
Abstract
Different approaches are in use to improve our knowledge about the causative agent of coronavirus disease (COVID-19). Cell culture-based methods are the better way to perform viral isolation, evaluate viral infectivity, and amplify the virus. Furthermore, next-generation sequencing (NGS) have been essential to analyze a complete genome and to describe new viral species and lineages that have arisen over time. Four naso-oropharyngeal swab samples, collected from April to July of 2020, were isolated and sequenced aiming to produce viral stocks and analyze the mutational profile of the found lineage. B.1.1.33 was the lineage detected in all sequences. Although the samples belong to the same lineage, it was possible to evaluate different mutations found including some that were first described in these sequences, like the S:H655Y and T63N. The results described here can help to elicit how the pandemic started to spread and how it has been evolving in south Brazil.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The enveloped RNA virus the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of COVID-19, a new disease whose first cases were reported in late 2019 in Wuhan, Hubei province, central China [1]. COVID-19 was characterized as a pandemic on March 11, 2020, by the World Health Organization (WHO). According to WHO (https://covid19.who.int/), there have been more than 480 million confirmed cases and more than 6 million deaths caused by COVID-19 globally ever since.
In the southernmost state of Brazil, Rio Grande do Sul (RS), the dispersion of different SARS-CoV-2 lineages was followed throughout the pandemic. From May to November of 2020, the lineages circulating in RS were classified as B.1.1, B.1.1.33, B.1.1.248, and B.1.1.49 [2, 3]. In late November 2020, there was an increase of P.2 (Zeta) cases [3], along with the description of one case of P.1 (Gamma), as well as other local and global variants. In late January 2021, a higher rate of Gamma variant cases was noticed, sparking a huge peak of SARS-CoV-2 new cases, hospitalizations, and deaths from February to the end of April 2021 [4]. In late June, a few cases of the B.1.617.2 variant (Delta) were reported, and in August of 2021, the Delta variant showed a wide dispersion, completely displacing the Gamma variant and its derived lineages on October [5]. In December of 2021, the first detections of the Omicron variant were described in RS state, but the Delta variant still predominated. This scenario quickly reversed in early January of 2022; the Delta variant was detected in only a few samples, and since then, the Omicron lineage has already dominated the sequenced samples (https://virological.org/t/rapid-establishment-of-omicron-variant-in-rio-grande-do-sul-state-brazil/783).
Here, we isolated and characterized four representative samples collected from the very early cases of COVID-19 that occurred in a highly populated area in Southern Brazil from April to July of 2020. These samples were isolated in VERO cells and sequenced for use as positive controls for both research and diagnosis. Mutational sequence profiles and signatures were also analyzed to better understand the lineage sequences. Moreover, one of the samples evaluated in this study was one of the first cases of COVID-19 that occurred in RS.
Material and methods
Molecular analysis
Preliminary diagnosis of COVID-19
Naso-oropharyngeal swab samples were received at Laboratório de Microbiologia Molecular (LMM) of Universidade Feevale for SARS-CoV-2 detection by RT-qPCR from suspected patients of COVID-19. The extraction of viral RNA was carried out by the commercial MagMAX™ CORE Nucleic Acid Purification Kit (Applied biosystems™, Thermo Fisher Scientific, Waltham, MA, USA) kit, using the automated equipment KingFisher™ Duo Prime (Thermo Fisher Scientific™). RT-qPCR was performed selectively for the envelope (E) gene according to the Charite Institute, Berlin, Germany, protocols [6] and using AgPath-ID One-Step RT-PCR Reagents (Thermo Fisher Scientific™).
Viral genome sequencing
Viral genome sequencing was carried out as previously described by Silva et al. (2021). Briefly, viral genome library preparation was carried out using the QIAseq SARS-CoV-2 Primer Panel paired for library enrichment and QIAseq FX DNA Library UDI Kit. Also, Illumina MiSeq platform (Foster City, CA, USA), using MiSeq Reagent Kit v3 (600-cycle) was used. The Geneious Prime™ suite was used for genome assembly and editing. FASTq reads were imported, trimmed to remove low-quality sequences and primers used for library generation (BBDuk 37.25), and mapped against the reference sequence hCoV-19/Wuhan/WIV04/2019 (EPI_ISL_402124) available at GISAID [7]. PANGO and Nextstrain lineage assignments were applied to characterize the consensus sequences. Mutational sequence profiles and signatures were also analyzed to better understand the lineage sequences. Moreover, to analyze single nucleotide polymorphisms (SNPs) and amino acid changes, Geneious Prime™ software was used to perform a variant calling approach between a FASTq dataset and their consensus sequences.
Phylogenetic analysis
Sequences generated in this study were aligned with 63 Brazilian SARS-CoV-2 complete genomes and the reference sequence (> 29 kb) that were retrieved from the GISAID database. Alignment using the reference sequence from Wuhan as the outgroup was performed using the Clustal Omega software. The maximum likelihood phylogenetic analysis under the general time-reversible model, allowing for a proportion of invariable sites and substitution rates were inferred empirically in IQ-TREE v2.1.2 Web server [8], especially for maximum likelihood (ML, applying 200 replicates and 1000 bootstraps).
Cell culture-based methods
Viral isolation
To isolate virus particles to be used as viral controls in later tests, clinical samples were subjected to the viral isolation technique detailed below. The activities were carried out in partnership with Centro de Pesquisas em Biologia Molecular e Funcional (CPBMF) of Pontifícia Universidade Católica do Rio Grande do Sul (PUCRS), which kindly granted us the facilities of the biosafety level 3 (BSL-3) laboratory. Cell cultures of Vero cells (CCL-81, ATCC, Manassas, VA, USA) were used for SARS-CoV-2 isolation. Cells were routinely maintained using Dulbecco’s modified Eagle medium (DMEM) low glucose containing 10% fetal calf serum, maintained in an incubator at 37 °C in an atmosphere of 5% CO2. Samples were previously filtered through 0.22-μm diameter membrane filters, thus preventing cell cytotoxicity. A total of 50 µL inoculum of each sample was placed in contact with cells in 25-cm2 cell culture flasks. The inoculum remained in contact with cell monolayers for 1 h for adsorption to occur. Afterward, the inoculum was removed, and a maintenance medium was added. Flasks of inoculated cells remained in the same incubator as described above under monitoring of 5-day passages. During the monitoring time, inoculated cells were compared to control cells under a light microscope to identify potential cytopathic effects (CPEs) of the viable virus. After that, the inoculated cells were inactivated in the BSL-3 lab to be sent to LMM where viral replication was confirmed through the increase of cycle threshold (CT) value using the same RT-qPCR protocol and sequenced using the high-performance Illumina MiSeq platform as previously described.
TCID 50 assay
Median tissue culture infectious dose (TCID50) was applied to quantify the number of infectious virus particles in two isolates of clinical samples that were used as controls in the lab. So, tenfold serial dilutions of samples were performed, being 10−1 to 10−8 the dilutions employed. VERO cells were routinely maintained as described above, and 96-well plates were used to carry out the assays. Briefly, 50 µL of each dilution was added to respective wells, and the same volume of DMEM was used as cell control. After that, 50 µL of VERO cell suspension was added to all wells. The plates were kept in an incubator for 5 days, observing the CPE under an optical microscope. The viral titration was expressed in TCID50/mL using the calculation described by Ramakrishnan (2016) [9].
Results
Eleven samples from the beginning of diagnostic activities at LMM were isolated in 2020. Among these samples, four were sequenced in order to identify the circulating lineage at the beginning of SARS-CoV-2 spreading in RS. The four samples were collected from April to July 2020, covering four cities of RS (Table 1). By inoculating the samples into VERO cells, we detected viral replication besides the cytopathic effect (Fig. 1), since RT-qPCR results were positive and CT values decreased in average by 4.27 already in the first passage, when compared to the CT detected for the patient’s clinical report. Furthermore, viral titration was applied to quantify the number of infectious virus particles in two isolates of clinical samples that were used as controls in the lab showing results of 3.16 × 104 and 5.62 × 104 TCID50/mL for LMM11934 and LMM14497, respectively (Table 1).

Images show viral isolation of SARS-CoV-2 in VERO cells being the cellular control (a) and cytopathic effect of the LMM14497 sample (b)
According to PANGO lineage assignment tool [10], these four sequences were classified as B.1.1.33 lineage (Fig. 2). Table 2 shows the amino acid changes with their frequency in specific genes of the four samples using the variant calling approach between FASTq reads and their consensus sequences. LMM6378 presented amino acid changes in genes N (2), ORF8 (1), S (2), and ORF1ab (6), being that the variant frequency ranged from 25.8 to 42.3%. LMM11934 showed only 2 mutations on ORF1ab (26.2–27.5%). LMM14497 was the sample that had more non-synonymous mutations in gene S (3) and ORF1ab (10) (25.4%—46.4%). In genes S (1) and ORF1ab (4), the sample LMM17042 also presented amino acid changes (26.4–38.2%).

Phylogenetic tree of SARS-CoV-2 complete genomes circulating in Brazil in the year 2020. Highlight in bold is the four isolated sequences of RS
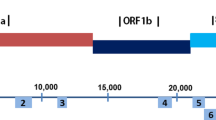
We identified seven nonsynonymous mutations in the S gene sequences from the four samples studied, when compared to the reference sequence hCoV-19/Wuhan/WIV04/2019 (EPI_ISL_402124) (Fig. 3). At least one nonsynonymous mutation was identified in each of the sequenced samples: LMM6378 (T63N and D614G), LMM11934 (D614G), LMM14497 (N74K, E96D, E484D, D614G, H655Y), and LMM17042 (T63N, N148K, D614G).

S gene translation alignment of the four samples with SARS-CoV-2 reference showing the mutations related. Highlighted in red, there is one sample with the specific mutation; in blue, two samples; and in green, all samples
Discussion
The B.1.1.33, known as a Brazilian lineage, has been detected in many countries. Brazil (84%), the USA (4%), Chile (3%), Canada (1%), and Argentina (1%) encompass most of the cases. Its earliest description date back to 2020–03-01, and the largest circulation in the world appears to have occurred between March 2020 and May 2021 (https://cov-lineages.org/lineage.html?lineage=B.1.1.33), the time interval in which our samples were collected.
The four sequenced samples belong to the B.1.1.33 lineage, which descends from lineage B.1. The latter is a large European lineage, whose origin, in turn, roughly corresponds to the Northern Italian outbreak early in 2020 (https://cov-lineages.org/lineage.html?lineage=B.1). On February 25, 2020, a Brazilian man, who traveled days before to Lombardy, Northern Italy, was confirmed as the first Brazilian case of COVID-19 [11]. Since the LMM6378 sample was one of the first COVID-19 positive samples in our laboratory and our diagnostic service started in a pioneering way in the region in March of 2020, we highlight that B.1.1.33 was one of the precursor lineages in the spread of SARS-CoV-2 in RS.
Through the analysis described, it was possible to evaluate all the amino acid changes in FASTq reads even though these changes were not frequent enough to compose the consensus sequences of their respective samples. Some of the nonsynonymous mutations found in these consensus sequences, when compared to the reference sample, corroborated the data from the variant calling approach, thus making it possible to observe the gradual genome shift in these sequences. As seen in the S gene (Table 2), the amino acid changes that occurred with higher frequency were N63T in LMM6378 (42.3%) and LMM17042 (38%), and D484E in LMM14497 (46.4%). Since these changes had not enough frequency, mutations T63N and E484D were seen in the S gene translation alignment (Fig. 3).
B.1.1.33 is characterized by mutations in genes ORF1b (P314L), S (D614G), ORF6 (I33T), ORF8 (S84L), and N (R203K, G204R, I292T) (https://outbreak.info/situation-reports?pango=B.1.1.33). Since we analyzed only the S gene sequence, S:D614G was the signature mutation found in our four sequences. Finding this mutation in all sequences was expected since it is already a B.1.1.33 lineage characteristic mutation that has been described as important for viral spread and higher infection titers [12].
Another mutation, which is not the lineage signature but has been detected worldwide in different lineages, is S:H655Y, which was found in sequence LMM14497. According to Escalera et al. (2022) [13], SARS-CoV-2 variants harboring S:655Y show an enhanced spike protein cleavage and fusogenic ability in vitro, increasing transmissibility in vivo, and an enhanced phenotype in the human primary airway model. Worldwide, 2,648,195 sequences with the S:H655Y mutation have been detected since its identification in January 2020. In Brazil, 71,726 sequences were described, being 1328 in RS (data from March 28, 2022: https://outbreak.info/). Here, we would like to highlight that our sequence (LMM14497), collected on July 9, 2020, corresponds to the earliest sample detected containing this mutation in RS.
The other mutations identified in our sequences have considerably low detection frequencies worldwide and in Brazil. S:T63N, the only mutation that was found in two of the sequenced samples in this study (LMM6378 and LMM17042), has been previously detected in 101 samples worldwide, being 13 of them from Brazil, with almost all of them (12) coming from RS (data from March 28, 2022: https://outbreak.info/). Interestingly, sample LMM6378 corresponds to the earliest isolate reported to contain mutation S:T63N in RS, being also one of the first positive samples for COVID-19 in the region. The detection of this mutation started with an increase but ended up not being broadly widespread.
In the state of RS, the other mutations identified (S:N74K, E96D, E484D, N148K) were described only in our sequences since then. LMM14497 showed the mutations: S:N74K (detected in 152 sequences worldwide being 5 in Brazil), S:E96D (detected in 2,243 sequences worldwide being 24 in Brazil), and S:E484D (299 sequences worldwide being 6 in Brazil). LMM17042 presented the S:N148K that has been detected in 210 sequences worldwide, being two sequences in Brazil (data from March 28, 2022: https://outbreak.info/). Through the sequencing and analysis of mutations found in introductory sequences to the pandemic in the region, it is possible to trace the changes in the viral genome that occurred within the same lineage. Therefore, it is possible to infer how mutations may have interfered with the evolution and viral dissemination, and how some became characteristic of the lineage due to adaptive advantage when compared to others that were not able to maintain their circulation.
The viral isolation method was effective to isolate the four samples with the possibility of increasing the viral titter after a passage in VERO cells, being possible its detection through qPCR and its titration. Therefore, cell culture-based methods can be used to produce stock in laboratories contributing to the detection and identification of the studied virus. Although these samples are representative of the same lineage, their genomes are different. Therefore, the analysis of the profiles and signatures of mutational sequences was approached in an important way to better understand the studied sequences. Thus, it was possible to detect in our samples important mutations that were identified for the first time in the southernmost state of Brazil.
In conclusion, we highlight the importance of studies that track the genome profile of the early SARS-CoV-2 lineages being possible to evaluate mutations that became more frequent over time showing their effect on viral fitness and, consequently, the viral evolution. Also, these data point out the importance of constant evaluation and monitoring of SARS-CoV-2 in the population, supporting efforts that help to delineate strategies in the face of possible adversities such as the emergence of new variants, vaccine immune escape, and increase in virulence.
Data Availability
Not applicable.
Code availability
Not applicable.
References
Zhou P, Lou Yang X, Wang XG et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 579(7798):270–273. https://doi.org/10.1038/s41586-020-2012-7
Franceschi VB, Caldana GD, de Menezes MA et al (2021) Genomic epidemiology of SARS-CoV-2 in Esteio, Rio Grande do Sul, Brazil. BMC Genom 22(1):1–17. https://doi.org/10.1186/s12864-021-07708-w
Francisco R S, Benites LF, Lamarca AP, et al. (2021) Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. 296(January). doi:https://doi.org/10.1016/j.virusres.2021.198345
da Silva MS, Demoliner M, Hansen AW et al (2021) Early detection of SARS-CoV-2 P.1 variant in Southern Brazil and reinfection of the same patient by P.2. Rev Inst Med Trop Sao Paulo. 63:1–8. https://doi.org/10.1590/s1678-9946202163058
Gularte JS, da Silva MS, Mosena ACS et al (2022) Early introduction, dispersal and evolution of Delta SARS-CoV-2 in Southern Brazil, late predominance of AY.99.2 and AY.101 related lineages. Virus Res. 311(January):198702. https://doi.org/10.1016/j.virusres.2022.198702
Corman V, Bleicker T, Brünink S et al (2020) Diagnostic detection of 2019-NCoV by real-time RT-RCR. https://virologie-ccm.charite.de/en/. Accessed Nov 2020
Shu Y, McCauley J (2017) GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance 22(13):2–4. https://doi.org/10.2807/1560-7917.ES.2017.22.13.30494
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32(1):268–274. https://doi.org/10.1093/molbev/msu300
Ramakrishnan MA (2016) Determination of 50% endpoint titer using a simple formula. World J Virol 5(2):85. https://doi.org/10.5501/wjv.v5.i2.85
Rambaut A, Holmes EC, O’Toole Á et al (2020) A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 5(11):1403–1407. https://doi.org/10.1038/s41564-020-0770-5
Rodriguez-Morales AJ, Gallego V, Escalera-Antezana JP, et al. (2020) COVID-19 in Latin America: The implications of the first confirmed case in Brazil. Travel Med Infect Dis 35(January). doi:https://doi.org/10.1016/j.tmaid.2020.101613
Plante JA, Liu Y, Liu J et al (2021) Spike mutation D614G alters SARS-CoV-2 fitness. Nature 592(7852):116–121. https://doi.org/10.1038/s41586-020-2895-3
Escalera A, Gonzalez-Reiche AS, Aslam S et al (2022) Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe 30(3):373–387. https://doi.org/10.1016/j.chom.2022.01.006
Acknowledgements
We thank the Brazilian Coordination for the Improvement of Higher-Level Personnel (CAPES), Foundation for Research Support of the State of Rio Grande do Sul (FAPERGS), and Brazilian National Council for Scientific Development (CNPq) for scholarships.
Funding
This work is an initiative of Rede Corona-ômica BR MCTI/FINEP affiliated to RedeVírus/MCTI (FINEP = 01.20.0029.000462/20, CNPq = 404096/2020–4).
Author information
Authors and Affiliations
Contributions
JSG, JDF, FRS: conceptualization. JSG, MSS, MF, MD, KS, AWH, VMAGP, FHH, VG, MNW, PRA, BLA, MD, PM, LAB, JDF, FRS: formal analysis and investigation. OAD, FRS: funding acquisition. JDF, FRS: supervision. FRS: project administration. JSG: writing—original draft. CVB, OAD, JDF, FRS: writing—review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval
Project approved by the Research Ethics Committee (CEP) at Feevale University. Process number: CAAE: 33202820.7.1001.5348.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Responsible Editor: Jônatas Abrahão
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gularte, J.S., da Silva, M.S., Filippi, M. et al. Viral isolation allows characterization of early samples of SARS-CoV-2 lineage B1.1.33 with unique mutations (S: H655Y and T63N) circulating in Southern Brazil in 2020. Braz J Microbiol 53, 1313–1319 (2022). https://doi.org/10.1007/s42770-022-00789-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-022-00789-z