
Abstract
The spliceosome, constituted by a protein set associated with small nuclear RNA (snRNA), is responsible for mRNA maturation through intron removal. Among snRNA genes, U1 is generally a conserved repetitive sequence. To unveil the chromosomal/genomic dynamics of this multigene family in grasshoppers, we mapped U1 genes by fluorescence in situ hybridization in 70 species belonging to the families Proscopiidae, Pyrgomorphidae, Ommexechidae, Romaleidae and Acrididae. Evident clusters were observed in all species, indicating that, at least, some U1 repeats are tandemly arrayed. High conservation was observed in the first four families, with most species carrying a single U1 cluster, frequently located in the third or fourth longest autosome. By contrast, extensive variation was observed among Acrididae, from a single chromosome pair carrying U1 to all chromosome pairs carrying it, with occasional occurrence of two or more clusters in the same chromosome. DNA sequence analysis in Eyprepocnemis plorans (species carrying U1 clusters on seven different chromosome pairs) and Locusta migratoria (carrying U1 in a single chromosome pair) supported the coexistence of functional and pseudogenic lineages. One of these pseudogenic lineages was truncated in the same nucleotide position in both species, suggesting that it was present in a common ancestor to both species. At least in E. plorans, this U1 snDNA pseudogenic lineage was associated with 5S rDNA and short interspersed elements (SINE)-like mobile elements. Given that we conclude in grasshoppers that the U1 snDNA had evolved under the birth-and-death model and that its intragenomic spread might be related with mobile elements.
Similar content being viewed by others


Introduction
In higher eukaryotes, most protein-coding transcripts contain multiple introns that need to be removed from the nascent RNA through splicing, an essential mechanism for mRNA maturation (West, 2012). The spliceosome consists of a set of proteins associated with U small nuclear RNAs (snRNAs), which are crucial components of this macromolecular complex. The major spliceosome complex is encoded by a multigene family including the U1, U2, U4, U5 and U6 snRNA genes (Gilbert, 1978; Busch et al., 1982; Bringmann and Lührmann, 1986; Nilsen, 2003; Valadkhan, 2005). Among U snRNA genes, the U1 snDNA is a multigene family showing variable number of repeats per genome and is conserved in sequence; however, distinct U1 snDNA intragenomic variants have been found in several organisms, such as toad (Forbes et al., 1984), fruit flies (Lo and Mount, 1990), pea (Hanley and Schuler, 1991), silk moths (Sierra-Montes et al., 2003), Oreochromis niloticus (Cabral-de-Mello et al., 2012) and humans (O’Reilly et al., 2012).
The U1 snRNA gene is tandemly arranged in some species, such as the toad Xenopus laevis (Zeller et al., 1984), the sea urchin Strongylocentrotus purpuratus (Yu et al., 1991) and the tapeworm Echinococcus multilocularis (Bretagne et al., 1991). In addition, the U1 snRNA is linked to other gene repeats (for example, 5S rDNA) in the crustacean Asellus aquaticus (Pelliccia et al., 2001), the fish Solea senegalensis (Manchado et al., 2006) and 10 razor shell species (Vierna et al., 2011). At the chromosomal level, the map** of U1 snRNA genes has been performed only in a few species, for instance, humans (Lund et al., 1983), mice (Lund and Nesbitt, 1988), crustaceans (Barzotti et al., 2003) and fishes (Cabral-de-Mello et al., 2012). Such endeavors have revealed that these genes are usually conserved with regard to location among related species, harbored on one or a few chromosome pairs.
Chromosomal studies in grasshopper species have revealed high karyotype conservatism, with most species showing 2n=23♂/24♀ acrocentric chromosomes and an X0♂/XX♀ sex chromosome system. However, derived karyotypes showing reductions in diploid number, derived sex chromosomes and supernumerary elements have also been reported (White, 1978; Hewitt, 1979; Mesa et al., 1982; Castillo et al., 2010). Current knowledge about repetitive DNA organization in grasshopper chromosomes is still scarce; concerning multigene families, only the 45S and 5S rDNAs and H3 and H4 histone genes have been mapped in multiple species, mainly in Acrididae representatives, which represent the most diverse family worldwide distributed, and together with Ommexechidae and Romaleidae are considered modern grasshoppers. In a lesser extent, chromosomal map** for rDNAs and/or distinct histone genes was also performed in closely sister families of Acrididae, that is, Romaleidae and Ommexechidae and in the earliest diverging basal lineage of Acridomorpha (Eumastacoidea) represented by Proscopiidae (Flook et al., 1999; Cabrero and Camacho, 2008; Cabrero et al., 2009; Cabral-de-Mello et al., 2011a, 2011b; Carvalho et al., 2011; Jetybayev et al., 2012; Leavitt et al., 2013; Neto et al., 2013; Palacios-Gimenez et al., 2013).
Grasshoppers show gigantic genomes, reaching 16.93 pg in Podisma pedestris (Westerman et al., 1987). The recent full genome sequencing in Locusta migratoria (genome size ca. 6 pg) has revealed that 60% of DNA in this species is repetitive (Wang et al. 2014), a figure which could be even higher in species with larger genomes. However, the chromosomal organization of such a major genomic component is mostly unknown despite being important information complementing full-genome sequencing projects. We show here that the integration of chromosomal and genomic analyses unveils new details on the organization and evolution of repetitive DNA families. We have thus performed fluorescence in situ hybridization (FISH) map** of the U1 snRNA gene in 70 species of grasshoppers belonging to five distantly related families, including the most representative groups, inferred from molecular phylogenetic studies (Leavitt et al., 2013): Proscopiidae, Ommexechidae, Pyrgomorphidae, Romaleidae and Acrididae (the latter including 10 subfamilies). We also analyzed U1 snDNA genomic organization in E. plorans, which is one of the species showing U1 clusters in seven chromosome pairs, using data obtained through 454 pyrosequencing, aiming to unveil possible causes for U1 snDNA intragenomic spread. In addition, we analyzed the U1 snDNA sequences in the recently published full genome of L. migratoria (Wang et al., 2014), a species showing only one U1 locus. This has revealed the colocalization of U1 snDNA with other repetitive DNA families being suggestive of possible spreading mechanism that could be generally operating in grasshopper genomes.
Materials and methods
Chromosomes, probes and FISH
Adult males and females of 63 species of grasshoppers belonging to families Proscopiidae (4 species), Ommexechidae (2 species), Pyrgomorphidae (2 species), Romaleidae (11 species) and Acrididae (44 species belonging to 10 subfamilies) were sampled from distinct localities in Spain, Paraguay, Argentina and Brazil (see Supplementary Material S1). Testes were fixed in 3:1 absolute ethanol:acetic acid and stored at −20 °C until use. Embryos of E. plorans were obtained as described by Camacho et al. (1991), and adult female gastric ceca were removed and fixed following Castillo et al. (2011). The different tissues were macerated in a drop of 50% acetic acid, and the slides were dried using a hot plate at 40–45 °C. In addition, previous results for Abracris flavolineata (Bueno et al., 2013) and six other species (Palacios-Gimenez et al., 2013) were also included for analysis.
The DNA probe for the U1 snDNA sequence was obtained by PCR using DNA of Rhammatocerus brasiliensis with the primers described by Cabral-de-Mello et al. (2012); 5S rDNA was obtained from a previously cloned fragment isolated from the E. plorans genome. The PCR product for U1 snDNA was previously sequenced, and the sequence was deposited in GenBank under the accession number KC896793 (Bueno et al., 2013). U1 snDNA and the 5S rDNA fragment were labeled with digoxigenin-11-dUTP (Roche, Mannheim, Germany) or biotin-14-dUTP (Invitrogen, San Diego, CA, USA) using PCR or nick translation. The FISH procedures were performed according to Pinkel et al. (1986), with some modifications (Cabral-de-Mello et al., 2010). Fiber FISH was performed as described in Muñoz-Pajares et al. (2011). In brief, cerebral ganglia were homogenized in 60% acetic acid and centrifuged for 10 min at 1000 r.p.m. After removing the supernatant, we added 500 μl of 1:3 acetic acid: ethanol and resuspended the cells. Fixed cells were then spread on clean moist slides and, before the complete evaporation of the fixative, the slides were placed in phosphate-buffered saline solution (pH=7) for 1 min. To relax chromatin, the slides were drained on a paper towel and immediately treated with 0.05 M NaOH in 30% ethanol as follows: 200 μl solution was placed on one end of a long coverslip that was then moved along the horizontal slide at an angle of about 30°. Then, we added a few drops of absolute ethanol (500 ml) and left it dry. Finally, to dehydrate the material, the slides were placed in a 70, 90 and 100% ethanol series and left it dry. Double FISH was performed as described in Cabrero et al. (2003). The probes were detected using anti-digoxigenin-rhodamine (Roche) or Streptavidin Alexa Fluor 488 conjugate (Invitrogen). The chromosomes were counterstained with 4,6-diamidino-2-phenylindole (DAPI) and mounted using Vectashield (Vector, Burlingame, CA, USA).
The chromosomes and signals were observed using an Olympus microscope (Tokyo, Japan) BX61 equipped with a fluorescence lamp and appropriate filters. The photographs were recorded using a DP70 cooled digital camera, and images were merged and optimized for brightness and contrast using Adobe Photoshop CS2. Finally, the signals were analyzed and organized into five categories: centromeric (c), proximal (pr), interstitial (i), subdistal (sd) and distal (d), as shown in the diagram in Supplementary Material S2. To facilitate the comparative analysis, we numbered the autosome pairs in each species in order of decreasing size, but this does not necessarily imply chromosome homology among the species.
Genomic organization of U1 snDNA in E. plorans and L. migratoria
As a first approach to unveiling the genomic organization of the U1 snDNA in grasshopper genomes, we analyzed two species showing very different patterns at the chromosomal level: E. plorans, showing U1 clusters in seven chromosome pairs, and L. migratoria, showing it in a single chromosome pair. For this purpose, we performed an analysis of the intragenomic variation of U1 snDNA in E. plorans by constructing a whole-genome sequencing library, which was sequenced using 3/8 of a 454 GS FLX Plus plate (accession numbers SRR1200829 and SRR1200835) generated by Macrogen Inc. (Seoul, Korea). We checked the quality of the reads with FASTQC (Andrews, 2012) and performed a quality trimming with the Roche’s 454 GS Assembler. After excluding reads shorter than 200 nt, we splitted the remaining reads in as many fragments of 200 nt as possible, for example, from a 537 nt long read we included in the analysis, a sequence corresponded to the 1–200 nt and another one to the 201–400. Then, we performed a graph-based clustering and assembly of these sequences to search for genes associated with U1 snDNA in the genome using the RepeatExplorer software (Novák et al., 2013). The contigs including the U1 snDNA were submitted to GenBank (accession numbers KJ606066–70). We then extracted the reads used to assemble each U1 carrier contig separately with SeqGrapheR (Novák et al., 2010) to map them against their contig and manually discard the U1 snDNA spacers to select the complete copies of U1 using Geneious v4.8 (Drummond et al., 2009).
In addition, we got copies of the U1 snDNA present in the assembled genome of L. migratoria recently published by Wang et al. (2014), which is available at GenBank with accession number AVCP000000000. We searched all the contigs matching one of the U1 snDNA copies previously found in E. plorans by a local BLASTN v2.2.28 search (Altschul et al., 1997) with default options. To extract only the U1 snDNA region in all the matched contigs, we searched the U1 snDNA copies of E. plorans using RepeatMasker (AFA Smit, R Hubley, P Green, unpublished data, current version: open-4.0.3 (RMLib: 20130422 & Dfam: 1.2)) and, with a custom Python script, we extracted the matching regions. We aligned the resulting sequences with MAFFT v7.157b (Katoh and Standley, 2013) with LINSI options organizing by identity. We then prepared a consensus sequence from this alignment of L. migratoria and repeated the previous procedure using this sequence as a reference. We considered all complete U1 copies and those truncated copies being found three or more times. To get representative lineages of complete copies, we performed a clustering analysis with UCLUST v6.0.307_linux32 (Edgar, 2010) using the ‘—cluster-fast’ option and 0.96 identity threshold, as this was the identity between the two functional lineages in E. plorans. We only considered as lineages those groups of sequences with two or more sequences, and we represented them by their consensus sequence. Truncated copies sharing the same deletion were included into the same lineage.
A minimum spanning tree was built with Arlequin v3.5 (Excoffier and Lischer, 2010) for the U1 snDNA sequences found in the contigs of both species. As an outgroup, we used the U1 sequence of the mountain pine beetle Dendroctonus ponderosae obtained in a local BLASTN search (Altschul et al., 1997) against NCBI NR database. The analysis for sequence diversity was conducted using DNASP v5.10 (Librado and Rozas, 2009) excluding positions with gaps in the alignments. The G+C content was calculated with the MEGA v5.0 (Tamura et al., 2011) software, and the prediction of secondary structure and Gibbs free energy (ΔG) for the different sequence lineages was performed with RNAFold (Hofacker, 2003), applying a constrained folding to avoid the pairing of the four conserved motifs, according to Vierna et al. (2013).
To find other repetitive elements associated with U1, in the contigs generated by RepeatExplorer in E. plorans and L. migratoria, we annotated them by means of a search in RepBase using the CENSOR software (Kohany et al., 2006) and also by means of a BLASTN search (Altschul et al., 1997) in the NCBI NR database. We considered only annotations obtained by both methods. Moreover, to visualize the relationship between the different contigs obtained from the E. plorans genome, including U1 snDNA and other repetitive sequences, we represented them graphically with the SeqGrapheR software (Novák et al., 2010). We also searched for repeated regions inside the selected contigs and compared each contig with itself applying a thereshold dotplot analysis integrated in Geneious v4.8 (Drummond et al., 2009).
Results
Chromosomal map** of U1 snDNA
The diploid chromosome number and sex chromosome system for the 70 species (some reported here for the first time) are shown in Supplementary Material S1. A total of 168 U1 snDNA sites were detected by FISH in the 70 species included, with an average of 2.4 sites per haploid genome. Most of these sites (162) were located on autosomes, whereas only 5 were on the X chromosome and 1 on a neo-Y chromosome. Most sites were located proximal to the centromere (60 sites, 35.7%), and fewer were interstitial (39, 23.2%), centromeric (35, 20.9%), subdistal (20, 11.9%) or distal (14, 8.3%; Table 1). Although most species carried U1 snDNA clusters on only one autosomal pair, variable patterns were also observed, even including two species with clusters on all chromosome pairs. Furthermore, in several species, one (for example, Ommexecha virens, Agriacris auripennis, Chorthippus apicalis, R. brasiliensis and Eumastusia koebelei koebelei) or two (for example, Omocestus bolivari, Omocestus burri, Omocestus panteli, and Stenobothrus festivus) chromosome pairs carried two or more clusters of U1 snDNA on the same chromosome (Table 1).
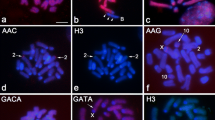
The five grasshopper families analyzed showed distinct patterns of distribution for the U1 snDNA clusters. In Proscopiidae, all four species analyzed showed a single interstitial cluster on the fourth largest autosome pair (Figures 1a and b). Similarly, the two Pyrgomorphidae species analyzed showed a single cluster proximally located on the third largest autosome pair (Figures 1c and d). The two Ommexechidae representatives also showed a coincident pattern but, in this case, showed three sites per species, with one of them proximally located on the largest autosome pair and the two other at centromeric and interstitial regions on the third largest autosome pair (Figure 1e).

FISH using the U1 snDNA as a probe in species of Proscopiidae (a and b), Pyrgomorphidae (c and d) and Ommexechidae (e), shown in metaphase I (a, c and e), metaphase II (b) and diplotene (d) cells. The chromosomes carrying U1 snDNA signals and the X chromosome are indicated. c centromere location. The inset in e corresponds to pair 3 from a metaphase II cell, showing the location of the two U1 snDNA clusters (centromeric and proximal). Bar=5 μm.
Among the Romaleidae, 7 out of the 11 species analyzed showed a single cluster of U1 snDNA on the third largest autosome pair, which was proximal in 6 species and subdistal in 1 species (Figure 2). However, A. auripennis showed two clusters (centromeric and proximal) on the eighth autosome pair (Figure 2a), and the two Chromacris species showed clusters on a single pair, namely, proximal on the fifth autosome of C. nuptialis and subdistal on the fourth autosome of C. speciosa (Figures 2c and d). Finally, Brasilacris gigas showed a single interstitial cluster on the seventh largest autosome pair (Figure 2b).

Chromosomal location of U1 snDNA in nine Romaleidae species, shown in metaphase I (a, b, d, f, h and i), metaphase II (c) and diplotene (e and g) cells. The chromosomes carrying U1 snDNA signals and the X chromosome are indicated. c centromere location. The inset in a corresponds to the eighth chromosome from an anaphase I cell, showing U1 clusters at centromeric and proximal/proximal locations. Bar=5 μm.
In the Acrididae family, the most representative sample analyzed with 51 species from 10 subfamilies, we found 144 U1 snDNA clusters, that is, 2.82 per haploid genome (Figures 3 and 4; Table 1). This analysis showed that 46 of these sites were proximal (32.0%), 34 were interstitial (23.6%), 32 were centromeric (22.2%), 18 were subdistal (12.5%) and 14 were distal (9.7%). About 40% of the species (21) showed U1 snDNA on a single chromosome pair, but some species showed it on two or more pairs, with two species (Oedipoda fuscocincta and Sphingonotus caerulans) carrying it on all chromosome pairs (see Figure 3l). In several species, two or more clusters were located on the same chromosome (Figure 4; Table 1).

Chromosome location of U1 snDNA in 12 species of Acrididae grasshoppers, belonging to seven subfamilies, shown in diplotene (a, c, d, f, i and l), female mitotic metaphase (b) and metaphase I (e, g, h, j and k) cells. The chromosomes carrying U1 snDNA signals and sex and B chromosomes are indicated. c centromere location. Chromosomes carrying small U1 snDNA clusters are shown enlarged within the insets. Note the extensive variation for the number and location of U1 snDNA clusters. Bar=5 μm.

Chromosome location of U1 snDNA clusters in eight Gomphocerinae (Acrididae) grasshoppers, shown in metaphase I (a, b, g and h) and diplotene (c, d and f) cells. (e and e’) Selected chromosome no. 1 from metaphase I of C. nevadensis showing DAPI pattern (e) and U1 snDNA location (e’). The chromosomes carrying U1 snDNA signals and the X and B chromosomes are indicated. c centromere location. Note the presence of multiple sites on the long (1 and 2) chromosomes of the 2n=17 species (a–e) and the presence of a 2n=23 species with a single U1 snDNA cluster (g) or four clusters on three chromosomes (h). Bar=5 μm.
The three best-represented acridid subfamilies were Leptysminae (7 species), Melanoplinae (13 species) and Gomphocerinae (16 species). The Leptysminae species showed 1–3 clusters that were predominantly interstitial (six clusters) or proximal (four) but rarely centromeric (one) and never subdistal or distal, with two species (Cylindrotettix obscurus and Stenopola sp) showing U1 snDNA clusters on the X chromosome (Figure 3e; Table 1). Almost all Melanoplinae species showed a single autosome pair carrying the genes for U1 snRNA, but they never showed a centromeric location (Table 1). Propedies auriculares was the only Melanoplinae species carrying two clusters on the same chromosome (Figure 3i; Table 1). Finally, Gomphocerinae species showed 1–6 U1 snDNA clusters, always on autosomes, with all types of location but higher frequency in proximal (21 clusters), interstitial (18) and subdistal (14) locations and lower frequency in centromeric (7) and distal (6) locations (Figure 4; Table 1). Remarkably, all Chorthippus and Omocestus species carried 2, 3 or more U1 snDNA clusters (see Figure 4) on the longest autosome, which could have been present in their common ancestor. The average number of U1 snDNA clusters per haploid genome was 1.6, 1.3 and 4.12 in the subfamilies Leptysminae, Melanoplinae and Gomphocerinae, respectively.
Finally, some acridid species carried B chromosomes, including E. plorans, C. attenuatus, Eu. k. koebelei, S. dorsalis, R. brasiliensis, A. flavolineata (Bueno et al., 2013), Orthoscapheus rufipes and Vilerna rugulosa, but no clusters of U1 snDNA were observed on any of these elements (see, for example, Figures 3f, j and k).
U1 snDNA and other repetitive elements in the E. plorans and L. migratoria genomes
Using graph-based clustering, we obtained a complex cluster in E. plorans with U1 snDNA in the central position and a multitude of ‘loops’ and ‘branches’ connected to it (Figure 5), as expected from its multiple locations in this genome. This cluster constituted 0.222% of the genomic sequences analyzed. The assembly of all DNA sequences included in the cluster resulted in a total of 63 contigs, 5 of them carrying U1 snDNA. On the basis of its coding region, we found complete U1 snRNA genes in two contigs (lineages 1 and 2), but there were several others showing different mutations, such as a deletion in the middle (lineage 3), a 5′ truncation of 34 nt (lineage 4) and a deletion and an insertion close to the 5′ end (lineage 5; Figure 6a). Nucleotide diversity was zero for lineages 1 and 3, but very high for lineages 2 (π=0.01536), 4 (π=0.03946) and 5 (π=0.01258; Table 2). We predicted the secondary structure of the five U1 snDNA consensus sequences, as this can be an indicator of its functionality or pseudogenicity (Figure 6b). Lineages 1 and 2 contained the four conserved sites described in Vierna et al. (2013) (Figure 6b), and they showed the same pattern, with four helices, as the consensus secondary structure of U1 snRNA reported in Vierna et al. (2013). The secondary structure of lineage 1 showed higher stability (ΔG=−56.0 kcal mol−1) than lineage 2 (ΔG=−54.6 kcal mol−1). In contrast, lineages 3, 4 and 5 showed secondary structures lacking the conserved motifs present in the two other lineages (Figure 6b). These results suggest the possibility that lineages 3–5 are pseudogenic. The number of sequences obtained for each lineage (shown in Table 2) indicates that 45% were functional and 55% were pseudogenic.

Schematic representation of the cluster containing five U1 snDNA lineages in the genome of E. plorans (shown in different colors). In addition to U1 snDNA, we found homology with several elements, such as incomplete 5S rDNA and SINEs (indicated as boxes). A full color version of this figure is available at the Heredity journal online.

(a) Alignment of the consensus sequence for the U1 snDNA lineages in E. plorans and in L. migratoria. (b) Minimum spanning tree (MST) built with the consensus sequence of the nine U1 lineages, also depicting their predicted secondary structure. Three types of changes are represented with different filled forms: circle, triangle and square, corresponding to substitutions, deletions and insertions, respectively (the number of involved nucleotides is also indicated for the latter two). Deletions and insertions are considered as single mutational events, and the number of involved positions in the alignment is also indicated beside the corresponding symbol. The mean Gibbs free energy (ΔG) for the U1 secondary structure, expressed in kcal mol−1, is depicted in brackets. In b, the conserved functional domains for lineages 1 and 2 are highlighted in light green. A full color version of this figure is available at the Heredity journal online.
In the case of L. migratoria, we found 130 U1 copies in 109 different contigs from the genome assembled by Wang et al. (2014), but only 55 (42.3%) were complete, with 47 of them grou** into four different lineages (Figure 6a) and the remaining 8 being singletons, which were discarded for subsequent analysis. All these 47 copies of U1 were found in differents contigs. In addition, we found 15 sequences showing the same truncation of 34 nt in the 5′ end, and they were considered a fifth lineage (Supplementary Material S3). The secondary structure of the lineages 1–4 (Figure 6b) suggested their functionality, with stability values (ΔG) ranging between −57.9 and −61.3 kcal mol−1. The truncation of lineage 5 makes it clearly pseudogenic (ΔG=−41.3 kcal mol−1; Figure 6b). The lowest nucletotide diversity was shown by lineage 2 (π=0.00781) and the highest one was observed in lineage 5 (π=0.15366; Table 2).
We built a minimum spanning tree with the five U1 snDNA lineages observed in E. plorans and the five ones observed in L. migratoria (Figure 6b). Because NCBI’s nucleotide database lacks full-length copies of U1 snDNA from Orthoptera, we searched the sequences of lineages 1 and 2 of E. plorans in the NR database of NCBI and chose the U1 snDNA sequence showing the lowest e-value for both lineages, which corresponded to that in the mountain pine beetle Dendroctonus ponderosae (accession number APGK01030738.1, positions 13746–13908). The minimum spanning tree shows lineage 1 in E. plorans as the most connected and directly joined to the outgroup, suggesting that it is ancestral in the genome of this species.
In addition to U1 snDNA, we found other identifiable repeated elements in both species. In E. plorans, the contig with the highest coverage included lineage 4 for U1 snDNA and also 5S rDNA. Both were present in the same orientation in the 3191-bp-long contig, and the dotplot analysis showed both ends as direct repetitions, suggesting a tandem distribution of these elements in the genome. After trimming one of the ends, the resulting sequence was 2879-bp long, with a spacer 1 of 1209 bp and a spacer 2 of 1421 bp. Spacer 1 also showed a tandemly duplicated region of ~350 bp. In addition, we found two more contigs, including a region annotated as 5S rDNA (Figure 5). This association of U1 snDNA with 5S rDNA repeats was corroborated by two-color FISH in mitotic metaphase and in distended fibers (fiber FISH; Figure 7). In addition to 5S rDNA, we found some regions that were annotated as SINE-like elements associated with the U1 snDNA cluster. For instance, one contig was annotated as tRNA (transfer RNA)-thr/pseudogenic and another showed homology with ALPINE/SINE (Figure 5).

FISH analysis for U1 snDNA (a–c) and 5S rDNA (b and c) in E. plorans chromosomes. (a) Mitotic metaphase, (b) selected chromosomes (pair 6) from a mitotic metaphase and (c) fiber FISH. Note the spread distribution for U1 snDNA on the chromosomes (a) and the colocalization (interspersed organization) of U1 snDNA and 5S rDNA (b and c). A full color version of this figure is available at the Heredity journal online.
In the L. migratoria genome, the contig with accession number AVCP010084704 showed a copy of lineage 2 U1 snDNA and also a copy of 5S rDNA in the same direction, and a direct repetition in both ends suggesting a tandem structure in the genome, likewise that for lineage 4 in E. plorans (see above). No other repetitive elements were found associated with the U1 snDNA in the L. migratoria genome, at least in the available contigs.
Discussion
U1 snDNA organization in grasshopper chromosomes
The occurrence of evident signals in all species studied indicates that, at least, part of U1 snDNA clusters among grasshoppers are tandemly arrayed, as observed in some other animals (Zeller et al., 1984; Bretagne et al., 1991; Yu et al., 1991; Cabral-de-Mello et al., 2012), this pattern being a common placement in the group. Although, the occurrence of dispersed (non-tandemly arrayed) sites, not detected through FISH could not be completely ruled out, as revealed in other eukaryotes through molecular analysis (see, for example, Mount et al., 2007; Marz et al., 2008; Vierna et al., 2011). A comparative analysis of the results for the five families indicated that the presence of a single cluster for U1 snDNA on one autosomal pair is the modal and median number, as it was found in approximately half of the species analyzed. Therefore, we hypothesize that this is the ancestral pattern in grasshoppers and that the divergent situations observed in specific groups could be the result of molecular mechanisms moving some U1 snRNA genes between non-homologous chromosomes, as observed for other multigene families (see discussion below). The ancestrality of a single U1 snDNA cluster per haploid genome is reinforced by the fact that this pattern was invariant in Proscopiidae, which is included in the superfamily Eumastacoidea and is considered the earliest diverging lineage of Acridomorpha, with a basal placement in molecular phylogenies (Leavitt et al., 2013). Similar to grasshoppers, the occurrence of a single site of U1 snDNA per haploid genome has also been reported in mice (Lund and Nesbitt, 1988), crustaceans (Barzotti et al., 2003) and fish (Cabral-de-Mello et al., 2012).
Grasshopper genomes typically consist of a gradual series of morphologically similar chromosomes slightly differing in size, such that it is difficult to identify each chromosome pair (except for the X and megameric bivalent chromosomes because of their heteropycnotic behavior during meiosis). This situation makes it impossible to determine whether chromosomes occupying the same size–order position (for example, the third) in two different species are actually homeologous. This implies that the chromosome numbers in Table 1 are only strictly valid per species and that extrapolation among species needs to be performed with extreme care. With this problem in mind, we can reach two main conclusions: (i) the single U1 snDNA cluster in the ancestor of the grasshopper species analyzed here was not located on the X chromosome because this pattern was extremely rare. (ii) It is likely that the ancestral location of the U1 snDNA cluster was on an autosome of a size being about the third in the genome because this was the size–order position found most frequently and in distinct distantly related families. In addition, adjacent positions (2nd and 4th) were also very frequently found (14 and 17 species); bearing in mind the uncertainty of determining homeology, this suggests that autosome 2 or 4 could actually be derived from ancestor chromosome 3 and that its size changed due to some genomic events, such as a higher proliferation for a given mobile element or other repetitive DNA. On this basis, chromosome 4 in Proscopiidae could be homologous to chromosome 3 in Romaleidae. Nonetheless, ascertaining precisely which of the chromosome pairs originally harbored the U1 snDNA cluster in ancestral grasshoppers requires a comparative analyses of shared synteny for multiple genetic markers among species. A first topic to investigate would be whether chromosome size–order is associated with genetic content.
For the family Proscopiidae, the conservative organization evidenced for U1 snRNA, that is, an interstitial cluster located on pair no. 4 and the occurrence of H3 histone and 5S rRNA genes on the same chromosome (Cabral-de-Mello et al., 2011b), indicate that this chromosome could have been maintained without gross rearrangements after the speciation process. In a similar way, at the family level, this conservative pattern for the distribution of U1 snDNA clusters was also found in Pyrgomorphidae (pair 3), Ommexechidae (pairs 1 and 3) and Romaleidae (pair 3). Furthermore, similarity was also observed at the genus level, such as in Ommexechidae (O. gracilis and O. virens), Romaleidae (Xyleus discoideus angulatus and Xy. d. discoideus) and Acrididae representatives, that is, Parascopas (P. obesus and P. sanguineus) and Schistocerca (S. flavofasciata and S. pallens), which may be indicative of a possible common descendent and conservation of U1 snRNA gene distribution in these genera.
In Acrididae, several species from some subfamilies showed clusters on one or two autosomal bivalents (for example, Acridinae, Catantopinae, Copiocerinae, Cyrtacanthacridinae and Ommatolampidinae), although no specific pattern of distribution on a given chromosome emerged in these groups. In general, the variation observed for the U1 snDNA number and location does not show a phylogenetic relationship beyond the genus level, except in the case of Gomphocerinae in which U1 snDNA spread throughout the genome was observed in most representatives (see below). Examples of this independent variation were most evident in the Oedipodinae subfamily, with Aiolopus strepens and Locusta migratoria showing U1 snDNA clusters restricted to one chromosome pair, whereas O. fuscocincta and S. caerulans carry clusters on all chromosomes. Similarly, each Ommatolampidinae species showed a specific pattern of U1 snDNA location.
Gomphocerinae constitutes one of the most diverse subfamilies within Acrididae (Contreras and Chapco, 2006) and includes a group of species with a reduction in diploid number to 2n=17 due to three centric fusion events (Hewitt, 1979). We also observed the highest number of U1 snDNA clusters per haploid genome (4.12) within Acrididae, which is comparatively much higher than those found in Leptysminae and Melanoplinae. Apparently, this capacity of movement and multiplication for U1 snDNA in Gomphocerinae is basal in the group and preceded the occurrence of diploid number reduction because multiple U1 clusters are also present in some species with 2n=23, such as R. brasiliensis. In addition, in the species with 2n=17, autosome pairs 1 and 2, which resulted from centric fusions, show multiple U1 snDNA clusters, suggesting either a higher spread rate in these metacentric chromosomes or that some of the acrocentric chromosomes involved in the fusions already carried multiple U1 snRNA clusters in the ancestor species.
Possible causes of U1 snDNA variation and its organization in the genome of E. plorans and L. migratoria
The high variation in the number of chromosome pairs carrying U1 snDNA clusters (1–12) clearly indicates that this DNA has spread between non-homologous chromosomes in many species, especially in Acrididae. This situation resembles the extensive variation in the number of 45S and 5S rDNA loci previously reported in Acrididae grasshoppers (Cabrero and Camacho, 2008; Cabral-de-Mello et al., 2011a). In all three cases, the spread was paralleled by an increase in the number of genomic clusters, suggesting that the mechanisms involved should entail duplication events. Therefore, ectopic recombination, the involvement of extrachromosomal circular DNA and the action of transposable elements have frequently been cited (Charlesworth et al., 1994; Coyne and Orr, 1998; Raskina et al., 2008; Nguyen et al., 2010; Cabral-de-Mello et al., 2011c; Panzera et al., 2012).
DNA sequencing in E. plorans and L. migratoria allowed a more precise analysis of the U1 snDNA genomic structure and evolution. Both species show similar association between U1 snDNA and 5S rDNA, even though one of them (L. migratoria) shows U1 genes in a single chromosome pair, whereas the other (E. plorans) carries them in seven chromosome pairs. The similar structure for U1 and 5S in both species suggests the possibility that this pattern is quite old in grasshoppers. In addition, the presence of SINE-like elements associated with U1 and 5S in the species where the U1 cluster has spread up to seven chromosomes (E. plorans) but their absence in the species where the U1 is restricted to one chromosomal pair (L. migratoria) suggests possible transposition events as responsible for such intragenomic spread, as previously suggested for the 5S rDNA (Drouin and Moniz de Sá, 1995; Cabral-de-Mello et al., 2011a). Supporting this possibility, there is evidence that pseudogenic copies of 5S rDNA could acquire SINE behavior (Kapitonov and Jurka, 2003; Gogolevsky et al., 2009). Genomic linkage for these multigene families has also been reported, for example, in crustaceans (Pelliccia et al., 2001), molluscs (Vierna et al., 2011) and fish (Manchado et al., 2006).
The remarkable similarity shown by the pseudogenic lineage 4 in E. plorans and lineage 5 in L. migratoria suggests their common ancestry, as the independent origin of so many sequences (21 in E. plorans and 15 in L. migratoria) with the same truncation is highly unlikely. Given that these two species belong to different Acridid subfamilies, we can conclude that this truncated (pseudogenic) lineage is quite old in grasshopper genomes and is probably present in other species too.
The extensive genomic variation observed for U1 snDNA in the two grasshopper genomes analyzed, even for putatively functional units, and the presence of pseudogenic copies in both species (55–58% of them), is consistent with the birth-and-death model for the evolution of gene families (Nei and Rooney, 2005), as previously observed in other insect genomes, for example, Aedes aegypti (Mount et al., 2007). Alternatively, the highly homogenized copies found for lineages 1 and 3 in E. plorans could suggest the occurrence of concerted evolution, but we believe that this could also be explained by recent intragenomic spread of these lineages. In addition, concerted evolution would hardly explain the long persistence of the truncated pseudogenic copies in both species. A mixed process of concerted evolution and birth-and-death evolution (Nei and Rooney, 2005) has been proposed for other multigene families, such as 5S rDNAs in distinct groups (Freire et al., 2010; Pinhal et al., 2011; Merlo et al., 2012; Vierna et al., 2013), but the U1 family in grasshoppers fits better to the birth-and-death model.
Conclusions
The analysis of five distinct families and some subfamilies from the most specious grasshopper family, that is, Acrididae, allowed a more precise depiction of the organization of U1 snDNA clusters in grasshoppers, providing a detailed knowledge of the karyotype organization of this sequence in the group as a whole. The high genomic dynamism of U1 snDNA clusters contrasts with the extensive conservation of macro-chromosomal structure in grasshoppers, indicating that the extensive variation observed in the number of U1 snDNA clusters per genome is not associated with major chromosomal rearrangements. As demonstrated by the analysis of the 454 sequences in the E. plorans genome, and that of the L. migratoria assembled genome, the high dynamism of this gene in the Acrididae could be the consequence of its association with transposable elements. The integration of these two kinds of data, that is, cytogenetic map** and genomic information, permitted a more detailed analysis for understanding patterns of U1 snDNA evolution. The existence of several U1 snDNA lineages in the same genome, including several pseudogenic lineages, opens new interesting questions. For instance, does the U1 snDNA cluster on a given chromosome show a single lineage or a mixture of several lineages, some functional and some pseudogenic? If the different clusters are homogeneous (that is, they contain either functional or pseudogenic copies), the search for the original ancestral location of the U1 cluster would be delimited to only those clusters showing functional U1 copies. Another interesting question is whether the pseudogenic lineages are more spread throughout the genome than the functional lineages. The fact that the analyzed cluster in E. plorans contains pseudogenic lineage 4 suggests this possibility, but the coverage of our analysis was actually very low. These questions can be technically approached, and similar genomic analysis in other species showing different degrees of U1 spread would help very much to unveil the genomic evolution of this gene.
Data archiving
Contigs including the U1 snDNA were obtained from sequenced genome of Eyprepocnemis plorans available from GenBank, accession numbers: KJ606066–KJ606070.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
Andrews S (2012) FastQC. a quality control tool for high throughput sequence data http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed 25 April 2012).
Barzotti R, Pelliccia F, Rocchi A (2003). Identification and characterization of U1 small nuclear RNA genes from two crustacean isopod species. Chromosome Res 11: 365–373.
Bretagne S, Robert B, Vidaud D, Goossens M, Houin R (1991). Structure of the Echinococcus multilocularis U1 snRNA gene repeat. Mol Biochem Parasitol 46: 285–292.
Bringmann P, Lührmann R (1986). Purification of the individual snRNPs U1, U2, U5 and U4/U6 from HeLa cells and characterization of their protein constituents. EMBO J 5: 3509–3516.
Bueno D, Palacios-Gimenez OM, Cabral-de-Mello DC (2013). Chromosomal map** of repetitive DNAs in Abracris flavolineata reveal possible ancestry for the B chromosome and surprisingly H3 histone spreading. PLoS One 8: e66532.
Busch H, Reddy R, Rothblum L, Choi YC (1982). SnRNAs, SnRNPs, and RNA processing. Annu Rev Biochem 51: 617–654.
Cabral-de-Mello DC, Moura RC, Martins C (2010). Chromosomal map** of repetitive DNAs in the beetle Dichotomius geminatus provides the first evidence for an association of 5S rRNA and histone H3 genes in insects, and repetitive DNA similarity between the B chromosome and A complement. Heredity 104: 393–400.
Cabral-de-Mello DC, Cabrero J, Loópez-Leoón MD, Camacho JPM (2011a). Evolutionary dynamics of 5S rDNA location in acridid grasshoppers and its relationship with H3 histone gene and 45S rDNA location. Genetica 139: 921–931.
Cabral-de-Mello DC, Martins C, Souza MJ, Moura RC (2011b). Cytogenetic map** of 5S and 18S rRNAs and H3 histone genes in 4 ancient Proscopiidae grasshopper species: contribution to understanding the evolutionary dynamics of multigene families. Cytogenet Genome Res 132: 89–93.
Cabral-de-Mello DC, Oliveira SG, Moura RC, Martins C (2011c). Chromosomal organization of the 18S and 5S rRNAs and histone H3 genes in Scarabaeinae coleopterans: insights into the evolutionary dynamics of multigene families and heterochromatin. BMC Genet 12: 88.
Cabral-de-Mello DC, Valente GT, Nakajima RT, Martins C (2012). Genomic organization and comparative chromosome map** of the U1 snRNA gene in cichlid fish, with an emphasis in Oreochromis niloticus. Chromosome Res 20: 279–292.
Cabrero J, Camacho JP (2008). Location and expression of ribosomal RNA genes in grasshoppers: abundance of silent and cryptic loci. Chromosome Res 16: 595–607.
Cabrero J, Bugrov A, Warcha1owska-Sliwa E, López-León MD, Perfectti F, Camacho JPM (2003). Comparative FISH analysis in five species of Eyprepocnemidine grasshoppers. Heredity 90: 377–381.
Cabrero J, López-Leoón MD, Teruel M, Camacho JP (2009). Chromosome map** of H3 and H4 histone gene clusters in 35 species of acridid grasshoppers. Chromosome Res 17: 397–404.
Camacho JPM, Cabrero J, Viseras E, Lopez-Leon MD, Navas-Castillo J, Alche JD (1991). G banding in two species of grasshopper and its relationship to C, N, and fluorescence banding techniques. Genome 34: 638–643.
Carvalho DB, Rocha MF, Loreto V, Silva AEB, Souza MJ (2011). Ommexecha virens (Thunberg, 1824) and Descampsacris serrulatum (Serville, 1831) (Orthoptera, Ommexechidae): karyotypes, constitutive heterochromatin and nucleolar organizing regions. Comp Cytogenet 5: 123–132.
Castillo ERD, Bidau CJ, Martí DA (2010). Neo-sex chromosome diversity in neotropical melanopline grasshoppers (Melanoplinae, Acrididae). Genetica 138: 775–786.
Castillo ER, Taffarel A, Martí DA (2011). Una técnica alternativa para el cariotipado mitótico en saltamontes: bandeo C y Fluorescente en Adimantus ornatissimus (Orthoptera: Acrididae). Rev Cienc Tecnol 16: 31–35.
Charlesworth B, Sniegowski P, Stephan W (1994). The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371: 215–220.
Contreras D, Chapco W (2006). Molecular phylogenetic evidence for multiple dispersal events in Gomphocerine grasshoppers. J Orthopt Res 15: 91–98.
Coyne JA, Orr HA (1998). The evolutionary genetics of speciation. Phil Trans R Soc Lond 353: 287–305.
Drouin G, Moniz de Sá M (1995). The concerted evolution of 5S ribosomal genes linked to the repeat units of other multigene families. Mol Biol Evol 12: 481–493.
Drummond AJ, Ashton B, Cheung M, Heled J, Kearse M (2009) Geneious 4.8. Biomatters, Auckland, New Zealand.
Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Excoffier L, Lischer HEL (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567.
Flook PK, Klee S, Rowell CHF (1999). Combined molecular phylogenetic analysis oft he Orthoptera (Arthropoda, Insecta) and implications for their higher systematics. Syst Biol 48: 233–253.
Forbes DJ, Kirschner MW, Caput D, Dahlberg JE, Lund E (1984). Differential expression of multiple U1 small nuclear RNAs in oocytes and embryos of Xenopus laevis. Cell 38: 681–689.
Freire R, Arias A, Insua AM, Méndez J, Eirın-López JM (2010). Evolutionary dynamics of the 5S rDNA gene family in the mussel Mytilus: mixed effects of birth-and-death and concerted evolution. J Mol Evol 70: 413–426.
Gilbert W (1978). Why genes in pieces? Nature 271: 501.
Gogolevsky KP, Vassetzky NS, Kramerov DA (2009). 5S rRNA-derived and tRNA-derived SINEs in fruit bats. Genomics 93: 494–500.
Hanley BA, Schuler MA (1991). Developmental expression of plant snRNAs. Nucleic Acids Res 22: 6319–6325.
Hewitt GM (1979). Animal Cytogenetics Vol. 3. Insecta 1. Orthoptera. Gebrüder Borntraeger: Berlin, Germany.
Hofacker IL (2003). Vienna RNA secondary structure server. Nucleic Acids Res 31: 3429–3431.
Jetybayev IE, Bugrov AG, Karamysheva TV, Camacho JPM, Rubtsov NB (2012). Chromosomal localization of ribosomal and telomeric DNA provides new insights on the evolution of Gomphocerinae grasshoppers. Cytogenet Genome Res 138: 36–45.
Kapitonov VV, Jurka J (2003). A novel class of SINE elements derived from 5S rRNA. Mol Biol Evol 20: 694–702.
Katoh K, Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780.
Kohany O, Gentles AJ, Hankus L, Jurka J (2006). Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7: 474.
Leavitt JR, Hiatt KD, Whiting MF, Song H (2013). Searching for the optimal data partitioning strategy in mitochondrial phylogenomics: a phylogeny of Acridoidea (Insecta: Orthoptera: Caelifera) as a case study. Mol Phylogenet Evol 67: 494–508.
Librado P, Rozas J (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Lo PC, Mount SM (1990). Drosophila melanogaster genes for U1 snRNA variants and their expression during development. Nucleic Acids Res 18: 6971–6979.
Lund E, Bostock C, Robertson M, Christie S, Mitchen JL, Dahlberg JE (1983). U1 small nuclear RNA genes are located on human chromosome1 and are expressed in mouse–human hybrid cells. Mol Cell Biol 3: 2211–2220.
Lund E, Nesbitt MN (1988). The embryonic and adult mouse U1 snRNA genes map to different chromosomal loci. Somat Cell Mol Genet 14: 143–148.
Manchado M, Zuasti E, Cross I, Merlo A, Infante C, Rebordinos L (2006). Molecular characterization and chromosomal map** of the 5S rRNA gene in Solea senegalensis: a new linkage to the U1, U2, and U5 small nuclear RNA genes. Genome 49: 79–86.
Marz M, Kirsten T, Stadler PF (2008). Evolution of spliceosomal snRNA genes in metazoan animals. J Mol Evol 7: 594–607.
Merlo MA, Pacchiarini T, Portela-Bens S, Cross I, Manchado M, Rebordinos L (2012). Genetic characterization of Plectorhinchus mediterraneus yields important clues about genome organization and evolution of multigene families. BMC Genetics 13: 33.
Mesa A, Ferreira A, Carbonell CS (1982). Cariología de los acridoideos neotropicales: estado actual de su conocimiento y nuevas contribuciones. Ann Soc Entomol Fr 18: 507–526.
Mount SM, Gotea V, Lin CF, Hernandez K, Makalowski W (2007). Spliceosomal small nuclear RNA genes in 11 insect genomes. RNA 13: 5–14.
Muñoz-Pajares AJ, Martínez-Rodríguez L, Teruel M, Cabrero J, Camacho JPM et al. (2011). A single, recent origin of the accessory B chromosome of the grasshopper Eyprepocnemis plorans. Genetics 187: 853–863.
Nei M, Rooney AP (2005). Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39: 121–152.
Neto MSR, Souza MJ, Loreto V (2013). Chromosomal evolution of rDNA and H3 histone genes in representative Romaleidae grasshoppers from northeast Brazil. Mol Cytogenet 6: 41.
Nguyen P, Sahara k, Yoshido A, Marec F (2010). Evolutionary dynamics of rDNA clusters on chromosomes of moths and butterflies (Lepidoptera). Genetica 138: 343–354.
Nilsen TW (2003). The spliceosome: the most complex macromolecular machine in the cell? Bioessays 25: 1147–1149.
Novák P, Neumann P, Macas J (2010). Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics 11: 378.
Novák P, Neumann P, Pech J, Steinhaisl J, Macas J (2013). RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next generation sequence reads. Bioinformatics 29: 792–793.
O’Reilly D, Dienstbier M, Cowley SA, Vazquez P, Drozdz M, Taylor S et al. (2012). Differentially expressed, variant U1 snRNAs regulate gene expression in human cells. Genome Res 23: 281–291.
Palacios-Gimenez OM, Castillo ER, Martí DA, Cabral-de-Mello DC (2013). Tracking the evolution of sex chromosome systems in Melanoplinae grasshoppers through chromosomal map** of repetitive DNA sequences. BMC Evol Biol 13: 167.
Panzera Y, Pita S, Ferreiro MJ, Ferrandis I, Lages C, Pérez R et al. (2012). High dynamics of rDNA cluster location in kissing bug holocentric chromosomes (Triatominae, Heteroptera). Cytogenet Genome Res 138: 56–67.
Pelliccia F, Barzotti R, Bucciarelli E, Rocchi A (2001). 5S ribosomal and U1 small nuclear RNA genes: a new linkage type in the genome of a crustacean that has three different tandemly repeated units containing 5S ribosomal DNA sequences. Genome 44: 331–335.
Pinkel D, Straume T, Gray JW (1986). Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci USA 83: 2934–2938.
Pinhal D, Yoshimura TS, Araki CS, Martins C (2011). The 5S rDNA family evolves through concerted and birth-and-death evolution in fish genomes: an example from freshwater stingrays. BMC Evol Biol 11: 151–164.
Raskina O, Barber JC, Nevo E, Belyayev A (2008). Repetitive DNA and chromosomal rearrangements: speciation-related events in plant genomes. Cytogenet Genome Res 120: 351–357.
Sierra-Montes JM, Pereira-Simon S, Freund AV, Ruiz LM, Szmulewicz MN, Herrera RJ (2003). A diversity of U1 small nuclear RNAs in the silk moth Bombyx mori. Insect Biochem Mol Biol 33: 29–39.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739.
Valadkhan S (2005). snRNAs as the catalysts of pre-mRNA splicing. Curr Opin Chem Biol 9: 603–608.
Vierna J, Jensen KT, Martínez-Lage A, González-Tizón AM (2011). The linked units of 5S rDNA and U1 snDNA of razor shells (Mollusca: Bivalvia: Pharidae). Heredity 107: 127–142.
Vierna J, Wehner S, Zu Siederdissen CH, Martínez-Lage A, Marz M (2013). Systematic analysis and evolution of 5S ribosomal DNA in metazoans. Heredity 111: 410–421.
Wang X, Fang X, Yang P, Jiang X, Jiang F, Zhao D et al. (2014). The locust genome provides insight into swarm formation and long-distance flight. Nat Commun 5: 2957.
West S (2012). The increasing functional repertoire of U1 snRNA. Biochem Soc Trans 40: 846–849.
Westerman M, Barton NH, Hewitt GM (1987). Differences in DNA content between two chromosomal races of the grasshopper Podisma pedestris. Heredity 58: 221–228.
White MJD (1978). Modes of Speciation. WH Freeman and Company: San Francisco, CA, USA.
Yu JC, Wendelburg B, Sakallah S, Marzluff WF (1991). The Ul snRNA gene repeat from the sea urchin (Strongylocentrotus purpuratus): the 70 kilobase tandem repeat ends directly 3' to a Ul gene. Nucleic Acids Res 19: 1093–1098.
Zeller R, Carri MT, Mattaj IW, De Robertis EM (1984). Xenopus laevis Ul snRNA genes: characterisation of transcriptionally active genes reveals major and minor repeated gene families. EMBO J 3: 1075–1081.
Acknowledgements
We are grateful to Dr Carlos Salvador Carbonell (Universidad de Montevideo, Uruguay), Dr Felipe Pascual (Universidad de Granada, Spain) and Dr Ricardo Gómez (Universidad de Castilla La Mancha, Spain) for the taxonomic identification of some specimens used here. This study was supported by the Programa Primeiros Projetos (PROPe, UNESP), Fundação de Amparo a Pesquisa do Estado de São Paulo-FAPESP (2011/19481-3), Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq (475308/2011-5), the Spanish Ministerio de Ciencia e Innovación (CGL2009-11917) and Plan Andaluz de Investigación (CVI-6649), and was partially performed by FEDER funds. AA and FJR-R were supported by scholarships from the Brazilian CNPq and the Spanish Junta de Andalucía, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article

{kind=link}
Cite this article
Anjos, A., Ruiz-Ruano, F., Camacho, J. et al. U1 snDNA clusters in grasshoppers: chromosomal dynamics and genomic organization. Heredity 114, 207–219 (2015). https://doi.org/10.1038/hdy.2014.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.87
- Springer Nature Switzerland AG