
Abstract
Schizophrenia is a debilitating psychiatric disorder characterized by positive, negative and cognitive symptoms. Despite more than a century of research, the neurobiological mechanism underlying schizophrenia remains elusive. White matter abnormalities and interneuron dysfunction are the most widely replicated cellular neuropathological alterations in patients with schizophrenia. However, a unifying model incorporating these findings has not yet been established. Here, we propose that myelination of fast-spiking parvalbumin (PV) interneurons could be an important locus of pathophysiological convergence in schizophrenia. Myelination of interneurons has been demonstrated across a wide diversity of brain regions and appears highly specific for the PV interneuron subclass. Given the critical influence of fast-spiking PV interneurons for mediating oscillations in the gamma frequency range (~30–120 Hz), PV myelination is well positioned to optimize action potential fidelity and metabolic homeostasis. We discuss this hypothesis with consideration of data from human postmortem studies, in vivo brain imaging and electrophysiology, and molecular genetics, as well as fundamental and translational studies in rodent models. Together, the parvalbumin interneuron myelination hypothesis provides a falsifiable model for guiding future studies of schizophrenia pathophysiology.
Similar content being viewed by others

Main
Schizophrenia is a chronically debilitating psychiatric disorder with a lifetime prevalence of ~1%.1 Patients with schizophrenia classically exhibit a constellation of positive, negative and cognitive symptoms.2 Although many theories have been proposed, the precise neurobiological mechanism underlying schizophrenia has remained elusive. The most widely described models have been the dopamine3 and glutamate hypotheses,44 although in recent years models regarding interneuron dysfunction5 and myelination abnormalities6 have gained increasing support.
In this Perspective, we hypothesize that previous observations of interneuron dysfunction and myelination abnormalities in schizophrenia might converge on the altered myelination of fast-spiking parvalbumin (PV) interneurons. First, we summarize the major evidence supporting interneuron dysfunction and myelination abnormalities in schizophrenia. Next, we summarize electron microscopy and immunofluorescence studies that convincingly demonstrate interneuron myelination, which frequently occurs on fast-spiking PV interneurons. Finally, we discuss how impairments in myelination of PV interneurons could lead to consequent abnormalities in gamma synchronization and ultimately give rise to the symptoms which define schizophrenia.
Parvalbumin interneuron dysfunction in schizophrenia
Deficits in GABAergic signaling have been widely proposed as a fundamental pathophysiological mechanism underlying schizophrenia.7 More specifically, several recent lines of evidence including human postmortem studies, genetics and in vivo electrophysiological recordings in patients and translational mouse models have identified fast-spiking PV interneurons as the major GABAergic cell-type affected in schizophrenia (Table 1).
Expression of GAD67—the predominant gamma-aminobutyric acid (GABA) synthesizing enzyme—has consistently been found to be reduced at both the messenger RNA and protein levels in several brain regions of patients with schizophrenia, a finding that has been well controlled for confounding factors.8, 9, 10, 11, 12, 13, 14 Downregulation of GAD67 messenger RNA has been reported in ~30% of dorsolateral prefrontal cortex interneurons15, 16 and entirely undetectable in ~50% of PV interneurons.17 Expression of PV messenger RNA18, 19, 20 and protein21 is also reduced in schizophrenia, while the neuronal density of cortical PV interneurons is unchanged22, 23, 24, 25 (but see also ref. 52). Since the expression of both PV and GAD67 are experience-dependent26—and GAD67 and PV expression are highly correlated26—their shared downregulation suggests a functional impairment of fast-spiking interneurons.27 Morphologically, PV cell inputs onto pyramidal neurons have no discernible alterations21 suggesting a primary functional abnormality of PV interneurons. Consistent with these neuropathological findings, in vivo positron emission tomography (PET) imaging has demonstrated widespread alterations of cortical GABA transmission in schizophrenia, a finding that was most prominent in the subset of patients who were antipsychotic-naïve.28 Altogether, these results provide compelling evidence of cortical PV interneuron dysfunction in schizophrenia.
PV interneurons are essential in generating cortical oscillations in the gamma range (~30–120 Hz), mediated by synchronized inhibition of large pyramidal cell ensembles.29, 30 Through rhythmic perisomatic inhibition onto surrounding pyramidal cells, synchronous ensembles of PV cells evoke high-frequency gamma oscillations in the cerebral cortex.31, 32, 33 Gamma synchrony has been shown to function critically across a range of cognitive functions, including working memory and attention34 with well-replicated abnormalities in schizophrenia.5, 35 Abnormalities in other frequency bands such as theta and alpha have also been reported in schizophrenia, but the neural mechanisms underlying these frequencies remain less well understood.35
Electroencephalographic studies in schizophrenia have shown a reduced amplitude and impaired phase locking of gamma band activity over frontal areas while assessing working memory and executive functioning tasks.35 Although some studies have observed concurrent increases in gamma band activity at rest, this finding has been less well replicated.35 Taken together, impairments of in vivo gamma oscillations in patients with schizophrenia are highly consistent with the PV interneuron abnormalities observed by postmortem histopathology.
The classical onset of schizophrenia occurs within a relatively narrow window of neurodevelopment, between ~18 and 25 years of age.2 This late adolescent age of onset has often been attributed to the ongoing functional maturation of the brain during this neurodevelopmental critical period.2 Specifically in late adolescence, rates of synaptic pruning and myelination become asymptotic for which impairments in these processes have been linked to the disease onset.2 Notably, maturation of gamma band synchrony also occurs during late adolescence35 which coincides developmentally with the clinical onset of schizophrenia.1
In addition to in vivo brain imaging, electroencephalographic recordings and postmortem histopathology, molecular genetic studies of schizophrenia have also revealed an important contribution of interneuron dysfunction to the pathophysiology of schizophrenia. A recent genetic study of copy number variation has now provided causal evidence for GABAergic dysfunction in the etiology of schizophrenia.36 In this study, Pocklington et al. performed a functional gene set analysis for enriched biological mechanisms using a large schizophrenia case-control dataset and found that copy number variations were significantly enriched in cases for genes responsible for inhibitory neurotransmission (in particular the GABAA receptor complex), glutamatergic neurotransmission, long-term synaptic plasticity and associative learning. The genetic variant with the highest known risk for schizophrenia is the 22q11 microdeletion which has a penetrance of ~40%.37, 38 Transgenic mouse models have been generated to investigate the underlying neurobiology conferred by 22q11 microdeletion. Df(16)A mice harboring a 27-gene microdeletion syntenic to a 1.5 Mb region of human 22q11.2 exhibit similar brain abnormalities as found in human 22q11 microdeletion carriers, including cortico-cerebellar, cortico-striatal and cortico-limbic circuits.39 Moreover, multiple different mouse models of 22q11 microdeletion have replicated a cell-type specific impairment in PV interneurons and disrupted local synchrony of neural activity, consistent with the deficit in gamma oscillations observed in schizophrenia.40, 41, 42
Evidence for interneuron dysfunction in schizophrenia has also been supported by a wide variety of non-genetic rodent models.43 The major examples include pharmacological NMDA receptor antagonism and neurodevelopmental immunological challenge, both of which consistently exhibit synaptic and network abnormalities reminiscent of schizophrenia pathophysiology. Specifically, these studies have identified electrophysiological changes in local microcircuit connectivity and synaptic plasticity, with alterations in excitation/inhibition balance and gamma band synchronization.
Taken together, the combination of genetic, postmortem, and in vivo electrophysiological and functional imaging results from human clinical studies of schizophrenia converge with translational rodent modeling to identify fast-spiking PV interneuron dysfunction as a major pathophysiological mechanism underlying schizophrenia etiology.
Myelination abnormalities in schizophrenia
Independent of PV interneuron alterations, myelination abnormalities have also been extensively implicated in schizophrenia through both in vivo brain imaging and postmortem assessments (Table 1). Numerous diffusion tensor imaging studies have been published for schizophrenia (reviewed in ref. 6), of which the overwhelming consensus has been the association of schizophrenia with globally decreased fractional anisotropy. Notably, the decrease in fractional anisotropy appears to become more severe with increasing age and illness duration.44 Many of the early brain imaging studies of schizophrenia were performed in cohorts with extensive histories of psychotropic medication, inpatient hospitalization, smoking and medical co-morbidities, which could have a confounding deleterious influence on white matter integrity. Thus, an important question has been whether myelination abnormalities are already present in drug-naïve patients with first-episode schizophrenia who have never received psychotropic medication. Recently, several diffusion tensor imaging studies have been performed in such cohorts,44, 45, 46, 47, 48, 132 Synaptic input from local GABAergic interneurons has been shown to dynamically regulate OPC differentiation to oligodendrocytes.133 OPCs receive strong GABAergic synaptic input from PV, and to a lesser extent from non-PV, interneurons.132 Notably, the peak neurodevelopmental period of interneuron-OPC connectivity (P10-P14) would thus position interneuron myelination precisely in the window following the initial onset of GABAergic burst firing, but before maturation of high-frequency gamma oscillations.134 This also closely aligns with the timing of human frontal cortex oligodendrocyte development which plateaus in early adulthood and is highly distinct from white matter development in which oligodendrocytes have already reached their maximum number by ~5 years of age.135 Moreover, in further contrast to white matter, frontal cortex gray matter exhibits a substantial turnover of oligodendrocytes and myelin that persists throughout adulthood.135 Analogously, rodent studies have demonstrated that OPCs exhibit important distinctions in their physiology, proliferation and differentiation between gray and white matter in rodents.136 Therefore, regional differences in human OPCs are also not unlikely.
Interestingly, direct contacts of interneurons onto OPCs137 are only locally distributed, reaching a typical maximum distance of 50–70 μm,132 which is highly similar to the estimate for the maximal length of OPC processes. An interesting question remains why interneurons have such a restricted spatial localization of their connectivity onto OPCs, since PV cells establish synaptic contacts with pyramidal cells across a distance approximately six times larger.138 One possibility is that OPCs utilize reciprocal synaptic input to regulate their proliferative drive. Alternatively, it may be that myelination preferentially occurs on proximal axonal segments, in close apposition to the observed localization of OPCs and allowing for rapid differentiation to oligodendrocytes with enhanced myelination plasticity.
Potential functions of interneuron myelination
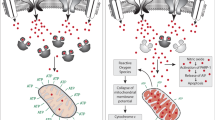
PV interneurons function to synchronize pyramidal cell ensembles, and thereby generate high-frequency oscillations.139 Since cortical PV axonal arborization is widely ramified and distributed over distances of up to 300 μm,138 there might be considerable benefits of myelination for optimizing the fidelity of fast action potential transmission. Indeed, computational modeling has suggested a unique contribution of (interneuron) conductance delays in the dynamics of gamma frequency oscillations.140 Evidence exists that nodes of Ranvier begin forming before the onset of myelination,117 a mechanism specific for GABAergic neurons, which enhances axonal conduction of action potentials without myelin. Thus, in addition to simply increasing the speed of action potential propagation, myelin could function to ensure the integrity of precisely timed action potentials, as has been proposed for myelinated excitatory axons.141 Myelin plasticity would then have the potential to support the local synchronization of action potentials necessary for generating high-frequency oscillations.142 Indeed, myelinated axons exhibit both higher conduction velocities and enhanced long-range coherence.143 Although non-PV cortical interneuron subtypes (e.g., somatostatin, VIP) exhibit synaptic connectivity across similar distances,138 their lack of influence in maintaining high-frequency oscillations is consistent with their absence of myelination. Furthermore, the activity-dependence of myelination144 might permit dynamically regulated influences on the fidelity of fast action potential transmission and high-frequency oscillations.
Furthermore, myelin could provide metabolic and trophic support for energetically costly PV cells. PV cell characteristics, including high-frequency spiking and rapid action potential kinetics, require a particularly high energy utilization through predominantly mitochondrial oxidative phosphorylation.145 Gamma band synchrony, closely linked to cognition, is highly sensitive to metabolic disruption. Furthermore, compared with pyramidal cells, PV cells exhibit high densities of mitochondria and expression of cytochrome c and cytochrome c oxidase, proteins crucial for the electron transport chain. Moreover, PV cell-specific disruption of cytochrome oxidase assembly leads to changes in PV cell intrinsic excitability, afferent synaptic input, and gamma/theta oscillations, as well as schizophrenia-related behavioral impairments in sensory gating and social behavior.146
During gamma oscillations, peak oxygen consumption approaches the demand observed during seizures and mitochondrial oxidative capacity operates near its functional limit.145 Metabolic and trophic support conferred by myelination147, 148 might therefore allow PV axons to optimize their energy utilization. Consistent with the importance of myelination in regulating axonal energy metabolism is the considerable discrepancy of mitochondria content (30-fold) in myelinated versus unmyelinated axons.149 Myelin has been proposed to regulate axonal energy metabolism via the monocarboxylate transporter 1 channel.147, 148 Furthermore, the high-peak oxygen consumption of PV cells during gamma band synchrony could require the additional lactate provided by oligodendrocytes.
Taken together, the electrophysiological dynamics of fast-spiking PV interneurons, their dense branching onto pyramidal neurons requiring finely tuned temporally synchronized inhibition, and their high-energy consumption are likely interdependent mechanisms governed by PV interneuron myelination.
Implications for schizophrenia
Both interneuron dysfunction and myelination abnormalities have been independently proposed as important contributors to the underlying pathophysiology of schizophrenia. These mechanisms have each amassed convincing support from postmortem histopathology, in vivo imaging and electrophysiology, genetics and neurodevelopment (Table 1). However, neither hypothesis is capable of accounting for the full set of clinical research findings in schizophrenia. In contrast, interneuron myelination brings together both of these models, explains a more comprehensive portion of the existing data, and offers a well-defined falsifiable model.
Impairments of PV interneuron myelination could directly contribute to schizophrenia through several mechanisms. Impaired action potential fidelity, energy restrictions during highly-demanding cognitive tasks, aberrant axonal branching and a higher occurrence of ectopic action potentials could each independently, or in combination, disrupt inhibitory network function. Such changes to PV interneurons would likely result in abnormalities of local gamma synchronization, with a potential further impact on the integrity of long-range thalamocortical and cortico-striatal circuits, and striatal dopamine signaling, ultimately giving rise to schizophrenia symptoms.
In this Perspective, we have proposed the novel hypothesis that altered myelination of PV interneurons might function prominently in the pathophysiology of schizophrenia. However, many questions remain to be answered. At what point during development does interneuron myelination occur and to what extent does this coincide with the clinical symptoms of schizophrenia? Does interneuron myelination vary across brain regions? Is cortical interneuron myelination truly reserved for fast-spiking PV interneurons, or are non-fast-spiking interneurons (e.g., somatostatin, VIP) myelinated as well? How does the plasticity of PV interneuron myelination compare with that of glutamatergic axons? And perhaps most importantly, to what extent might PV interneuron myelination represent an etiological pathophysiology and therapeutic target for schizophrenia?
Future studies to examine the parvalbumin interneuron myelination hypothesis could be approached through a variety of methods. In particular, the most important experiments would include: (a) detailed histological assessments of subtype-specific interneuron axonal myelination in postmortem brain tissue from patients with schizophrenia, (b) corresponding functional studies in rodent models of schizophrenia to directly assess the causality of alterations in parvalbumin interneuron myelination on behavioral and electrophysiological phenotypes, (c) electrophysiological studies of rodent models with temporally-controlled and cell-type specific disruption of myelination and (d) functional genomic studies on the effect of schizophrenia risk variants on (interneuron) myelination, for example, by utilizing human induced pluripotent stem cells or genetically modified mice.
References
Owen MJ, Sawa A, Mortensen PB . Schizophrenia. Lancet 2016; 388: 86–97.
Insel TR . Rethinking schizophrenia. Nature 2010; 468: 187–193.
Carlsson A . The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1988; 1: 179–186.
Moghaddam B, Javitt D . From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012; 37: 4–15.
Gonzalez-Burgos G, Cho RY, Lewis DA . Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry 2015; 77: 1031–1040.
Fitzsimmons J, Kubicki M, Shenton ME . Review of functional and anatomical brain connectivity findings in schizophrenia. Curr Opin Psychiatry 2013; 26: 172–187.
Lewis DA, Curley AA, Glausier JR, Volk DW . Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 2012; 35: 57–67.
Gonzalez-Burgos G, Hashimoto T, Lewis DA . Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr Psychiatry Rep 2010; 12: 335–344.
Hashimoto T, Arion D, Unger T, Maldonado-Avilés JG, Morris HM, Volk DW et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry 2008; 13: 147–161.
Thompson M, Weickert CS, Wyatt E, Webster MJ . Decreased glutamic acid decarboxylase(67) mRNA expression in multiple brain areas of patients with schizophrenia and mood disorders. J Psychiatr Res 2009; 43: 970–977.
Woo TU, Walsh JP, Benes FM . Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry 2004; 61: 649–657.
Impagnatiello F . A decrease of reelin expression as a putative vulnerability factor. Proc Natl Acad Sci USA 1998; 95: 15718–15723.
Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry 2000; 57: 1061–1069.
Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN et al. Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am J Psychiatry 2011; 168: 921–929.
Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 1995; 52: 258–266.
Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA . Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry 2000; 57: 237–245.
Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 2003; 23: 6315–6326.
Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA . Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am J Psychiatry 2008; 165: 479–489.
Mellios N, Huang H-S, Baker SP, Galdzicka M, Ginns E, Akbarian S . Molecular determinants of dysregulated GABAergic gene expression in the prefrontal cortex of subjects with schizophrenia. Biol Psychiatry 2009; 65: 1006–1014.
Fung SJ, Webster MJ, Sivagnanasundaram S, Duncan C, Elashoff M, Weickert CS . Expression of interneuron markers in the dorsolateral prefrontal cortex of the develo** human and in schizophrenia. Am J Psychiatry 2010; 167: 1479–1488.
Glausier JR, Fish KN, Lewis DA . Altered parvalbumin basket cell inputs in the dorsolateral prefrontal cortex of schizophrenia subjects. Mol Psychiatry 2014; 19: 30–36.
Woo TU, Miller JL, Lewis DA . Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am J Psychiatry 1997; 154: 1013–1015.
Beasley CL, Zhang ZJ, Patten I, Reynolds GP . Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry 2002; 52: 708–715.
Tooney PA, Chahl LA . Neurons expressing calcium-binding proteins in the prefrontal cortex in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2004; 28: 273–278.
Pantazopoulos H, Woo TU, Lim MP, Lange N, Berretta S . Extracellular matrix-glial abnormalities in the amygdala and entorhinal cortex of subjects diagnosed with schizophrenia. Arch Gen Psychiatry 2010; 67: 155–166.
Donato F, Rompani SB, Caroni P . Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 2013; 504: 272–276.
Brown JA, Ramikie TS, Schmidt MJ, Báldi R, Garbett K, Everheart MG et al. Inhibition of parvalbumin-expressing interneurons results in complex behavioral changes. Mol Psychiatry 2015; 20: 1499–1507.
Frankle WG, Cho RY, Prasad KM, Mason NS, Paris J, Himes ML et al. In vivo measurement of GABA transmission in healthy subjects and schizophrenia patients. Am J Psychiatry 2015; 172: 1148–1159.
Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 2009; 459: 663–667.
Sohal VS, Zhang F, Yizhar O, Deisseroth K . Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009; 459: 698–702.
Hormuzdi SG, Pais I, LeBeau FE, Towers SK, Rozov A, Buhl EH et al. Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36-deficient mice. Neuron 2001; 31: 487–495.
Traub RD, Kopell N, Bibbig A, Buhl EH, LeBeau FE, Whittington MA . Gap junctions between interneuron dendrites can enhance synchrony of gamma oscillations in distributed networks. J Neurosci 2001; 21: 9478–9486.
Whittington MA, Traub RD, Jefferys JG . Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature 1995; 373: 612–615.
Senkowski D, Gallinat J . Dysfunctional prefrontal gamma-band oscillations reflect working memory and other cognitive deficits in schizophrenia. Biol Psychiatry 2015; 77: 1010–1019.
Uhlhaas PJ, Singer W . Oscillations and neuronal dynamics in schizophrenia: the search for basic symptoms and translational opportunities. Biol Psychiatry 2014; 77: 1001–1009.
Pocklington AJ, Rees E, Walters JTR, Han J, Kavanagh DH, Chambert KD et al. Novel findings from CNVs implicate inhibitory and excitatory signaling complexes in schizophrenia. Neuron 2015; 86: 1203–1214.
Karayiorgou M, Morris MA, Morrow B, Shprintzen RJ, Goldberg R, Borrow J et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci USA 1995; 92: 7612–7616.
Lindsay EA, Morris MA, Gos A, Nestadt G, Wolyniec PS, Lasseter VK et al. Schizophrenia and chromosomal deletions within 22q11.2. Am J Hum Genet 1995; 56: 1502–1503.
Ellegood J, Markx S, Lerch JP, Steadman PE, Genç C, Provenzano F et al. Neuroanatomical phenotypes in a mouse model of the 22q11.2 microdeletion. Mol Psychiatry 2014; 19: 99–107.
Chun S, Westmoreland JJ, Bayazitov IT, Eddins D, Pani AK, Smeyne RJ et al. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science 2014; 344: 1178–1182.
Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA . Impaired hippocampal–prefrontal synchrony in a genetic mouse model of schizophrenia. Nature 2010; 464: 763–767.
Meechan DW, Tucker ES, Maynard TM, LaMantia A-S . Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc Natl Acad Sci USA 2009; 106: 16434–16445.
Rosen AM, Spellman T, Gordon JA . Electrophysiological endophenotypes in rodent models of schizophrenia and psychosis. Biol Psychiatry 2015; 77: 1041–1049.
White T, Magnotta VA, Bockholt HJ, Williams S, Wallace S, Ehrlich S et al. Global white matter abnormalities in schizophrenia: a multisite diffusion tensor imaging study. Schizophr Bull 2011; 37: 222–232.
Alvarado-Alanis P, León-Ortiz P, Reyes-Madrigal F, Favila R, Rodríguez-Mayoral O, Nicolini H et al. Abnormal white matter integrity in antipsychotic-naïve first-episode psychosis patients assessed by a DTI principal component analysis. Schizophr Res 2015; 162: 14–21.
Cheung V, Cheung C, McAlonan GM, Deng Y, Wong JG, Yip L et al. A diffusion tensor imaging study of structural dysconnectivity in never-medicated, first-episode schizophrenia. Psychol Med 2008; 38: 877–885.
Filippi M, Canu E, Gasparotti R, Agosta F, Valsecchi P, Lodoli G et al. Patterns of brain structural changes in first-contact, antipsychotic drug-naive patients with schizophrenia. Am J Neuroradiol 2013; 35: 30–37.
Gasparotti R, Valsecchi P, Carletti F, Galluzzo A, Liserre R, Cesana B et al. Reduced fractional anisotropy of corpus callosum in first-contact, antipsychotic drug-naive patients with schizophrenia. Schizophr Res 2009; 108: 41–48.
Guo W, Liu F, Liu Z, Gao K, **ao C, Chen H et al. Right lateralized white matter abnormalities in first-episode, drug-naive paranoid schizophrenia. Neurosci Lett 2012; 531: 5–9.
Pérez-Iglesias R, Tordesillas-Gutiérrez D, Barker GJ, McGuire PK, Roiz-Santiañez R, Mata I et al. White matter defects in first episode psychosis patients: a voxelwise analysis of diffusion tensor imaging. Neuroimage 2010; 49: 199–204.
Mandl RCW, Rais M, van Baal GCM, van Haren NEM, Cahn W, Kahn RS et al. Altered white matter connectivity in never-medicated patients with schizophrenia. Hum Brain Mapp 2013; 34: 2353–2365.
Melicher T, Horacek J, Hlinka J, Spaniel F, Tintera J, Ibrahim I et al. White matter changes in first episode psychosis and their relation to the size of sample studied: a DTI study et al. White matter abnormalities in schizophrenia and schizotypal personality disorder. Schizophr Bull 2015; 41: 300–310.
Lener MS, Wong E, Tang CY, Byne W, Goldstein KE, Blair NJ et al. White matter abnormalities in schizophrenia and schizotypal personality disorder. Schizophr Bull 2015; 41: 300–310.
Lyu H, Hu M, Eyler LT, ** H, Wang J, Ou J et al. Regional white matter abnormalities in drug-naive, first-episode schizophrenia patients and their healthy unaffected siblings. Aust N Z J Psychiatry 2015; 49: 246–254.
Liu X, Lai Y, Wang X, Hao C, Chen L, Zhou Z et al. A combined DTI and structural MRI study in medicated-naïve chronic schizophrenia. Magn Reson Imaging 2014; 32: 1–8.
Sun H, Lui S, Yao L, Deng W, **ao Y, Zhang W et al. Two patterns of white matter abnormalities in medication-naive patients with first-episode schizophrenia revealed by diffusion tensor imaging and cluster analysis. JAMA Psychiatry 2015; 72: 678–686.
Yao L, Lui S, Deng W, Wu M, Chen L, **ao Y et al. Association of white matter deficits with clinical symptoms in antipsychotic-naive first-episode schizophrenia: an optimized VBM study using 3 T. MAGMA 2014; 27: 283–290.
Wang Q, Cheung C, Deng W, Li M, Huang C, Ma X et al. White-matter microstructure in previously drug-naive patients with schizophrenia after 6 weeks of treatment. Psychol Med 2013; 43: 2301–2309.
Do KQ, Cuenod M, Hensch TK . Targeting oxidative stress and aberrant critical period plasticity in the developmental trajectory to schizophrenia. Schizophr Bull 2015; 41: 835–846.
Hermoye L, Saint-Martin C, Cosnard G, Lee S-K, Kim J, Nassogne M-C et al. Pediatric diffusion tensor imaging: normal database and observation of the white matter maturation in early childhood. Neuroimage 2006; 29: 493–504.
Lebel C, Walker L, Leemans A, Phillips L, Beaulieu C . Microstructural maturation of the human brain from childhood to adulthood. Neuroimage 2008; 40: 1044–1055.
Lebel C, Gee M, Camicioli R, Wieler M, Martin W, Beaulieu C . Diffusion tensor imaging of white matter tract evolution over the lifespan. Neuroimage 2012; 60: 340–352.
Miller DJ, Duka T, Stimpson CD, Schapiro SJ, Baze WB, McArthur MJ et al. Prolonged myelination in human neocortical evolution. Proc Natl Acad Sci USA 2012; 109: 16480–16485.
Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB et al. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 2003; 362: 798–805.
Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 2001; 98: 4746–4751.
Barley K, Dracheva S, Byne W . Subcortical oligodendrocyte- and astrocyte-associated gene expression in subjects with schizophrenia, major depression and bipolar disorder. Schizophr Res 2009; 112: 54–64.
Katsel P, Davis KL, Gorman JM, Haroutunian V . Variations in differential gene expression patterns across multiple brain regions in schizophrenia. Schizophr Res 2005; 77: 241–252.
Aberg K, Saetre P, Jareborg N, Jazin E . Human QKI, a potential regulator of mRNA expression of human oligodendrocyte-related genes involved in schizophrenia. Proc Natl Acad Sci USA 2006; 103: 7482–7487.
Schmitt A, Wilczek K, Blennow K, Maras A, Jatzko A, Petroianu G et al. Altered thalamic membrane phospholipids in schizophrenia: a postmortem study. Biol Psychiatry 2004; 56: 41–45.
Matthews PR, Eastwood SL, Harrison PJ . Reduced myelin basic protein and actin-related gene expression in visual cortex in schizophrenia. PLoS One 2012; 7: e38211.
Aston C, Jiang L, Sokolov BP . Microarray analysis of postmortem temporal cortex from patients with schizophrenia. J Neurosci Res 2004; 77: 858–866.
Harris LW, Lockstone HE, Khaitovich P, Weickert CS, Webster MJ, Bahn S . Gene expression in the prefrontal cortex during adolescence: implications for the onset of schizophrenia. BMC Med Genomics 2009; 2: 28.
Hof PR, Haroutunian V, Copland C, Davis KL, Buxbaum JD . Molecular and cellular evidence for an oligodendrocyte abnormality in schizophrenia. Neurochem Res 2002; 27: 1193–1200.
Hof PR, Haroutunian V, Friedrich VL, Byne W, Buitron C, Perl DP et al. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol Psychiatry 2003; 53: 1075–1085.
Uranova NA, Kolomeets NS, Vigreva OV, Zimina IS, Rachmanova VI, Orlovskaya DD . Ultrastructural pathology of myelinated fibers in schizophrenia. Zh Nevrol Psikhiatr Im S S Korsakova 2013; 113: 63–69.
Uranova NA, Vostrikov VM, Vikhreva OV, Zimina IS, Kolomeets NS, Orlovskaya DD . The role of oligodendrocyte pathology in schizophrenia. Int J Neuropsychopharmacol 2007; 10: 537–545.
Vostrikov VM, Uranova NA, Rakhmanova VI, Orlovskaia DD . Lowered oligodendroglial cell density in the prefrontal cortex in schizophrenia. Zh Nevrol Psikhiatr Im S S Korsakova 2004; 104: 47–51.
Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI . Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr Res 2004; 67: 269–275.
Schmitt A, Steyskal C, Bernstein H-G, Schneider-Axmann T, Parlapani E, Schaeffer EL et al. Stereologic investigation of the posterior part of the hippocampus in schizophrenia. Acta Neuropathol 2009; 117: 395–407.
Kerns D, Vong GS, Barley K, Dracheva S, Katsel P, Casaccia P et al. Gene expression abnormalities and oligodendrocyte deficits in the internal capsule in schizophrenia. Schizophr Res 2010; 120: 150–158.
Williams MR, Marsh R, Macdonald CD, Jain J, Pearce RKB, Hirsch SR et al. Neuropathological changes in the nucleus basalis in schizophrenia. Eur Arch Psychiatry Clin Neurosci 2013; 263: 485–495.
Byne W, Tatusov A, Yiannoulos G, Vong GS, Marcus S . Effects of mental illness and aging in two thalamic nuclei. Schizophr Res 2008; 106: 172–181.
Mauney SA, Pietersen CY, Sonntag KC, Woo TU . Differentiation of oligodendrocyte precursors is impaired in the prefrontal cortex in schizophrenia. Schizophr Res 2015; 169: 374–380.
Williams MR, Galvin K, O’Sullivan B, MacDonald CD, Ching EWK, Turkheimer F et al. Neuropathological changes in the substantia nigra in schizophrenia but not depression. Eur Arch Psychiatry Clin Neurosci 2014; 264: 285–296.
Williams MR, Hampton T, Pearce RKB, Hirsch SR, Ansorge O, Thom M et al. Astrocyte decrease in the subgenual cingulate and callosal genu in schizophrenia. Eur Arch Psychiatry Clin Neurosci 2013; 263: 41–52.
Chambers JS, Perrone-Bizzozero NI . Altered myelination of the hippocampal formation in subjects with schizophrenia and bipolar disorder. Neurochem Res 2004; 29: 2293–2302.
Bernstein H-G, Smalla K-H, Dürrschmidt D, Keilhoff G, Dobrowolny H, Steiner J et al. Increased density of prohibitin-immunoreactive oligodendrocytes in the dorsolateral prefrontal white matter of subjects with schizophrenia suggests extraneuronal roles for the protein in the disease. Neuromolecular Med 2012; 14: 270–280.
Williams MR, Harb H, Pearce RKB, Hirsch SR, Maier M . Oligodendrocyte density is changed in the basolateral amygdala in schizophrenia but not depression. Schizophr Res 2013; 147: 402–403.
Miyakawa T, Sumiyoshi S, Deshimaru M, Suzuki T, Tomonari H . Electron microscopic study on schizophrenia. Mechanism of pathological changes. Acta Neuropathol 1972; 20: 67–77.
Uranova NA, Vikhreva OV, Rachmanova VI, Orlovskaya DD . Ultrastructural alterations of myelinated fibers and oligodendrocytes in the prefrontal cortex in schizophrenia: a postmortem morphometric study. Schizophr Res Treatment 2011; 2011: 325789.
Toritsuka M, Makinodan M, Kishimoto T . Social experience-dependent myelination: an implication for psychiatric disorders. Neural Plast 2015; 2015: 465345.
Duncan LE, Holmans PA, Lee PH, O’Dushlaine CT, Kirby AW, Smoller JW et al. Pathway analyses implicate glial cells in schizophrenia. PLoS One 2014; 9: e89441.
Goudriaan A, de Leeuw C, Ripke S, Hultman CM, Sklar P, Sullivan PF et al. Specific glial functions contribute to schizophrenia susceptibility. Schizophr Bull 2014; 40: 925–935.
Xu K, Schadt EE, Pollard KS, Roussos P, Dudley JT . Genomic and network patterns of schizophrenia genetic variation in human evolutionary accelerated regions. Mol Biol Evol 2015; 32: 1148–1160.
Chavarria-Siles I, White T, de Leeuw C, Goudriaan A, Lips E, Ehrlich S et al. Myelination-related genes are associated with decreased white matter integrity in schizophrenia. Eur J Hum Genet 2016; 24: 381–386.
Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427.
McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM . Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science 2005; 309: 2222–2226.
Somogyi P, Soltész I . Immunogold demonstration of GABA in synaptic terminals of intracellularly recorded, horseradish peroxidase-filled basket cells and clutch cells in the cat’s visual cortex. Neuroscience 1986; 19: 1051–1065.
Somogyi P, Kisvárday ZF, Martin KA, Whitteridge D . Synaptic connections of morphologically identified and physiologically characterized large basket cells in the striate cortex of cat. Neuroscience 1983; 10: 261–294.
Somogyi P, Freund TF, Wu JY, Smith AD . The section-Golgi impregnation procedure. 2. Immunocytochemical demonstration of glutamate decarboxylase in Golgi-impregnated neurons and in their afferent synaptic boutons in the visual cortex of the cat. Neuroscience 1983; 9: 475–490.
Chung K, Wallace J, Kim S-Y, Kalyanasundaram S, Andalman AS, Davidson TJ et al. Structural and molecular interrogation of intact biological systems. Nature 2013; 497: 332–337.
Ong WY, Yeo TT, Balcar VJ, Garey LJ . A light and electron microscopic study of GAT-1-positive cells in the cerebral cortex of man and monkey. J Neurocytol 1998; 27: 719–730.
Seress L, Gulyás AI, Ferrer I, Tunon T, Soriano E, Freund TF . Distribution, morphological features, and synaptic connections of parvalbumin- and calbindin D28k-immunoreactive neurons in the human hippocampal formation. J Comp Neurol 1993; 337: 208–230.
Hinova-Palova DV, Edelstein L, Landzhov BV, Braak E, Malinova LG, Minkov M et al. Parvalbumin-immunoreactive neurons in the human claustrum. Brain Struct Funct 2014; 219: 1813–1830.
Sloper JJ, Powell TP . A study of the axon initial segment and proximal axon of neurons in the primate motor and somatic sensory cortices. Philos Trans R Soc Lond B Biol Sci 1979; 285: 173–197.
Hendry SH, Houser CR, Jones EG, Vaughn JE . Synaptic organization of immunocytochemically identified GABA neurons in the monkey sensory-motor cortex. J Neurocytol 1983; 12: 639–660.
DeFelipe J, Jones EG . Vertical organization of gamma-aminobutyric acid-accumulating intrinsic neuronal systems in monkey cerebral cortex. J Neurosci 1985; 5: 3246–3260.
DeFelipe J, Hendry SH, Jones EG . A correlative electron microscopic study of basket cells and large GABAergic neurons in the monkey sensory-motor cortex. Neuroscience 1986; 17: 991–1009.
Takasu N, Nakatani T, Arikuni T, Kimura H . Immunocytochemical localization of gamma-aminobutyric acid in the hypoglossal nucleus of the macaque monkey, Macaca fuscata: a light and electron microscopic study. J Comp Neurol 1987; 263: 42–53.
Ralston DD, Milroy AM . Inhibitory synaptic input to identified rubrospinal neurons in Macaca fascicularis: an electron microscopic study using a combined immuno-GABA-gold technique and the retrograde transport of WGA-HRP. J Comp Neurol 1992; 320: 97–109.
**no S, Klausberger T, Marton LF, Dalezios Y, Roberts JDB, Fuentealba P et al. Neuronal diversity in GABAergic long-range projections from the hippocampus. J Neurosci 2007; 27: 8790–8804.
Peters A, Proskauer CC . Smooth or sparsely spined cells with myelinated axons in rat visual cortex. Neuroscience 1980; 5: 2079–2092.
De Biasi S, Amadeo A, Arcelli P, Frassoni C, Spreafico R . Postnatal development of GABA-immunoreactive terminals in the reticular and ventrobasal nuclei of the rat thalamus: a light and electron microscopic study. Neuroscience 1997; 76: 503–515.
Conti F, Zuccarello LV, Barbaresi P, Minelli A, Brecha NC, Melone M . Neuronal, glial, and epithelial localization of gamma-aminobutyric acid transporter 2, a high-affinity gamma-aminobutyric acid plasma membrane transporter, in the cerebral cortex and neighboring structures. J Comp Neurol 1999; 409: 482–494.
Gartner U, Hartig CAW, Brauer K, Brückner G, Arendt T . Electron microscopic evidence for different myelination of rat septohippocampal fibres. Neuroreport 2001; 12: 17–20.
Gartner U, Brauer K, Hartig W, Brückner G, Arendt T . Different myelination of rat septohippocampal fibres as revealed by immunofluorescence double-labelling. Brain Res 2000; 878: 188–193.
Freeman SA, Desmazières A, Simonnet J, Gatta M, Pfeiffer F . Acceleration of conduction velocity linked to clustering of nodal components precedes myelination. Proc Natl Acad Sci USA 2015; 112: E321–E328.
Mize RR . Immunocytochemical localization of gamma-aminobutyric acid (GABA) in the cat superior colliculus. J Comp Neurol 1988; 276: 169–187.
Wouterlood FG, Härtig W, Brückner G, Witter MP . Parvalbumin-immunoreactive neurons in the entorhinal cortex of the rat: localization, morphology, connectivity and ultrastructure. J Neurocytol 1995; 24: 135–153.
Sawyer SF, Martone ME, Groves PM . A GABA immunocytochemical study of rat motor thalamus: light and electron microscopic observations. Neuroscience 1991; 42: 103–124.
Roberts RC . An electron microscopic study of GABAergic neurons and terminals in the central nucleus of the inferior colliculus of the rat. J Neurocytol 1987; 16: 333–345.
Katsumaru H, Kosaka T, Heizmann CW, Hama K . Immunocytochemical study of GABAergic neurons containing the calcium-binding protein parvalbumin in the rat hippocampus. Exp Brain Res 1988; 72: 347–362.
Kita H, Kosaka T, Heizmann CW . Parvalbumin-immunoreactive neurons in the rat neostriatum: a light and electron microscopic study. Brain Res 1990; 536: 1–15.
Brauer K, Härtig W, Gärtner U, Brückner G, Arendt T . Different myelination of rat septohippocampal fibres as revealed by immunofluorescence double-labelling. Brain Res 2000; 878: 188–193.
Micheva KD, Wolman D, Mensh BD, Pax E, Buchanan J, Smith SJ et al. A large fraction of neocortical myelin ensheathes axons of local inhibitory neurons. Elife 2016; 5: e15784.
Aoki C, Pickel VM . Neuropeptide Y in cortex and striatum. Ultrastructural distribution and coexistence with classical neurotransmitters and neuropeptides. Ann N Y Acad Sci 1990; 611: 186–205.
Cipolloni PB, Keller A . Thalamocortical synapses with identified neurons in monkey primary auditory cortex: a combined Golgi/EM and GABA/peptide immunocytochemistry study. Brain Res 1989; 492: 347–355.
Mizukawa K, McGeer PL, Vincent SR, McGeer EG . The distribution of somatostatin-immunoreactive neurons and fibers in the rat cerebral cortex: light and electron microscopic studies. Brain Res 1987; 426: 28–36.
De Lima AD, Morrison JH . Ultrastructural analysis of somatostatin-immunoreactive neurons and synapses in the temporal and occipital cortex of the macaque monkey. J Comp Neurol 1989; 283: 212–227.
DiFiglia M, Aronin N . Ultrastructural features of immunoreactive somatostatin neurons in the rat caudate nucleus. J Neurosci 1982; 2: 1267–1274.
Hinova-Palova DV, Landzhov B, Dzhambazova E, Minkov M, Edelstein L, Malinova L et al. Neuropeptide Y immunoreactivity in the cat claustrum: A light- and electron-microscopic investigation. J Chem Neuroanat 2014; 61–62: 107–119.
Orduz D, Maldonado PP, Balia M, Vélez-Fort M, de Sars V, Yanagawa Y et al. Interneurons and oligodendrocyte progenitors form a structured synaptic network in the develo** neocortex. Elife 2015; 4: e06953.
Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL et al. GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat Neurosci 2015; 18: 674–682.
Le Magueresse C, Monyer H . GABAergic interneurons shape the functional maturation of the cortex. Neuron 2013; 77: 388–405.
Yeung MSY, Zdunek S, Bergmann O, Bernard S, Salehpour M, Alkass K et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell 2014; 159: 766–774.
Hill RA, Patel KD, Medved J, Reiss AM, Nishiyama A . NG2 cells in white matter but not gray matter proliferate in response to PDGF. J Neurosci 2013; 33: 14558–14566.
Lin S, Bergles DE . Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat Neurosci 2004; 7: 24–32.
Fino E, Packer AM, Yuste R . The logic of inhibitory connectivity in the neocortex. Neuroscientist 2013; 19: 228–237.
Hu H, Gan J, Jonas P . Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science 2014; 345: 1255263.
Jadi MP, Margarita Behrens M, Sejnowski TJ . Abnormal gamma oscillations in N-Methyl-D-Aspartate receptor hypofunction models of schizophrenia. Biol Psychiatry 2015; 79: 716–726.
Yamazaki Y, Fujiwara H, Kaneko K, Hozumi Y, Xu M, Ikenaka K et al. Short- and long-term functional plasticity of white matter induced by oligodendrocyte depolarization in the hippocampus. Glia 2014; 62: 1299–1312.
Pajevic S, Basser PJ, Fields RD . Role of myelin plasticity in oscillations and synchrony of neuronal activity. Neuroscience 2014; 276: 135–147.
Salami M, Itami C, Tsumoto T, Kimura F . Change of conduction velocity by regional myelination yields constant latency irrespective of distance between thalamus and cortex. Proc Natl Acad Sci USA 2003; 100: 6174–6179.
Fields RD . A new mechanism of nervous system plasticity: activity-dependent myelination. Nat Rev Neurosci 2015; 16: 756–767.
Kann O, Papageorgiou IE, Draguhn A . Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J Cereb Blood Flow Metab 2014; 34: 1270–1282.
Inan M, Zhao M, Manuszak M, Karakaya C, Rajadhyaksha A, Pickel VM et al. Energy deficit in parvalbumin neurons leads to circuit dysfunction, impaired sensory gating and social disability. Neurobiol Dis 2016; 93: 35–46.
Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012; 487: 443–448.
Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012; 485: 517–521.
Kasthuri N, Hayworth KJ, Berger DR, Schalek RL, Conchello JA, Knowles-Barley S et al. Saturated reconstruction of a volume of neocortex. Cell 2015; 162: 648–661.
Pietersen CY, Mauney SA, Kim SS, Passeri E, Lim MP, Rooney RJ et al. Molecular profiles of parvalbumin-immunoreactive neurons in the superior temporal cortex in schizophrenia. J Neurogenet 2014; 28: 70–85.
Hu H, Jonas P . A supercritical density of Na(+) channels ensures fast signaling in GABAergic interneuron axons. Nat Neurosci 2014; 17: 686–693.
Acknowledgements
Funding was provided by ZonMw Vidi 017.106.384 from the Netherlands Organization for Scientific Research and NeuroBasic PharmaPhenomics consortium to SAK, and the Dutch Technology Foundation STW applied science division of NWO and the Technology Programme of the Ministry of Economic Affairs (Project 12197) to SAK and JS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Stedehouder, J., Kushner, S. Myelination of parvalbumin interneurons: a parsimonious locus of pathophysiological convergence in schizophrenia. Mol Psychiatry 22, 4–12 (2017). https://doi.org/10.1038/mp.2016.147
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2016.147
- Springer Nature Limited
This article is cited by
-
Improved functional map** of complex trait heritability with GSA-MiXeR implicates biologically specific gene sets
Nature Genetics (2024)
-
Functional myelin in cognition and neurodevelopmental disorders
Cellular and Molecular Life Sciences (2024)
-
Association between enhanced carbonyl stress and decreased apparent axonal density in schizophrenia by multimodal white matter imaging
Scientific Reports (2023)
-
Fast-spiking parvalbumin-positive interneurons in brain physiology and Alzheimer’s disease
Molecular Psychiatry (2023)
-
Spatial Distribution of Parvalbumin-Positive Fibers in the Mouse Brain and Their Alterations in Mouse Models of Temporal Lobe Epilepsy and Parkinson’s Disease
Neuroscience Bulletin (2023)