
Abstract
Industries from different business sectors are facing challenges against global competitions for the development of sustainable and renewable products in the twenty-first century. Likewise, constant effort from pulp and paper manufacturers in minimizing paper cost with better quality in the active field of filler modification technology is much appreciated. In the present study, chitosan has been explored as a surface modifier alternative to conventional starch for precipitated calcium carbonate (PCC) to design chitosan/PCC composite filler. Two different dissolution mediums, hydrochloric and acetic acid, for chitosan have been seen to affect PCC crystals. Commercial PCC comprises mostly aragonite polymorphs along with some calcite crystals as implied by FTIR, XRD and FE-SEM images. It is interesting to note that surface treatment of PCC with 4.5% chitosan can successfully induce crystal transformation of PCC from aragonite to calcite polymorphs. Further, the deposition of chitosan was estimated from TOC measurements and the presence of deposited amount was validated from TGA analysis. Moreover, the introduction of chitosan (dissolved in HCl) to PCC dispersion was found to raise the zeta potential from − 14.43 to − 11.3 mv. Finally, the tensile strength of handsheets increased by 8.2% with 20% enhancement in ash with chitosan/PCC composite filler compared to the unmodified PCC. Therefore, bio-based PCC composites which proved to be promising for the development of high ash paper without compromising essential properties may result in saving wood pulp and production cost. Thus, implementing such seafood waste as a value-added additive is beneficial both to the industries and the environment because of its biodegradability and eco-friendliness.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Recent trends of substituting non-renewable petroleum-based resources with renewable have attracted papermaking industries in develo** value-added sustainable and biodegradable products. Therefore, both wood and agro residues derived ligno-cellulosic feed-stocks are utilized to produce paper-based materials containing three-dimensional cellulose networks. Though high strength paper can be achieved with 100% load-bearing cellulose fibers, substituting these fiber factions with mineral fillers is a common strategy to reduce the cost and energy associated with papermaking. The most naturally occurring mineral fillers include clay, talc, ground calcium carbonate (GCC) and precipitated calcium carbonate (PCC) which are added to furnish prior to the headbox section in a conventional papermaking process. Among these fillers, PCC has gained a lot of attention worldwide due to its manufacturing flexibility with controllable chemical and physical properties and unique crystal habits. The application of fillers contributes certain functional properties to the sheet such as dimensional stability, light scattering efficiency, printability and writability [1]. These desired characteristics of the sheets are determined by critical parameters including shape, particle size distribution, aspect ratio, average particle size, aggregate size and density during the manufacturing of fillers. It was found that the smallest particles are detrimental to the paper strength at constant shape. However, the size of PCC is comparatively smaller than that of cellulose fibers resulting in poorer first-pass ash retention in the wire section [1, 2]. Furthermore, the traditional route of stochastic incorporation of fillers in the cellulose network weakens the fiber–fiber bonding at high filler addition level as they may partially reside at fiber interstice which deteriorates the ultimate strength of individual fibers as well as paper. Since papermakers crave for high ash content paper, different classical routes were explored in integrating these pigment fillers with pulp fibers [2,3,4,5,6]. Pre-flocculation or surface modification of PCC through coating or encapsulation among preceding techniques has been an impressive approach to mitigate negative effects on mechanical properties of paper at high ash level [2,3,4, 7]. Industrial low-cost native starch or cationic starch is a widely accepted biopolymer in filler modification among various researchers as this hydrogen bonding polymer can develop strong bonds with pulp fibers when dried after starch/mineral composite fillers are added to the furnish. Another approach to fabricate these bio-based composite fillers with paper sheet is in situ crystallization of calcium carbonate in the presence of biopolymers [8]. But there are certain limitations associated with starch such as consumption of extra chemicals during the coating process, loss of cationicity at alkaline pH, sensitivity to water and its prone to microbial attack [9]. In addition to starch, several other synthetic or biodegradable polymers were considered for filler modification by adsorbing these polyelectrolytes onto them to confer the enhanced bonding ability between fillers and fibers. Thus, polymer-induced aggregation leads to a larger particle size of the composite fillers which can then be sufficiently retained in the sheet. In paper technology, synthetic polymeric materials are vastly applied as retention and strength aids [10, 11]. Processing with such synthetic polymers results in environmental pollution, serious health issues due to their carcinogenicity and creates problems in recycling or composting paper materials reported by consumers and government bodies. This might have motivated researchers to look for an alternate approach to utilizing biodegradable resources. Since the processing of crustaceans in the seafood industries constitutes 60% of shell waste according to Environmental Protection Agency (EPA), it should be a major concern for the industries to look for solid waste management. Hence, interest lies in the extraction of value-added products from such wastes and their potential application in diverse fields [9, 12, 13]. Chitosan is that biogenic environmental-friendly polymer having natural cationic charge density, biodegradability, non-toxicity, antibacterial and antifungal properties. Not only in medical science and other sectors, chitosan has also emerged as a value-added potential additive among paper industries. Although expensive compared to conventional wet-end additives, its unique property of flocculation ability both at acidic and alkaline pH makes it a better retention-drainage aid in stock preparation [14,15,16]. In contrast to conventional starch, 30% faster drainage rates, higher internal bond strength, wet/dry strength with no tendency of slime formation, and desired surface and optical properties were notable for chitosan in terms of good machine runnability and energy consumption. It was anticipated that this would minimize the overall cost of production to manufacture an economically feasible paper with high-quality specifications as per customers’ demand [17, 18]. This polymer was found to be effective both for surface coating and wet-end section in develo** specialty grade or packaging papers. The NH2 group in the polymer backbone not only provides additional bonding ability with cellulose but also imparts water resistance to the paper when made at alkaline conditions as revealed in these studies. So there is an urge to modify filler surface with this polymer proving possibilities to widen the scope of its application [14, 15, 19]. In the year 2012, a US patent disclosed that PCC’s inherent alkalinity enables chitosan’s precipitation that can effectively functionalize the PCC surface for the development of paper-based composite materials. However, the patent did not reveal either the effect of functionalization on PCC structure or illustrate any mechanism for acidic chitosan-basic PCC reaction [20].
Synthetically produced PCC can exist in three anhydrous polymorphs namely calcite, aragonite and vaterite. Calcite, being the most thermodynamically stable has become a choice to the papermakers. Under different process conditions, it exhibits different morphologies which can be either rhombohedral, scalenohedral or prismatic [3]. Crystallization of calcium carbonate starts with nucleation and crystal growth takes place either by aggregation or by phase transformation mechanism [21]. Thus, crystallization of PCC particles is tailored by manipulating supersaturation, reactant feed, temperature or with some additives to acquire desired properties required for paper [8]. The templating action of many polymers in controlling the calcium carbonate crystal growth and their orientation has been discussed extensively [22,23,24]. Stabilization of phase, growth rate and ultimate crystal morphology depends on the degree of inorganic/additive interactions. Biomimetic approaches of mineralization show the presence of different polymorphs which can certainly be elucidated by stereochemical complementary between calcium ions and active groups in the polymer chain as well as geometry matching. Chitosan-mediated bio-mineralization has gained a lot of attention and has been observed that phase transformation is indeed a result of molecular reorganization between polymer and mineral [21, 22].
Many researchers have already evaluated chitosan as a filler modifier for papermaking application but the influence of chitosan on structural changes in PCC during the modification process and their effect on paper properties was given less importance in the reported literature. In the present work, a simple route of integrating chitosan with mineral filler PCC was established for develo** chitosan/PCC composite filler. Detailed characterization of crystal growth and morphology of PCC in the presence of this polymer were stressed so as to evaluate the performance of the corresponding composite as strength and filler retention additive in cellulose integrated paper material.
2 Experimental methods
2.1 Materials
Chitosan from shrimp shells with deacetylation > 75% was procured from Hi-media, India. Precipitated Calcium carbonate (PCC) and bleached Hardwood pulp were obtained from Paper mill, India. Hydrochloric acid (HCl) and acetic acid used in the whole study were of analytical grade. The filler suspension slurry was alkaline with pH 10.4.
2.2 Method of preparing modified PCC fillers
1 g of powder filler was dispersed in 50 ml of distilled water with a mechanical stirrer for at least 2 h before modification. 1% chitosan was dissolved in an aqueous solution of 0.1% HCl or 1% acetic acid under constant stirring at 60 °C prior to the experiments [25]. Then, 0.045 g chitosan in solution form was loaded onto the filler slurry followed by mixing for at least 30 min in water bath shaker at room temperature. The polymer percentage calculated on the basis of oven-dried (OD) PCC was 4.5%. No other auxiliary chemical was used to maintain the pH for precipitation of chitosan on PCC particles. After that, the samples were centrifuged at 3000 rpm for 10 min. The supernatant was collected for calculating the sorbed amount of chitosan by analyzing the total organic carbon (TOC) of the samples through the TOC analyzer (TOC-LCPH, Shimadzu). Deposited amount DA was determined by the following equation
C1 and C2 are the added and residual amounts of chitosan after treatment. M is the mass of PCC. All the units are expressed in g. In TOC analyzer, liquid supernatant containing residual chitosan was introduced into the combustion tube which was filled with an oxidation catalyst, generally platinum catalyst and heated to 680 °C. The sample was burnt in the combustion tube and as a result the TC (total carbon) components of the sample were converted into carbon dioxide which was then detected by NDIR (non-dispersive infrared) gas analyzer. Similarly, inorganic carbon IC was determined by acidifying the sample to evolve carbon-di-oxide. In both the cases, calibration curves are generated with IC and TC standard solutions to evaluate TOC (TC–IC) of the samples. The obtained filler pellets were then dried at 30 °C for their subsequent characterization
2.3 Characterization of fillers
The chemical group analysis was done by FTIR (Fourier transform infrared spectrophotometry) through the Perkin Elmer spectrophotometer in a transmission mode conducting 16 scans at a resolution of 4 cm−1 to acquire absorption spectra between 4000 and 500 cm−1. Samples were diluted with KBr (potassium bromide) to form pressed pellets. Further phase transformation phenomenon was studied by examining the crystal structure through WAXD [RIGAKU ULTIMA IV Wide-angle X-ray diffractometer at a scan rate of 4°/min over a range of 2θ = 5°–80°. The operating voltage was 40 kV with a current 30 mA. The radiation source was Nickel filtered CuKα (λ = 1.5406 Å). Next, the thermal stability of modified and unmodified PCC was observed by thermogravimetric analysis (TGA) by TA instrument, USA in a temperature range between 40 and 900 °C at 10 °C/min in a nitrogen atmosphere with 60 ml/min gas flow rate. The weight of the samples varied from 10 to 20 mg. Further, particle size distribution was obtained from the PSD analyzer (LA960, Horiba Ltd., Kyoto, Japan). The principle is based on static light scattering Mie theory. Approximately, 0.1 g of fillers was dispersed in 100 ml distilled water and ultra-sonicated for at least 30 min prior to zeta potential measurements by ZetaPlus zeta potential analyzer (Brookhaven, 205 H2oltsville, NY, USA) [26]. Image analysis of flocs was also carried out by the optical microscope (Motic B1 Series). At first, modified and unmodified filler slurries were diluted to 0.2%, and one drop of the suspension was placed on the glass slides. The glass slides were then air-dried for further investigation. Crystal morphology was examined by TESCAN MIRA 3 Field Emission Scanning Electron Microscope. A very little amount of sample was gold sputtered for 1 min prior to imaging. Images were taken at a magnification of 25 kX, 5kX and 2 kX with an accelerating voltage 10 kV.
2.4 Handsheet preparation and characterization
Dried pulps were first beaten to a level of 38°SR and then disintegrated at a pulp consistency of 2% for 10 min at 3000 rpm. Pulp slurry was then diluted to 0.4% prior to the addition of unmodified and modified PCCs. The addition level of filler was 15% based on OD pulp. The stock suspension was then mechanically stirred at 400 rpm for 5 min and transferred to a batch laboratory sheet Former so as to achieve a target basis weight of 70 g m−2 according to the TAPPI test method T 205 sp-02. The handsheets were then conditioned in a humidity chamber at 23 °C ± 1 °C and relative humidity 50 ± 2% before testing. For determining the filler retention, ash in the paper was estimated by incinerating paper sheets at 525 °C as per the TAPPI standard method T 211 om-93. Thus, filler retention was calculated from the following equation
Note that control means ash associated with unfilled sheets.
Tensile strength was evaluated according to the TAPPI test methods T 494 om-01 by the tensile tester (SE 060, L&W). Other physical properties such as tear and bust were obtained from tear tester (SE009) and burst tester (SE180), respectively. In another experiment, handsheets were subjected to spectrophotometer CM 3630 (Konica Minolta). The morphology of the paper samples incorporated with fillers was analyzed by FE-SEM.
3 Results and discussion
Both PCC and modified PCC were characterized by different analytical tools, FTIR, XRD and TGA to detect the chemical groups, phases present in the dried powders and thermal behavior of the composite fillers, respectively. Thus, spectral data obtained from FTIR reveal valuable information regarding the polymorphs of the fillers distinguished by their different carbonate vibration modes. Our commercial PCC comprises mainly aragonite phases assigned by the following carbonate absorption peaks at 1081, 856, 1494, 713 and 700 cm−1.
These peaks correspond to symmetric stretching (ѵ1), out-of-plane bending (ѵ2); doubly degenerate planar asymmetric stretching (ѵ3) and doubly degenerate planar bending (ѵ4), respectively. These characteristic peaks of aragonite polymorph have been reported by several researchers [24]. CO32− ions remain inactive in the infrared region and that the band at 1081 cm−1 cannot be seen in the pure calcite phase of calcium carbonate has already been verified in the previous literature [27] [28]. Apart from stretching vibrations, out-of-plane and in-plane bending of C-O bond resembling absorption band at 873 and 712 cm−1 are attributed to calcite phases in the sample [28]. The IR spectra of industrially manufactured PCC used in this study have similar absorption bands as per the published literature data revealed from Fig. 1 [29, 30]. The presence of CH and OH species in the unmodified PCC depends on various synthesis routes or due to the interaction of some organic additives as dispersing agents during the manufacturing process [30,31,32]. In comparison to the unmodified one, those modified with chitosan displaying a slight displacement of peak around 1494 cm−1 toward lower absorption band at 1458 cm−1 is due to the differences in the crystal structure of calcite and aragonite. Rather it is assigned to the specific band for calcite [28]. The spectra display a broader peak at 3431 cm−1 mainly assigned to the OH or NH stretching vibrations related to chitosan. Moreover, the absorption band at 2919 and 2874 cm−1 designated for C–H stretch has become broadened due to the overlap** C–H bond associated with chitosan [33, 34]. A weak intensified peak at 1150 cm−1 and a shoulder at 1030 cm−1 could be assigned to an asymmetric stretch of C–O–C bridge and C–O stretch, respectively, revealing the presence of the polymer in the modified samples [34]. Another interesting feature in the modified PCC spectrum observed is that the peaks at 1082 and 856 cm−1 have sufficiently decreased. In fact, a more pronounced peak at 873 cm−1 and disappearance of peak at 700 cm−1 confirms the transformation of aragonite polymorphs to calcite phases. Note that peaks relayed to calcite phases are more intensified when chitosan was dissolved in acetic acid for the modification of the PCC surface. The crystal structure of unmodified and modified PCC was further studied by XRD and shows a good correlation with FTIR data. The coexistence of both calcite and aragonite phases can be detected from the diffraction pattern of pure PCC as noted from Fig. 2.

FTIR of A Commercial PCC and PCC modified with chitosans and B distinguished peaks of aragonite and calcite

XRD patterns of A PCC, B PCC + chitosan (HCl) and C PCC + chitosan (acetic acid) black filled circle calcite, red filled circle aragonite
The diffraction peaks positioned at 2θ of 23.0° (112), 29.4° (104), 35.9° (110), 47.5° (018), and 28.5° (116) are associated with calcite polymorph whereas 2θ of 26.2° (111), 27.2° (021), 33.1° (012), 37.9° (112), 38.4° (130), 45.8° (221), 50.3° (132), and 52.5° (023) are related to aragonite phase [35]. After introducing chitosan in PCC dispersion, the intensity of peaks at 2θ = 26.2° and 27.2° corresponding to the planes (111) and (021) of aragonite has been sufficiently reduced with an increase in the peak intensity at 2θ = 29.4° corresponding to (104) planes of calcite. Furthermore, the calcite and aragonite content quantified from respective intensities based on previously reported methods were found to be 21% and 79%, respectively, in the initial PCC sample [36]. On the other hand, chitosan-modified PCC is indexed as a mixture of the majority of calcite with small traces of aragonite. In particular, the dissolution of chitosan in acetic acid and sorption of the polymer on PCC surfaces produce a larger quantity of calcite crystals (97%) compared to hydrochloric acid (92%). The respective aragonite quantity estimated was 3% and 8% [36]. However, traces of chitosan could not be detected from XRD patterns of the modified samples. This can be supported with the earlier reports of XRD where authors did not define the presence of chitosan acting as oriented support for the growth of calcium carbonate polymorphs [37]. It was observed that when an acidic solution of chitosan (pH 5.5 in case of both acids) was subjected to the alkaline environment of PCC (pH 10.4), chitosan started precipitating out of the solution resulting in co-aggregation of PCC and polymeric macromolecules with a final solution pH of 7.8–7.9 at equilibrium. The principle of chitosan regeneration in alkaline medium from acidic solution is governed by the screening of electrostatic repulsion between the protonated amine groups above its pKa value [38]. At this stage, the stiff rod-like structure of chitosan conforms more into a random coil followed by deposition on PCC surfaces [39]. Therefore, we have discussed the probable mechanism behind this polymorph transformation by analyzing the data from FTIR and XRD. Generally, calcium carbonate exists in the form of three anhydrous crystal polymorphs among which vaterite is the least stable and calcite being the most thermodynamically stable [40]. The findings from the recent studies suggest that nucleation and growth of resultant crystalline polymorphs (vaterite, aragonite and calcite) occur via dissolution–re-precipitation mechanism through an intermediate hydrated amorphous calcium carbonate (ACC) precursors. [21, 35, 40, 41]. It was inferred that controlling of critical process variables like change of solution pH by acids and organic additives affect the yield of particular crystal polymorphs and its stabilization. pH-dependent dissolution rates of ACC lead to different crystallization pathways that ultimately define a particular calcium carbonate polymorph [42,43,44,45,46]. Ca2+ and CO3− binding strength and equilibrium carbonate species CO3−/, HCO3− at different pH are the determining factors for ACC structures that mimic certain local polymorphs which further transform into calcite, aragonite or vaterite. The driving force for the conversion of one polymorph to other was postulated to be the HCO3− ions as calcium carbonate growth rate was proportional to the ion activity product of Ca 2+ and HCO3− [47]. On the other hand, chitosan as an additive has been studied as a template for the nucleation and growth of different polymorphic structures based on the charge density and solution pH [21, 22]. This near-neutral macromolecule at alkaline pH was thought to gather Ca2+ ions on its surface providing an interface between this organic–inorganic composite and the surrounding liquid for active nucleation site of calcite crystallization [48]. Therefore, as per our system concerned, the transformation of aragonite crystals to polycrystalline calcite aggregates can be explained on the principle of (1) dissolution–recrystallization mechanism and (2) sorption of chitosan on the crystal surface. In our opinion, the fast dissolution of metastable aragonite at this pH might have released dissolved inorganic HCO3− and Ca2+ ions with co-precipitation of chitosan on aragonite surface [49].
At this point, interaction of chitosan with precipitated calcium carbonate would be difficult to explain since electrostatic forces would not be very strong because of deprotonated amine groups of chitosan in weak alkaline condition [48, 50]. However, chitosan described as a hydrogel in near neutral pH solution could be able to promote calcium carbonate polymorphism. Therefore, we can conclude that chitosan with its amine and OH groups deposited on the crystal surface and served as a template for the recrystallization of calcite aggregates [48]. Inhibition to the growth of free single crystal could be due to the polymeric chains of the macromolecule since it has the ability to form bridges between calcium carbonate particles. Moreover, from the work of Busenberg and Plummer 1987 percentage transformation of more soluble aragonite to the less soluble calcite in contact with water depends on solution pH, solution chemistry, temperature and precipitation time. Various models established for this mechanistic behavior state that the rate of dissolution of primary crystals is equal to the growth of secondary mineral crystals [51]. Several authors have explained that electronegative charge on specific anionic additives likely to adsorb Ca2+ ions which further attracts the HCO3/–CO3− ions to facilitate nucleation of calcium carbonate species [42, 52]. So, in our case, the appearance of a larger number of calcite crystals in the presence of chitosan/acetic acid system may be expected due to an additional effect of this organic acid adsorption on the polymorph rearrangements. Previously, it was observed that carboxylated organic acids with their deprotonated negatively charged sites (COO–) at 7–8 adsorbed on the surface of aragonite crystals inducing their recrystallization to calcite [42]. Although further investigations are necessary to better understand the mechanism of active nucleation pathway to control of rearrangements of PCC polymorphs. The presence of chitosan in the modified PCC was further confirmed by the thermal behavior as observed from TGA-DTG curves in Fig. 3A). Unmodified PCC exhibits single-step degradation profile when the temperature is increased from 40 to 900 °C. This is in agreement with the reported literature [53, 54]. The degradation begins at Tonset = 511 °C and the decomposition completes at temperature 683 °C since this temperature range accelerates the formation of CaO and CO2 [32]. In comparison, it is seen that the thermal degradation profile for modified PCCs has changed with two consecutive degradation steps. Besides weight loss due to PCC, an additional weight loss is seen to take place between 210 and 400 °C. This is due to the dehydration of the saccharide ring accompanied by a random chain breakdown favoring complete decomposition of the chitosan molecule [55]. There is no evidence of accompanying curves of OH ions in the DTG graph (Fig. 3B) which further ensures that water is not present in the samples.

A TGA and B DTG curves of PCC and chitosan-modified PCC
Moreover, the mass loss for samples PCC + Chitosan (Acetic acid) and PCC + Chitosan (HCl) is around 47% which is relatively higher than the unmodified one whose mass loss was 44% indicating the existence of sorbed bio modifier on PCC surface [56]. Additionally, it is noticeable that though Tonset for calcium carbonate decomposition is almost analogous in all the samples. Maximum weight loss temperature Tmax of modified PCCs deviates from the actual Tmax of the unmodified one as observed from the DTG plot. Tmax for sample PCC, PCC + Chitosan (Acetic acid) and PCC + Chitosan (HCl) is 657 °C, 647 °C and 662 °C, respectively, implying that chitosan precipitated from HCl on PCC surface makes the filler more thermal stable compared to acetic acid. It is possible that acetic acid-mediated less thermal stable composites are attributed to the occurrence of chitosonium acetate originated from residual acids between chitosan chains in the dried composites [57].
The sorption mechanism of chitosan on the PCC surface was then illustrated through the TOC analysis, and the dissolving medium considered was HCl since organic acid from acetic acid interfered with TOC of chitosan as exhibited in Fig. 4A. There was a significant increment in TOC in the presence of acetic acid as the concentration of chitosan was raised. This initial examination aided us to eliminate the usage of acetic acid since it would be difficult to evaluate the actual concentration of deposited chitosan from the chitosan-acetic acid standard curve. Figure 4B describes the deposition of chitosan onto PCC. Depending on the degree of deacetylation, the intrinsic pKa of chitosan from shrimp is almost close to 6.5 [33]. Hence, the polymer is completely soluble at pH below its pKa value with 90% of its amine group protonated in the glucosamine units [58]. The polymer possesses a cationic behavior in the acidic medium with electrostatic repulsion between its chains [33]. This is in agreement with our result showing that the charge density of chitosan in HCl (pH 5.5) is + 7.3 meq/g. In our study, the high alkalinity of PCC makes the amine group in chitosan deprotonated. The deprotonation means that at pH greater than neutrality chitosan starts becoming insoluble as discussed in previous sections with much of a coiled entangled structure rather than a stiff rod-like structure resulting in significantly higher adsorption on PCC [55]. Similar conclusions were drawn for the adsorption of chitosan on quartz at pH 9 [38]. It is evident from the graph in Fig. 4B that the deposition increases with an increasing amount of chitosan and the maximum value of the deposited amount is found to be 10.7% to reach the saturation level at an added dose of 11% of PCC. When subjected to alkaline pH 10.4 exerted by PCC in CO2 free water, the chitosan might have lost its enough cationic charge to neutralize the colloidal anionic charge on PCC [20]. This hypothesis can be related to our charge density measurement data where we could observe that, initially, PCC had an anionic charge density of − 5.49 μeq/g. Even PCC functionalized with 4.5% chitosan, anionic charge density decreased to − 4.4 μeq/g but no charge reversal or charge nullification could be detected at this point. We, therefore, propose that the dominant mechanism was flocculation of PCC by the polymer rather than coagulation. Roussy and Van Vooren observed that solubility and insolubility of chitosan at different pH strongly affects the coagulation/flocculation behavior of mineral colloids [58]. They concluded that at high pH the neutralized amine groups of polymer chains physically entrap the mineral colloids in its network by bridging mechanism leading to instability of the colloids. Similar trends are also observed in our study. The final pH measured at a chitosan dose of 11% of PCC after adsorption decreased to 7.2. The amount of chitosan on PCC remained almost constant with a further increase in the dose of chitosan. With 4.5% addition of chitosan to PCC, the deposited chitosan amount quantified was 4.3%. An excessive amount of chitosan might have led to re-stabilization of the suspension indicated by the high value of TOC in the supernatant. A similar explanation was given by Roussy and Van Vooren when residual turbidity increased due to excess addition of chitosan for decantation of colloid particles at natural water pH [39]. The increase in the zeta potential of PCC from − 14.3 to − 11.3 mv indicates that adsorbed uncharged chitosan just moved the slip surface of the electrical double layer with some electrostatic interactions. Otherwise, the surface potential of PCC might have been close to zero or reversed [38].

A Total organic carbon (TOC) versus chitosan concentration in 0.1% HCl and 1% acetic acid. B Sorption of chitosan onto PCC at ambient temperature for 30 min when HCl was used as dissolving medium for chitosan. A Macroscopic optical photographs of unmodified and modified filler slurries taken with a digital camera
The crystal morphology and surface morphology of the PCC particles were studied by optical microscope and FE-SEM images as depicted in Figs. 5 and 6, respectively. The micrographs obtained under different magnification revealed that aggregated mineral flocs in both modified and unmodified PCC have become dark due to light opaqueness. More aggregated polymorphism was observed with modified one representing surface treatment of PCC either by encapsulation, flocculation, and adsorption/precipitation. Recrystallization of aragonite particles in the presence of chitosan was further examined by FE-SEM.

Optical microscopic images (magnification 10X and 40X) of A unmodified PCC. B, C Chitosan-modified PCC. B Represents chitosan dissolved in acetic acid. C Represents chitosan dissolved in HCl

FE-SEM images of unmodified PCC magnified A at 25 kx, B at 5 kx and C at 2 kx. Chitosan-modified PCC magnified D at 25 kx E at 5 kx and F at 2 kx, D–F represent chitosan dissolved in acetic acid. Chitosan-modified PCC magnified G at 25 kx, H at 5 kx and I at 2 kx, G–I represent chitosan dissolved in HCl
The coexistence of aragonite and calcite is evident from Fig. 6A with a predominance of aragonite crystals in the native PCC. The rod-shaped aragonite crystals are arranged in bundles in a semi-circular structure [43]. As discussed previously in the above sections of the study there are possibilities that interaction between NH2 and OH groups in chitosan with carbonate ions can affect the ultimate crystal habit leading to the formation of a scalenohedral type of calcite in the presence of chitosan [3] [59]. The prevalence of calcite with traces of aragonite is observed in Fig. 6G which can relate well with the increased intensity of peak corresponding to (104) plane. The aggregation phenomenon can further be corroborated with optical microscope images revealing irregular shaped dense and bigger flocculated particles in modified PCCs. Several other authors reported a morphological change of different grade of fillers preflocculating or modifying with different polymers [6, 14, 60]. The median particle size of native PCC was around 11 µm which increased dramatically to 104 µm after treatment with chitosan when HCl was considered as a dissolution medium. Individual PCC particles were being aggregated to larger flocs in modified PCC through a bridging mechanism by precipitated polymeric chains either as a result of weak electrostatic interaction between chitosan and Ca2+ ions or by physical entrapment of the minerals in its coiled structure.
Additionally, Fig. 7 shows that modified PCC exhibits much broader size distribution compared to the native one indicating nonuniform aggregation of filler particles as also observed in FE-SEM images. Similar particle size distribution is not only obtained by papermakers in producing pre-flocculated filler particles [61] but also can be observed in other industries where flocculation is an important mechanism to optimize a system. The encapsulation mechanism is proposed in Fig. 8. As prepared filler slurries were then employed to analyze the effect of chitosan modified filler particles on tensile strength and filler retention calculated from residual ash in handsheets. It is a well-known fact that incorporating filler at high ash level interferes with fiber–fiber bonding resulting in decreased paper strength. Paper strength generally depends on individual fiber strength, inter-fiber contact area as well as bonding strength.

Particle size distribution of A unmodified PCC, B Chitosan-modified PCC. B Represents chitosan dissolved in HCl

Schematic representation of encapsulation of PCC with chitosan
Figure 9A reveals that at 15% filler dose, the tensile strength of paper decreases by 21% in the case of unmodified fillers compared to unfilled paper. This finding is in consistent with the reported works [4]. Interestingly, handsheets fabricated with chitosan-induced aggregated fillers are seen to improve tensile strength by 8.2% compared to native ones. Enhancement or negative impact of paper strength with different pre-flocs or bio-based composite mineral fillers is well verified by many authors [4, 8, 60].

A Tensile index, B Ash and filler retention of unfilled, filled and modified filler at 15% filler dose based on OD pulp. MPCC represents modified PCC
According to our proposed mechanism instead of depositing on one single particle, chitosan seems to encapsulate several particles bounded to one another and enmeshed within the polymer chains. When mixed with pulp, amine groups (NH2) of chitosan protruding from the surface of modified PCC establish hydrogen bonds with the cellulose fibers, thereby increasing in paper strength when dried. A similar trend for bursting strength of handsheets can be observed in the case of modified filler as evident in Fig. 10A. Higher improvement in tensile and burst with their corresponding correlation was reported by many while applying functionalized polymers in filler modification technology to achieve higher ash content [14, 60, 62].

A Optical properties, B tear, burst and apparent density of unfilled, filled and modified filler at 15% filler dose based on OD pulp. MPCC represents modified PCC
Therefore, it is expected that the bonding potential of chitosan may be superior when it is anchored on the surface of PCC fillers rather than stochastically distributed between fibers and fillers. However, papers containing modified PCC exhibit almost the same tear index noticed in Fig. 10A. Though authors have not emphasized much on the behavior of tear strength of paper while using chitosan as a filler modifier [14, 19], a similar effect was observed when PCC-cellulose hybrid composites were incorporated in sheet [63]. On the other hand, at the same ash level, filler retention of unmodified and modified PCC calculated is 38% and 50%, respectively. It is commonly known that the filler retention depends upon its particle size, shape and chemical nature. Therefore, higher retention achieved with the aggregated PCC particles may be due to the larger floc sizes. Presumably, a slight decrease in surface potential of PCC may allow the chitosan to adsorb on cellulose fibers effectively ensuring higher retention of modified fillers [60]. It is worth noting that the ash level studied under the experimental conditions does not have a significant impact on the optical properties of the paper in case of modified filler as seen in Fig. 10B. However, one can expect that the marginal increase in brightness around 1% at higher ash level is due to higher retention of larger particles of modified PCC [15]. Further, an increase in filler retention as a result of successful surface encapsulation of PCC with chitosan polymer is evident from Fig. 11 which shows networks of cellulose fibers incorporated with mineral fillers. Clearly, SEM analysis reveals that modified PCC not only fills the voids but also creates linkages between fibers by adsorbing sufficiently onto them. This signifies the inevitable adhesive nature of chitosan and its promising end-use application in papermaking.

FE-SEM images of cellulose networks filled with A unmodified fillers, B modified fillers
3.1 An outlook for production cost
Approximately, the cost of cellulose pulp (CP) is INR 65/kg (1 USD = INR 75) and PCC is INR 14/kg. Therefore, the estimated cost of base/raw materials for manufacturing PCC-filled paper shall be INR 67100/t considering PCC charge of 150 kg/t of CP and that of for manufacturing modified PCC-filled paper shall be INR 71150/t when 6.70 kg chitosan was applied per tonne of CP for PCC modification. The cost of chitosan is roughly around INR 600/kg. It was found in this study that even such a low dose of chitosan consumed for PCC modification can able to increase PCC retention in pulp fibers by 20% compared to the paper filled with unmodified PCC. It is to be also noted that an increase in ash content reduced the tensile strength loss without mitigating other physical, structural and optical properties. Therefore, replacing cellulose fiber with a higher amount of PCC can be expected to have faster drainage rates and lower energy consumption in paper machines, thus minimizing the overall cost of production. Furthermore, chitosan in its native state was enough to induce PCC flocculation at alkaline pH and function as a potential retention aid without any additional auxiliary chemicals or quarternization. Nevertheless, its utilization from shrimp waste in filler modification technology provides new opportunities for the development of sustainable and eco-friendly products. Overall, a 6% increase in base/raw materials cost with chitosan biopolymer shall be compensated with reduced energy cost in production lines, its value addition to the paper products and its subsequent eco-friendly environmental impact at industrial scale.
4 Conclusions
In the present work, an attempt was made to modify the precipitated calcium carbonate (PCC) with chitosan. The notable change in the crystal structure morphology was observed by FTIR, XRD and FE-SEM, respectively. The principle of transition from aragonite crystals from native PCC to prismatic calcite crystals of modified PCC may be proposed based on the dissolution–recrystallization mechanism in the presence of chitosan that leads to stabilization and orientation of polymorphs, thus altering the ultimate crystal habit. Presumably, the polymer acts as a template for the growth of polycrystalline calcites by adsorbing on the surface of aragonite and facilitates bridging of calcium carbonate particle with its coiled like chains. Moreover, the higher intensity at 2θ = 29.4° corresponding to (104) plane of the XRD pattern in case of PCC modified with chitosan dissolved in acetic acid implies a higher fraction of calcite compared to HCl used as dissolving medium. Chitosan-induced aggregation of PCC fillers observed under optical images and FE-SEM was well corroborated to the particle size distribution data. The existence of chitosan chains in modified PCC was confirmed by TGA and TOC analysis. The deposition efficiency of chitosan was above 90% as correlated with measured TOC values. When incorporated with pulp fibers, these polysaccharide/mineral composites showed enhanced filler retention compared to the unmodified one without negatively affecting the physical strength of paper. However, at present ash level, optical properties did not significantly change. Thus, the comprehensive characterization of the composite filler in accordance with our study may be beneficial for the development of high ash paper with desired properties. Such practice of converting seafood wastes into value-added additives are favorable not only to the paper industries but also to the environment leading to an efficient approach to reduce exploitation of forest resources and improve waste management. Further, work related to the factors governing the phase transformation of PCC polymorphs should be thoroughly studied in the future.
References
Gaudreault R, Di Cesare N, Van De Ven TGM, Weitz DA (2015) Structure and strength of flocs of precipitated calcium carbonate induced by various polymers used in papermaking. Ind Eng Chem Res 54(24):6234–6246
Kumar P, Negi YS, Singh SP (2011) Filler loading in the lumen or/and cell wall of fibers—a literature review Pradeep Kumar, Yuvraj Singh Negi, and Surendra Pal Singh. BioResources 6:3526–3546
Subramanian R, Fordsmand H, Paulapuro H (2007) “Precipitated calcium carbonate (pcc)—cellulose composite fillers; effect of pcc particle structure on the production and properties of uncoated fine paper. Bioresources 2(1):91–105
Shen J, Song Z, Qian X, Yang F (2010) Carboxymethyl cellulose/alum modified precipitated calcium carbonate fillers: preparation and their use in papermaking. Carbohyd Polym 81(3):545–553
Huang X, Sun Z, Qian X, Li J, Shen J (2014) Starch/sodium oleate/calcium chloride modified filler for papermaking: impact of filler modification process conditions and retention systems as evaluated by filler bondability factor in combination with other parameters. Ind Eng Chem Res 53(15):6426–6432
Huang X, Qian X, Li J, Lou S, Shen J (2015) Starch/rosin complexes for improving the interaction of mineral filler particles with cellulosic fibers. Carbohyd Polym 117:78–82
Fan J et al (2017) Interaction between two oppositely charged starches in an aqueous medium containing suspended mineral particles as a basis for the generation of cellulose-compatible composites. Ind Crops Prod 97:417–424
Kuusisto JE, Maloney TC (2015) The effect of carbonation conditions on the properties of carbohydrate-calcium carbonate hybrid pigments. BioResources 10(2):3277–3292
Srinivasa PC, Tharanathan RN (2007) Chitin/chitosan—safe, ecofriendly packaging materials with multiple potential uses. Food Rev Int 23(1):53–72
Song X, Hubbe MA (2014) Enhancement of paper dry strength by carboxymethylated β-D-glucan from oat as additive. Holzforschung 68(3):257–263
Ahrenstedt L, Oksanen A, Salminen K, Brumer H (2008) Paper dry strength improvement by xyloglucan addition: wet-end application, spray coating and synergism with borate. Holzforschung 62(1):8–14
Sun G et al (2008) Preparation of H-oleoyl-carboxymethyl-chitosan and the function as a coagulation agent for residual oil in aqueous system. Front Mater Sci Chin 2(1):105–112
Wang H et al (2017) Environmentally benign chitosan as reductant and supporter for synthesis of Ag/AgCl/chitosan composites by one-step and their photocatalytic degradation performance under visible-light irradiation. Front Mater Sci 11(2):130–138
Yu Y, Xue G, Gu C, Lou J, Li S (2013) Preparation of chitosan modified talc and its application in high filler content paper. J Appl Polym Sci 129(5):2692–2698
Chen Z, Li C, Song Z, Qian X (2014) Modification of precipitated calcium carbonate filler for papermaking with adsorption of cationically derivatized chitosan and carboxymethyl chitosan. BioResources 9(4):5917–5927
Amit S, Ghosh UK (2018) An approach for phycoremediation of different wastewaters and biodiesel production using microalgae. Environ Sci Pollut Res 25(19):18673–18681
Miranda R, Nicu R, Bobu E, Blanco A (2016) Efficiency of chitosan and their combination with bentonite as retention aids in papermaking. BioResources 11(4):10448–10468
Hamzeh Y, Sabbaghi S, Ashori A, Abdulkhani A, Soltani F (2013) Improving wet and dry strength properties of recycled old corrugated carton (OCC) pulp using various polymers. Carbohyd Polym 94(1):577–583
Shen J, Song Z, Qian X, Song C (2008) Chitosan coated papermaking grade PCC filler prepared by alkali precipitation : properties and application. In: The second international papermaking and environment conference, pp 659–664
Soane DS, Berg MC, Willium MA (2012) Functionalization of paper components U.S. Patent Application No. 13/369,392
Declet A, Reyes E, Suárez OM (2016) Calcium carbonate precipitation: a review of the carbonate crystallization process and applications in bioinspired composites. Rev Adv Mater Sci 44(1):87–107
Chen X, Shao Z, Cheng C, Yao J, Wu Y (2011) Crystallization of calcium carbonate on chitosan substrates in the presence of regenerated silk fibroin. Langmuir 27(6):2804–2810
Ritchie AW et al (2013) Reversed crystal growth of rhombohedral calcite in the presence of chitosan and gum arabic. CrystEngComm 15(47):10266–10271
Shen Q et al (2005) Crystallization and aggregation behaviors of calcium carbonate in the presence of poly(vinylpyrrolidone) and sodium dodecyl sulfate. J Phys Chem B 109(39):18342–18347
Czechowska-Biskup R, Wojtasz-Pająk A, Sikorski J, Henke A, Ulanski P, Rosiak J (2007) Solution of hydrochloric acid as simple solvent of chitosan for viscosity and light-scattering based molecular weight. Polish Chitin Society, pp 87–94
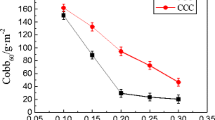
Ghosh I, Sharma C, Tandon R (2018) Role of additives to improve adsorption of xylan on precipitated calcium carbonate. Cellulose 25(3):1583–1598
Chong KY, Chia CH, Zakaria S, Sajab MS, Chook SW, Khiew PS (2015) CaCO3-decorated cellulose aerogel for removal of Congo Red from aqueous solution. Cellulose 22(4):2683–2691
Toffolo M, Regev L, Dubernet S, Lefrais Y, Boaretto E (2019) FTIR-based crystallinity assessment of aragonite-calcite mixtures in archaeological lime binders altered by diagenesis. Minerals 9(2):121
Jimoh OA, Otitoju TA, Hussin H, Ariffin KS, Baharun N (2017) Understanding the precipitated calcium carbonate (PCC) production mechanism and its characteristics in the liquid-gas system using milk of lime (MOL) suspension. S Afr J Chem 70:1–7
Shi X, Rosa R, Lazzeri A (2010) On the coating of precipitated calcium carbonate with stearic acid in aqueous medium. Langmuir 26(11):8474–8482
Barzagli F, Giorgi C, Mani F, Peruzzini M (2017) CO2 capture by aqueous Na2CO3 integrated with high-quality CaCO3 formation and pure CO2 release at room conditions. J CO2 Util 22:346–354
Patowary M, Pathak K, Ananthakrishnan R (2015) A facile preparation of superhydrophobic and oleophilic precipitated calcium carbonate sorbent powder for oil spill clean-ups from water and land surfaces. RSC Adv 5(97):79852–79859
Boudouaia N, Bengharez Z, Jellali S (2019) Preparation and characterization of chitosan extracted from shrimp shells waste and chitosan film: application for Eriochrome black T removal from aqueous solutions. Appl Water Sci 9(4):1–12
Queiroz MF, Melo KRT, Sabry DA, Sassaki GL, Rocha HAO (2015) Does the use of chitosan contribute to oxalate kidney stone formation? Mar Drugs 13(1):141–158
Chong K-Y, Chia C-H, Zakaria S (2014) Polymorphs calcium carbonate on temperature reaction. In: AIP conference proceedings, vol 52, February 2015, pp 52–56
Locock A (2012) Recommended method for semi-quantitative measurement of aragonite—calcite—dolomite mixtures by powder X-ray diffraction
Díaz-Dosque M et al (2008) Use of biopolymers as oriented supports for the stabilization of different polymorphs of biomineralized calcium carbonate with complex shape. J Cryst Growth 310(24):5331–5340
Feng B, Peng J, Zhu X, Huang W (2017) The settling behavior of quartz using chitosan as flocculant. J Mater Res Technol 6(1):71–76
Roussy J, Van Vooren M, Guibal E (2005) Influence of chitosan characteristics on coagulation and flocculation of organic suspensions. J Appl Polym Sci 98(5):2070–2079
Tateno N, Kyono A (2014) Structural change induced by dehydration in ikaite. J Mineral Petrol Sci 109:157–168
Ogino T, Suzuki T, Sawada K (1987) The formation and transformation mechanism of calcium carbonate in water. Geochim Cosmochim Acta 51:2757–2767
Kawano M, Tokonamib M (2014) Effect of organic acids on the precipitation rate and polymorphism of calcium carbonate minerals in solutions with mg2 + ions. Clay Sci 18:1–10
Ma Y, Feng Q (2015) A crucial process: organic matrix and magnesium ion control of amorphous calcium carbonate crystallization on β-chitin film. CrystEngComm 17(1):32–39
Falini G et al (2007) Polymeric admixtures effects on calcium carbonate crystallization: relevance to cement industries and biomineralization. CrystEngComm 9(12):1162–1170
Verch A, Antonietti M, Gebauer BD, Co H (2009) The multiple roles of additives in CaCO3 crystallization : a quantitative case study, pp 435–439
Shaw S, Bots P, Benning LG (2012) The role of pH and Mg on the stability and crystallization of amorphous calcium carbonate. J Alloy Compd 536:S477–S479
Falini G, Manuel J (2016) Role of CaCO °3 neutral pair in calcium carbonate crystallization. Cryst Growth Des 16:4173–4177
Hiranya T, Nawarathna K, Nakashima K, Kawasaki S (2019) Chitosan enhances calcium carbonate precipitation and solidi fi cation mediated by bacteria. Int J Biol Macromol 133:867–874
Busenberg E, Plummer LN (1987) A comparative study of the dissolution and crystal growth kinetics of calcite and aragonite. US Geolog Surv Bull 1578:139–168
Nilsen-nygaard J, Strand SP, Vårum KM, Draget KI, Nordgård CT (2015) Chitosan: gels and interfacial properties. Polymers 7:552–579
Perdikouri C, Kasioptas A, Geisler T, Schmidt BC, Putnis A (2011) Experimental study of the aragonite to calcite transition in aqueous solution. Geochimica et Cosmochimica 75:6211–6224
Saraya MEI, Hassan H, Rokbaa AE (2017) Formation and stabilization of vaterite calcium carbonate by using natural polysaccharide. Adv Nanopart 6(4):158–182
Patel D, Kuperkar KC (2018) electrokinetic potential, thermal and microscopic investigation of surfactant-polymer templates on precipitated calcium carbonate morphology with dynamic simulation. Chem Sel 3(16):4382–4386
Li XG, Lv Y, Ma BG, Wang WQ, Jian SW (2017) Decomposition kinetic characteristics of calcium carbonate containing organic acids by TGA. Arab J Chem 10(November):S2534–S2538
Lim BY, Poh CS, Voon CH, Salmah H (2015) Rheological and thermal study of chitosan filled thermoplastic elastomer composites. Appl Mech Mater 754–755(April):34–38
Wilson OC, Hull JR (2008) Surface modification of nanophase hydroxyapatite with chitosan. Mater Sci Eng C 28(3):434–437
Ferfera-Harrar H, Dairi N (2014) Green nanocomposite films based on cellulose acetate and biopolymer-modified nanoclays: studies on morphology and properties. Iran Polym J (Engl Ed) 23(12):917–931
Roussy J, Van Vooren M, Guibal E (2005) Chitosan for the coagulation and flocculation of mineral colloids. J Dispers Sci Technol 25(5):663–677
Gamelas JAF, Lourenco AF, Ferreira PJ (2011) New modified filler obtained by silica formed by sol-gel method on calcium carbonate. J Sol Gel Sci Technol 59(1):25–31
Chauhan VS, Bhardwaj NK (2013) Enhancing paper properties by preflocculation of talc using amphoteric starch. Nord Pulp Pap Res J 28(2):248–256
Seo D (2012) The effects of process variables for GCC pre-flocculation on floc and handsheet properties. Nordic Pulp Pap Res J 27(2):382–387
Peng Y, He B, Zhao L (2015) Lime mud CaCO3 for use as a filler material in papermaking: impact of its preflocculation with cationic polyacrylamide. J Appl Polym Sci 132(11):41640
Mohamadzadeh-Saghavaz K, Resalati H, Ghasemian A (2014) Cellulose-precipitated calcium carbonate composites and their effect on paper properties. Chem Pap 68(6):774–781
Acknowledgement
The research grant for the present work was supported by the Ministry of Human Resource Development (MHRD), Government of India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ghosh, I., Sharma, C. & Tandon, R. Structural evaluation of chitosan-modified precipitated calcium carbonate composite fillers for papermaking applications. SN Appl. Sci. 2, 1577 (2020). https://doi.org/10.1007/s42452-020-03313-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-03313-w