 min and washed with ice-cold phosphate-buffered saline before flow cytometry analysis. The data were analyzed by Summit5.2 (Beckman Coulter, Indianapolis, IN, USA). Ten thousand cells were seeded in triplicate in 48-well plates and allowed to settle for 24 h. One hundred nanograms of luciferase reporter plasmids or the control-luciferase plasmid, plus 5 ng of pRL-TK renilla plasmid (Promega, Madison, WI, USA), were transfected into pancreatic adenocarcinoma cells using the Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendation. Luciferase and renilla signals were measured 48 h after transfection using the Dual Luciferase Reporter Assay Kit (Promega) according to a protocol provided by the manufacturer. Three independent experiments were performed, and the data are presented as mean±s.d. Statistical tests for data analysis included Fisher’s exact test, log-rank test, chi-square test and Student’s two-tailed t-test. Multivariate statistical analysis was performed using a Cox regression model. Statistical analyses were performed using the SPSS 11.0 statistical software package for Windows SPSS Inc. (Chicago, IL, USA). Data represent mean±s.d. P<0.05 was considered statistically significant.Plasmids, retroviral infection and transfection
Immunohistochemistry
Sphere formation assay
Chemical reagents
Xenografted tumor
Flow cytometric analysis
Luciferase assay
Statistical analysis
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136: E359–E386.
Bosetti C, Bertuccio P, Negri E, La Vecchia C, Zeegers MP, Boffetta P . Pancreatic cancer: overview of descriptive epidemiology. Mol Carcinog 2012; 51: 3–13.
Ilmer M, Boiles AR, Regel I, Yokoi K, Michalski CW, Wistuba II et al. RSPO2 enhances canonical Wnt signaling to confer stemness-associated traits to susceptible pancreatic cancer cells. Cancer Res 2015; 75: 1883–1896.
Siegel R, Naishadham D, Jemal A . Cancer statistics, 2013. CA Cancer J Clin 2013; 63: 11–30.
Singh SK, Chen NM, Hessmann E, Siveke J, Lahmann M, Singh G et al. Antithetical NFATc1-Sox2 and p53-miR200 signaling networks govern pancreatic cancer cell plasticity. EMBO J 2015; 34: 517–530.
Stathis A, Moore MJ . Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol 2010; 7: 163–172.
Li J, Wientjes MG, Au JL . Pancreatic cancer: pathobiology, treatment options, and drug delivery. AAPS J 2010; 12: 223–232.
Oberstein PE, Olive KP . Pancreatic cancer: why is it so hard to treat? Therap Adv Gastroenterol 2013; 6: 321–337.
Pattabiraman DR, Weinberg RA . Tackling the cancer stem cells-what challenges do they pose? Nat Rev Drug Discov 2014; 13: 497–512.
McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci USA 2012; 109: E2939–E2948.
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100: 672–679.
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007; 1: 313–323.
Lee HJ, You DD, Choi DW, Choi YS, Kim SJ, Won YS et al. Significance of CD133 as a cancer stem cell markers focusing on the tumorigenicity of pancreatic cancer cell lines. J Korean Surg Soc 2011; 81: 263–270.
Jimeno A, Feldmann G, Suarez-Gauthier A, Rasheed Z, Solomon A, Zou GM et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther 2009; 8: 310–314.
Li Y, Dong X, Yin Y, Su Y, Xu Q, Zhang Y et al. BJ-TSA-9, a novel human tumor-specific gene, has potential as a biomarker of lung cancer. Neoplasia 2005; 7: 1073–1080.
Cipriano R, Miskimen KL, Bryson BL, Foy CR, Bartel CA, Jackson MW . Conserved oncogenic behavior of the FAM83 family regulates MAPK signaling in human cancer. Mol Cancer Res 2014; 12: 1156–1165.
Liu L, Liao GQ, He P, Zhu H, Liu PH, Qu YM et al. Detection of circulating cancer cells in lung cancer patients with a panel of marker genes. Biochem Biophys Res Commun 2008; 372: 756–760.
Liu L, Ma C, Xu Q, Cheng L, **ao L, Xu D et al. A rapid nested polymerase chain reaction method to detect circulating cancer cells in breast cancer patients using multiple marker genes. Oncol Lett 2014; 7: 2192–2198.
Lee SY, Meier R, Furuta S, Lenburg ME, Kenny PA, Xu R et al. FAM83A confers EGFR-TKI resistance in breast cancer cells and in mice. J Clin Invest 2012; 122: 3211–3220.
Grant S . FAM83A and FAM83B: candidate oncogenes and TKI resistance mediators. J Clin Invest 2012; 122: 3048–3051.
Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 2001; 7: 1028–1034.
Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010; 12: 468–476.
Oshimori N, Oristian D, Fuchs E . TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015; 160: 963–976.
Goode EL, Chenevix-Trench G, Song H, Ramus SJ, Notaridou M, Lawrenson K et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet 2010; 42: 874–879.
Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014; 512: 82–86.
Ding J, Huang S, Wu S, Zhao Y, Liang L, Yan M et al. Gain of miR-151 on chromosome 8q24.3 facilitates tumour cell migration and spreading through downregulating RhoGDIA. Nat Cell Biol 2010; 12: 390–399.
Mahlamaki EH, Barlund M, Tanner M, Gorunova L, Hoglund M, Karhu R et al. Frequent amplification of 8q24, 11q, 17q, and 20q-specific genes in pancreatic cancer. Genes Chromosomes Cancer 2002; 35: 353–358.
Dean M, Fojo T, Bates S . Tumour stem cells and drug resistance. Nat Rev Cancer 2005; 5: 275–284.
Clevers H . The cancer stem cell: premises, promises and challenges. Nat Med 2011; 17: 313–319.
Wang Y, Liu Y, Malek SN, Zheng P, Liu Y . Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011; 8: 399–411.
Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012; 149: 1284–1297.
** L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE . Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 2006; 12: 1167–1174.
Lee S, Heinrich EL, Lu J, Lee W, Choi AH, Luu C et al. Epidermal growth factor receptor signaling to the mitogen activated protein kinase pathway bypasses Ras in pancreatic cancer cells. Pancreas 2016; 45: 286–292.
Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012; 22: 304–317.
Adjei AA . Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst 2001; 93: 1062–1074.
Brennan KR, Brown AM . Wnt proteins in mammary development and cancer. J Mammary Gland Biol Neoplasia 2004; 9: 119–131.
Tan AR, Alexe G, Reiss M . Transforming growth factor-beta signaling: emerging stem cell target in metastatic breast cancer? Breast Cancer Res Treat 2009; 115: 453–495.
Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 2006; 66: 6063–6071.
Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA et al. TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 2010; 18: 655–668.
Mishra L, Derynck R, Mishra B . Transforming growth factor-beta signaling in stem cells and cancer. Science 2005; 310: 68–71.
van Amerongen R, Bowman AN, Nusse R . Developmental stage and time dictate the fate of Wnt/beta-catenin-responsive stem cells in the mammary gland. Cell Stem Cell 2012; 11: 387–400.
Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011; 145: 926–940.
Klein T, Heremans Y, Heimberg H, Pipeleers D, Madsen OD, Serup P et al. Investigation and characterization of the duct cell-enriching process during serum-free suspension and monolayer culture using the human exocrine pancreas fraction. Pancreas 2009; 38: 36–48.
Yamaguchi H, Kojima T, Ito T, Kimura Y, Imamura M, Son S et al. Transcriptional control of tight junction proteins via a protein kinase C signal pathway in human telomerase reverse transcriptase-transfected human pancreatic duct epithelial cells. Am J Pathol 2010; 177: 698–712.
Liu L, Wu J, Ying Z, Chen B, Han A, Liang Y et al. Astrocyte elevated gene-1 upregulates matrix metalloproteinase-9 and induces human glioma invasion. Cancer Res 2010; 70: 3750–3759.
Acknowledgements
This work was supported by the Science and Technology Program of Guangdong Province (no 2014A020212155 and 2014A030313008), the PhD Start-up Fund of Natural Science Foundation of Guangdong Province (no 2015A030310052), the Natural Science Foundation of China (81325013 and 91529301), the Medical Scientific Research Foundation of Guangdong Province (no B2013127), the Natural Science Foundation of Guangdong Province (no 2015A030313044).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogenesis website
Rights and permissions
Oncogenesis is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Chen, S., Huang, J., Liu, Z. et al. FAM83A is amplified and promotes cancer stem cell-like traits and chemoresistance in pancreatic cancer. Oncogenesis 6, e300 (2017). https://doi.org/10.1038/oncsis.2017.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2017.3
- Springer Nature Limited
This article is cited by
-
Novel genome-wide DNA methylation profiling reveals distinct epigenetic landscape, prognostic model and cellular composition of early-stage lung adenocarcinoma
Journal of Translational Medicine (2024)
-
Promotion of stem cell-like phenotype of lung adenocarcinoma by FAM83A via stabilization of ErbB2
Cell Death & Disease (2024)
-
Diagnostic value of immune-related biomarker FAM83A in differentiating malignant from benign pleural effusion in lung adenocarcinoma
Discover Oncology (2024)
-
Pancreatic cancer stemness: dynamic status in malignant progression
Journal of Experimental & Clinical Cancer Research (2023)
-
Regulation of early diagnosis and prognostic markers of lung adenocarcinoma in immunity and hypoxia
Scientific Reports (2023)