
Abstract
The renal secretion of many drugs is facilitated by membrane transporters, including organic cation transporter 2, multidrug and toxin extrusion protein 1/2-K and organic anion transporters 1 and 3. Inhibition of these transporters can reduce renal excretion of drugs and thereby pose a safety risk. Assessing the risk of inhibition of these membrane transporters by investigational drugs remains a key focus in the evaluation of drug–drug interactions (DDIs). Current methods to predict DDI risk are based on generating in vitro data followed by a clinical assessment using a recommended exogenous probe substrate for the individual drug transporter. More recently, monitoring plasma-based and urine-based endogenous biomarkers to predict transporter-mediated DDIs in early phase I studies represents a promising approach to facilitate, improve and potentially avoid conventional clinical DDI studies. This perspective reviews the evidence for use of these endogenous biomarkers in the assessment of renal transporter-mediated DDI, evaluates how endogenous biomarkers may help to expand the DDI assessment toolkit and offers some potential knowledge gaps. A conceptual framework for assessment that may complement the current paradigm of predicting the potential for renal transporter-mediated DDIs is outlined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Renal transporter mediated drug–drug interactions (DDIs) can cause severe adverse events. |
During drug development, conservative guideline-derived decision criteria frequently require dedicated DDI studies to assess the DDI risk for the key renal transporters organic cation transporter 2 (OCT2), multidrug and toxin extrusion proteins (MATEs) and organic anion transporters 1 and 3 (OAT1 and OAT3). |
Several endogenous biomarkers for OCT2, MATE1/2-K, and OAT1 and OAT3 renal transporter inhibition have been identified to inform the risk for potential renal transporter DDIs. |
We propose a new decision criteria process to support the use of endogenous biomarkers in early-phase clinical trials to assess the potential for renal transporter-mediated DDIs. |
1 Introduction
Drug–drug interactions (DDIs) may occur when two (or more) drugs are co-administered to a patient, resulting in the altered efficacy or safety of one or both drugs. DDIs may be mediated by drug-metabolising enzymes or drug transporters, leading to a rise in the plasma concentration of the drug whose metabolism or transport is inhibited. Evaluation of the DDI risk is an essential element of establishing benefit–risk profiles of a new molecular entity (NME) during drug development [1, 2].
The kidneys play an essential role in the elimination of drugs and metabolites into the urine from the circulation. In addition to passive glomerular filtration, drugs are eliminated into the urine by active drug transport systems (involving tubular secretion and reabsorption) in the proximal renal tubules [3]. Solute carrier (SLC) membrane transporters play an important role in the metabolism and excretion of small-molecule drugs [4, 5]. Important renal transporters involved in this process include the organic cation transporters (OCTs), multidrug and toxin efflux proteins (MATEs) and organic anion transporters (OATs) (Fig. 1).

Schematic of the kidney (A), nephron with major blood vessels (B) and renal proximal tubule cells (C). In (B), the basic physiological mechanisms of handling fluid and electrolytes by the nephron, filtration, secretion, reabsorption and excretion are labelled. In (C), the major renal membrane transporters expressed on renal proximal tubule cells and their potential endogenous biomarkers are shown. The transporters located in the basolateral plasma membrane include organic anion transporter (OAT) 1/3 and organic cation transporter (OCT) 2. Transporters located in the apical membrane include multidrug and toxin efflux protein 1 (MATE) 1/2-K. GCDCA-s glycochenodeoxycholate-3-sulphate, HVA homovanillic acid, NMN N1-methylnicotinamide, m1A N1-methyladenosine, PDA pyridoxic acid
The OCT2 transporter (from the SLC22 gene family) is predominantly expressed on the basolateral membrane of tubular epithelial cells and is responsible for the renal uptake of water-soluble cationic compounds such as metformin and cisplatin [6,7,8]. The subsequent efflux (secretion) of these drugs into urine is mediated by MATE transporters, expressed on the apical side of the tubules. Human MATEs have two isoforms, MATE1 and MATE2-K (from the SLC47 family), which can transport various substances, including creatinine, corticosteroids, metformin, cimetidine, and certain antibiotics [9]. Cimetidine, levofloxacin, and pyrimethamine are potent inhibitors of MATE transporters [10, 11]. Together, OCT2 and MATE1/2-K are the major transporters for the secretion of cationic drugs into the urine [12]. Metformin, a well-characterised substrate of these transporters, serves as a typical probe for assessing potential OCT2 and/or MATE1/2-K-associated DDIs [13].
Drugs inhibiting MATE1/2-K and OCT2 transporters can decrease the elimination of metformin and thereby increase its plasma concentrations (Table S1 of the Electronic Supplementary Material [ESM]), leading to an elevated risk of metformin-associated lactic acidosis, a rare but potentially fatal adverse event [14]. Metformin-associated lactic acidosis is due to the accumulation of lactate through the inhibition of hepatic glucose production from lactate molecules. When the first signs of metformin-associated lactic acidosis develop (e.g. severe vomiting and diarrhoea), metformin administration is stopped and urgent medical attention is given [14]. In a similar way, inhibition of the MATE1/2-K transporters can cause drug accumulation, and possibly nephrotoxicity. Such a scenario is thought to explain cisplatin-induced nephrotoxicity [3, 15]. As OCT2 and MATE1/2-K are involved in the disposition and elimination of a variety of drugs (Table S1 of the ESM), evaluation of the inhibitory potential of a drug towards OCT2, MATE1 or MATE2-K is an integral part of drug development.
In addition to OCT2 and MATEs, the OAT1, OAT2 and OAT3 transporters from the SLC22 family, located on the basolateral membrane of the proximal tubule cells, play an important role in mediating the uptake of drugs from the blood [3, 15]. OAT1 and OAT3 preferentially handle the active tubular secretion of anionic substances in the kidneys. Several commonly used drugs have recognised interactions with the OAT1 and OAT3 transporters. For example, OAT1 and OAT3 are involved in the renal clearance (CLR) of methotrexate, a chemotherapeutic agent used to treat autoimmune diseases. Decreased methotrexate elimination and associated methotrexate toxicity can manifest as myelosuppression, hepatotoxicity and mucositis [16]. Non-steroidal anti-inflammatory drugs (NSAIDs) and probenecid both diminish OAT1-mediated and OAT3-mediated tubular secretion, resulting in clinically decreased CLR and consequently increased systemic exposure of several anionic drugs including methotrexate, furosemide, cefaclor, cefonicid and ciprofloxacin [17] (Table S1 of the ESM). In addition, NSAIDs can inhibit OAT-mediated renal secretion of diuretics, thereby reducing their effectiveness [18]. This interaction can result in decreased diuretic efficacy, with potential for fluid retention or exacerbation of heart failure in susceptible patients [19]. Further examples of renal transporter-mediated DDIs can be found in comprehensive reviews by Ivanyuk et al. [9] and Łapczuk-Romańska et al. [20].
To ensure patient safety, NMEs with major transporters in the proximal tubules that could lead to transporter-mediated severe DDIs with other co-administered drugs are routinely assessed early in drug development by mechanism-based static approaches [15, 21]. Typically, this involves predicting the DDI risk based on in vitro data followed by a clinical assessment using a recommended exogenous probe substrate for the individual drug transporter [1, 21,22,23]. However, there are several issues with this approach, which is discussed below. We performed a comprehensive review of the available in vitro and in vivo data to support endogenous biomarker use to evaluate DDIs mediated by renal transporters. Using this information, we propose a conceptual framework (from a pharmaceutical industry perspective) to integrate endogenous biomarkers in early clinical development to streamline the assessment of renal transporter-mediated DDIs.
2 Current Pharmaceutical Industry Approaches to Assess the DDI Risk Mediated by Renal Transporters
As potential life-threatening adverse events associated with metformin and other concomitant medications are a significant concern for health authorities, current regulatory guidelines from the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) underscore the importance of conducting dedicated DDI (probe) studies, using a stepwise decision tree to evaluate the risk of DDIs for NMEs that inhibit renal transporters (Fig. 2A) [2, 24]. To understand the DDI potential, each NME is assessed to see if it is a substrate or inhibitor of various enzymes and transporters. Subsequent in vitro and clinical assessment is then performed, evaluating whether the NME inhibits any of the renal transporters known to be involved in clinically relevant in vivo DDIs. At present, OCT2, MATE1/2-K, OAT1 and OAT3 are the key transporters evaluated.

Current approach (A) [4, 26, 27] and proposed biomarker-informed approach (B) decision process to streamline the renal transporter (organic cation transporter [OCT] 2, multidrug and toxin extrusion protein [MATE] 1, MATE2-K) drug–drug interaction (DDI) risk assessment. AUC area under the plasma concentration–time curve, CLR renal clearance, EMA European Medicines Agency, FDA US Food and Drug Administration, NME new molecular entity, OAT organic anion transporter, PoCP proof-of-clinical principle, SRD single rising dose, MRD multiple rising dose, ↑ increased, ↓ decreased, *significant increase in AUC is greater than 1.25-fold; *baseline (predose) urine or plasma biomarker concentrations are required for an accurate assessment of biomarker exposure (AUC, CLR) changes; ***significant decrease in CLR is less than 0.80-fold. A PoCP study shows that a candidate drug results in a biological and/or clinical change associated with the disease and the mechanism of action. A PoCP study is most critical when develo** novel innovative compounds, and less relevant for less innovative compounds developed in a pre-determined linear manner where there are fewer uncertainties and risks
Transporter in vitro inhibition potency parameters (half-maximal inhibitory concentration [IC50] or inhibition constant [Ki] values) are routinely generated for each NME in respect of the uptake of a known substrate for the different renal transporters in cells overexpressing these transporters [2] along with the unbound maximum plasma concentration (Cmax,u). The ratio of Cmax,u to IC50 or Ki provides a quantitative reflection of in vivo inhibition potency. Regulatory-guided “static” decision trees are then applied to assess the DDI risk and the need for a formal clinical DDI study using a probe substrate drug (Fig. 2A). If the Cmax,u/IC50 ratio is below a cut-off value (0.02 for the EMA [24] and 0.1 for the FDA [2]), no additional follow-up assessment of DDIs is recommended. If the ratio is above the cut-off value, a clinical DDI trial is recommended [2, 25] to support the enrolment of patients for proof-of-clinical principle (PoCP) or phase II studies. Alternatively, the affected drug based on in vitro assessment may be excluded from the subsequent clinical studies to ensure the safety of the patients in the current practice (Fig. 2A). Based on the existing regulatory guidelines, pharmaceutical industry approaches to evaluate renal transporter-mediated DDIs currently involve conducting a stand-alone probe or cocktail DDI study or excluding patients taking the affected drug from subsequent clinical trials (Table 1).
3 Limitations with Current Assessment Approaches
The forementioned current approaches have associated limitations (Table 1). First, current cut-off values that might be overly conservative can limit robust in-vitro-to-in-vivo extrapolation, reducing the reliability of solely relying on in vitro DDI study outcomes for a prospective DDI risk assessment. Recent analyses highlight a need to refine the current approach to evaluate these in vitro studies that determine whether an NME is likely to inhibit drug transporters to a clinically significant extent [25]. Mathialagan et al., assessed the performance of the existing Cmax,u/IC50 ratio cut-off values used to assess potential OCT2 and MATE1/2-K transporter interactions [26]. Based on EMA criteria, high false-positive predictions were observed for the drug candidates that may inhibit OCT2 transporters (a positive predictive value of 64% was reported) and candidates that inhibit MATE1/2-K transporters (positive predictive value = 47%) [26]. A separate analysis reported high false-positive predictions (positive predictive value = 52%) using EMA criteria for drugs that inhibit OAT 1/3 transporters [28]. The current cut-off values likely result in a higher number of clinical DDI probe studies being performed. The cost and time delay implications of conducting these dedicated probe studies to assess the potential NME renal transporter-mediated DDI potential are substantial. These studies can take several months from study protocol design to data analysis and reporting, cost hundreds of thousands of dollars, can delay the clinical development programme by up to 6 months and waste significant resources, especially in view of the recognised high false-positive predictions mentioned above.
Second, there are issues with the alternative option to exclude potentially affected patients from future PoCP or phase II studies (Fig. 2A). Here, consideration is required regarding the expected number of patients taking a medication associated with a potential renal transporter-mediated DDI. For example, the number of patients taking metformin and furosemide can be as high as ~ 25–50% and ~ 25–35%, respectively, depending on the indication (Boehringer Ingelheim, unpublished data). Excluding these patients from phase II PoCP studies can slow recruitment and result in an enrolled trial population that is not representative of the target patient population [27]. This in turn can cause delays in regulatory approval, restrictive labelling and post-marketing sponsor commitments [27].
4 Use of Endogenous Biomarkers to Assess the DDI Risk
In recent years, there has been considerable interest in the evaluation of changes in the concentrations of endogenous biomarkers to assess the potential for a drug to inhibit transporters [22, 25]. Some endogenous biomarkers are substrates of clinically relevant drug transporters, meaning that changes in their concentrations, in conjunction with other available information, may more reliably predict the transporter inhibitory potential of a drug in vivo. Changes in the concentrations of endogenous biomarkers early in drug development can also inform concomitant medication strategies for subsequent efficacy and safety trials and inform the DDI assessment strategy.
Endogenous biomarkers can be used in a variety of ways during the clinical development process. First, the kinetics of endogenous biomarkers can be evaluated during initial phase I trials, thereby guiding further clinical development to potentially avoid a dedicated DDI study if there is no change in the kinetics of the endogenous biomarker in the presence of the NME [29]. Factors to consider when selecting each biomarker include their selectivity, sensitivity, specificity, predictivity, robustness and ease of accessibility [22, 30,31,32]. Second, the change in the kinetics of each endogenous biomarker should ideally reflect the interaction of the inhibitor with the activity of a single transporter, and the extent of change should be similar to one of the clinically used DDI probe drugs [22, 32]. In addition, biomarkers should be well characterised regarding their kinetics, endogenous synthesis, active transport, metabolic transformation and influence on a disease state. Intrinsic and dietary confounding factors that can influence their concentrations should be minimised [22, 33]. This stringent set of criteria means that only a handful of endogenous transporter biomarkers have been identified. Below, we review the key endogenous biomarkers proposed for OCT2, MATEs, and OAT1/3 renal uptake transporters.
4.1 Endogenous Biomarkers for OCT2, MATE1 and MATE2-K Transporters
For the renal organic cation secretion axis, represented by the OCT2 and MATE renal transporters, creatinine, N1-methylnicotinamide (NMN) and N1-methyladenosine (m1A) have been identified as potential endogenous biomarkers for a potential transporter interaction assessment (Table 2). Creatinine, a metabolite of muscle creatine, is mainly excreted passively via glomerular filtration, with 10–40% actively secreted [34], mainly by OCT2, MATE1/2-K [35]. Creatinine is commonly utilised as a biomarker for renal function. N1-methylnicotinamide (NMN), a metabolite of niacin, is metabolised, passively cleared via glomerular filtration [34] and actively transported into urine by OCT2 and MATE1/2-K [36, 37]. Unchanged NMN CLR accounts for ~35% of total clearance, encompassing the entirety of the renal elimination pathways, including filtration, secretion and reabsorption, thereby providing a surrogate view of the renal handling of NMN [34]. It is an endogenous substrate of OCT2 and MATE1/2-K [36], and a potential endogenous biomarker for assessing activity of these transporters [21, 37, 38]. Finally, m1A is an endogenous purine nucleoside derived from transfer RNA, that is an endogenous substrate of OCT2 and MATE1/2-K and undergoes significant tubular secretion in the kidneys of humans [8, 21, 39].
4.1.1 In Vitro Biomarker Potency
In vitro potency of m1A, NMN and creatinine was assessed by uptake ratios, Ki and intrinsic clearance-mediated uptake using data retrieved from studies conducted in human OCT2, and MATE1/2-K transfected human embryonic kidney (HEK) cells (Table S2 of the ESM). Uptake ratios for NMN and creatinine were all ≥ 2-fold higher in transporter-transfected HEK cell lines versus wild-type HEK cells, indicating they are endogenous substrates for the OCT2 and MATE1/2-K transporters. Similarly, uptake ratios for m1A confirm that it is a substrate of OCT2 and MATE2-K and can be used as a quantitative biomarker for OCT2 and MATE2‐K‐mediated DDIs. An in vitro evaluation, based on Ki and/or IC50 values, shows potential inhibitory activity of prototypical inhibitor, pyrimethamine for NMN, m1A and creatinine. Representative studies show Ki values are similar for both MATE1 (0.083–0.125 µM) [21, 36] and MATE2-K (0.056–0.22 µM) [8, 36] whereas for OCT2 (0.47–41.2 µM) [21], the values are higher. The limited available data for m1A and creatinine show predicted renal uptake clearance values for OCT2 of 0.3 [8] and 0.1–5.9 [54, 55] µL/min/mg protein, respectively, suggesting a predominant contribution of OCT2. For the MATE1/2-K transporters, no renal uptake clearance data have been reported for m1A and NMN.
4.1.2 In Vivo Biomarker Studies
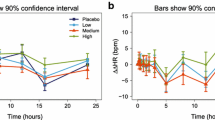
Several healthy volunteer studies have evaluated the potential of different biomarkers to assess renal transporter-mediated DDIs (Fig. 3). In 2015, Müller et al., investigated the effect of co-administering trimethoprim, a known OCT and MATE inhibitor, with metformin (850 mg dose) on NMN levels [37]. This was a pioneering study that measured the correlation between the CLR ratios of metformin and NMN. The relatively strong correlation (coefficient 0.73) suggested the potential utility of NMN as a biomarker.

Correlation (R2 values) between renal clearance (CLR) ratio (adjusted gMean) of biomarkers (N1-methyladenosine [m1A], N1-methylnicotinamide [NMN] and creatinine) versus metformin when administered with or without various potential renal transport inhibitors (trimethoprim [37], pyrimethamine [21], aboricitinib [41], cimetidine [42] and bevurogant [43]) in healthy volunteers. *Metformin 10 mg was given as part of transporter cocktail comprising digoxin 0.25 mg, furosemide 1 mg, metformin 10 mg and rosuvastatin 10 mg [56]. Bid twice daily, MD multiple doses, qd, once daily, qid four times daily, SD single dose
Fast forwarding to 2021, Miyake et al. evaluated the viability of three endogenous MATEs substrates, m1A, NMN and creatinine, as potential biomarkers for MATEs transporters [21]. Metformin (500 mg), the reference probe drug, was administered alone or in combination with escalating single doses of pyrimethamine (10, 25 or 75 mg), a recognised and potent MATE inhibitor (Tables S3 and S4 of the ESM). Of the three biomarkers, m1A showed the strongest correlation (R2 = 0.65) compared with NMN (R2 =0.53) and creatinine (R2 = 0.11). In this study, the impact of metformin on the pharmacokinetics of the biomarkers, m1A, NMN and creatinine was explored by comparing conditions with (control) and without (baseline) metformin administration. A differential response in CLR and exposure (area under the plasma concentration–time curve [AUC]) was noted between NMN and m1A following administration of metformin. While both biomarkers exhibited decreased CLR, there was an increase in the AUC of NMN compared with m1A. This differential response in the exposure of these biomarkers suggests that the effects of metformin extend beyond mere competition at shared renal transporters (OCT2, MATE1/2-K). The increase in the AUC with NMN may be linked to the inhibitory effect of metformin on glycerol-3-phosphate dehydrogenase 2 [57], which could increase the availability of nicotinamide adenine dinucleotide hydrogen, potentially altering NMN’s metabolic processing and renal excretion. This mechanism may also help explain the stronger correlation seen with m1A and metformin compared with that between NMN and metformin.
The utility of NMN and creatinine as endogenous substrates for OCT2 and MATEs transporters was further evaluated in another DDI study by Müller et al., where metformin (10-mg and 500-mg doses) was administered with or without multiple doses of cimetidine, a strong OCT2 and MATE inhibitor [42]. A decrease in CLR of NMN and creatinine was observed over 24 hours. The correlation between the CLR ratio of each biomarker versus 500 mg of metformin over 12 hours was stronger for NMN (R2 = 0.87) versus creatinine (R2 = 0.71) (Fig. 3, Table S5 and Fig. S1 of the ESM). In contrast, almost no correlation was observed for NMN and creatinine versus metformin (10 mg) when it was administered as part of a cocktail (R2 = 0.20 and 0.37, respectively). This trend for creatinine to be a less reliable biomarker was consistent with the findings observed by Miyake et al., [21].
Building upon these initial findings, two subsequent healthy volunteer studies examined the value of NMN as a biomarker in real-world drug development scenarios. In a study assessing the potential for MATE1/2-K inhibition with the oral Janus kinase 1 inhibitor, aboricitinib, there was a good correlation between the CLR ratio of NMN versus metformin 500 mg (R2 = 0.64) [41]. Finally, in a study assessing the potential for OCT2/MATEs inhibition with the retinoic acid-related orphan receptor gamma t antagonist bevurogant (BI 730357), a relatively high correlation was observed between the CLR ratio of NMN versus metformin (10 mg) over 12 hours (R2 = 0.70) [43]. Taken together, a variety of studies performed under inhibiting and non-inhibiting conditions show that NMN displays a relatively good correlation versus metformin (R2 range = 0.53–0.87) [21, 37, 41,42,43].
4.2 Endogenous Biomarkers for OAT1 and OAT3 Transporters
For the renal organic anion secretion axis, represented by OAT1 and OAT3 renal transporters, 4-pyridoxic acid (PDA), homovanillic acid (HVA), glycochenodeoxycholate-3-sulphate (GCDCA-s), taurine, kyneuric acid and 6β-hydroxycortisol have been identified as potential endogenous biomarkers to investigate potential interactions (Table 2). Shen and colleagues identified PDA and HVA as promising endogenous biomarkers of OAT1 and OAT3 [17, 46]. Subsequent experiments using transporter-overexpressing cell models confirmed that PDA and HVA are substrates for human OAT1 and OAT3, as well as OAT2 (HVA), but are not substrates for OCT2 and MATEs transporters [46]. Tsuruya and colleagues further identified taurine and GCDCA-s, as biomarkers of OAT1 and OAT3, respectively [49]. Taurine is an endogenous OAT1 substrate, whereas 6β-hydroxycortisol and GCDCA-s are substrates of OAT3; they may, therefore, potentially serve as endogenous biomarkers for assessing DDIs with these transporters [32, 58]. Preclinical in vitro and in vivo data suggest that kynurenic acid is an emerging endogenous biomarker for OAT1/3-mediated DDIs [52]. Kynurenic acid is a substrate of OAT1/3 and OAT2, but not OCT2, or MATE1/2-K, and shares comparable affinities between OAT1 and OAT3.
4.2.1 In Vitro Biomarker Potency
In vitro assessment of PDA, HVA and kyneuric acid show uptake ratios that are all ≥2-fold higher in OAT1 and OAT3 transporter-transfected HEK cell lines, indicating they are endogenous substrates for these transporters (Table S6 of the ESM). Similarly, uptake ratios for taurine in OAT1, GCDCA-s and 6β-hydroxycortisol in OAT3 showed a greater than 2-fold increase, indicating their potential use as specific quantitative biomarkers for OAT1 and OAT3 transporter‐mediated DDI, respectively. In vitro evaluation (using Ki and/or IC50 values) shows potential inhibitory activity of prototypical inhibitor, probenecid for OAT1 and OAT3. Representative studies show Ki values of 9.5 µM for taurine in OAT1, and 7.4 and 12.1 µM for GCDCA-s and 6β-hydroxycortisol in OAT3 [49]. The available data show predicted renal uptake clearance of kyneuric acid in OAT1 and OAT3 to be 40 and 25 µL/min/mg of protein respectively, suggesting a predominant contribution of OAT1 [52].
4.2.2 In Vivo Biomarker Studies
The utility of plasma and urine PDA and HVA as biomarkers for OAT1/3 transporters was evaluated in a randomised crossover DDI study where single doses of probenecid 1000 mg alone, furosemide 40 mg alone or furosemide 1 hour after probenecid (40 and 1000 mg orally) were administered to healthy volunteers on days 1, 8 and 15, respectively [17]. Administration of probenecid (a strong OAT inhibitor) with furosemide (an accepted probe substrate for OAT function) significantly increased exposure (AUC) of PDA and HVA by 3.2-fold and 2.1-fold, respectively. Increases in PDA and furosemide exposure (AUC) were similar (3.1-fold and 3.3-fold, respectively), while those for HVA were smaller (2.1-fold) [Table S7 of the ESM]. Renal clearance of PDA and HVA were decreased by probenecid to a smaller but similar extent (0.40 and 0.23, respectively) compared with furosemide and probenecid (0.67). The increase in PDA exposure (AUC) following OAT1/3 inhibition by probenecid treatment (3.2-fold) was more pronounced than that of HVA (2.1-fold), indicating that plasma PDA is a promising endogenous biomarker for OAT1/3 function, with plasma exposure responding in a similar manner to furosemide (3.3-fold).
The utility of PDA, HVA, GCDCA-s and taurine as biomarkers for OAT1/3 transporters was evaluated in a DDI study where multiple doses of probenecid 500 mg every 6 hours were administered to healthy female subjects [29] (Table S7 of the ESM). PDA and HVA were the most sensitive biomarkers based on their significant increase in exposure (AUC) following administration of probenecid (3.7-fold and 2.1-fold increases, respectively), with a corresponding decrease in the CLR of GCDCA-s, PDA and HVA (Table S7 of the ESM). GCDCA-s was the most sensitive OAT biomarker based on urine levels. PDA has affinity towards multiple renal transporters, whereas GCDCA-s has higher selectivity towards the OAT3 transporter. However, given that GCDCA-s is also a substrate of organic anion transporting polypeptides (OATP)1B [59], it may not be a good biomarker for assessing a NME that is known to inhibit both OAT3 and OATP1B. In this situation, selecting an alternative endogenous biomarker that does not inhibit OATP1B is advisable. Measurement of plasma PDA in the early phase I studies is recommended for a compound suspected to be an OAT inhibitor. Combined monitoring of PDA and GCDCA-s in urine and plasma is then recommended to tease out the involvement of OAT1/3 in the inhibition interaction [29].
Modelling and simulation results further support the utility of PDA as a selective endogenous biomarker for investigating weak-to-strong OAT1/3-mediated DDIs [47]. PDA is a more robust OAT1/3 biomarker than HVA. Taken together, the in vitro and in vivo data, along with the modelling and simulation results, suggest that plasma PDA is the most promising biomarker for the evaluation of DDI mediated by the OAT1/3 transporter.
The substrates for OAT1 and OAT3 largely overlap but are not identical. OAT3 shows a preference for bulkier and more lipophilic organic anions, such as penicillin G and baricitinib, compared with OAT1, which favours smaller and more hydrophilic anions such as the antiviral agent’s acyclovir, lamivudine and tenofovir [11, 15, 60]. There is an ongoing need to identify endogenous biomarkers that can clinically differentiate between OAT1 and OAT3 inhibition However, the current absence of specific inhibitors for OAT1 or OAT3 [11] precludes validation of endogenous biomarker specificity for OAT1 or OAT3.
5 Factors to Consider While Utilising Endogenous Biomarkers to Assess Renal Transporter-Mediated DDI Risk
Endogenous biomarker levels can be altered by disease, nutrients, drugs and other intrinsic factors. Figure 4 highlights the complex dynamics involved in the renal transporter and endogenous biomarker landscape and the relationships within the clinical and pharmacological domains. Central to this framework are the renal transporters and endogenous biomarkers, which are influenced by multiple factors that range from ethnic differences and genetic variations [61] to external factors such as food and drugs. Intrinsic factors and an array of renal and malignant disorders can also influence renal transporter disposition. Notably, the use of endogenous biomarkers, further illuminated by metabolomics, offers an innovative avenue to assess drug interaction potential. The integration of these elements underscores the importance of understanding these dynamics to ensure the consideration of factors that may affect fluctuations in biomarker baseline levels.

Interplay of factors influencing the measurement of endogenous biomarkers for assessing renal transporter-mediated drug–drug interactions (DDIs) in clinical drug development. The dotted lines (and the respective arrows) indicate the connection between each of the components shown in the different boxes on the figure. Exogenous and endogenous factors and various diseases (several examples shown) on the left of the figure can all influence the assessment of endogenous biomarkers levels within the context of the relevant renal transporter. The exogenous, endogenous and disease impact factors, along with endogenous biomarkers both influence the drug interaction assessment and thereby influence the clinical development process shown on the right of the figure. Examples of diseases that can influence endogenous biomarker levels are shown by the solid green line. ADHD attention-deficit hyperactivity disorder, RA rheumatoid arthritis. aRefers to phase I clinical trials (in healthy volunteers or in oncology patients) conducted prior to a phase I DDI study. bOnce the drug label (package insert) is approved and available, pharmacists and healthcare providers use it to manage medication therapy (pharmacotherapy). The drug label serves as the written rule, with medication therapy being the actual implementation of that rule. Together, these two parts ensure safe and effective treatment
Two important factors to consider in the assessment of endogenous biomarkers for renal transporter-mediated DDIs are ethnicity and genetic variation. Sato et al., used a model-based meta-analysis to determine the effect of ethnic differences between Japanese and Western populations on the oral clearance of 81 drugs [61]. A multi-layered error model was developed to account for the variability in drug properties according to ethnic background. Classifying drugs in phase I studies according to their mechanism of clearance followed by the use of a model-based statistical analysis was shown to be useful for understanding ethnic differences in the pharmacokinetics and clearance of NMEs.
Emerging pharmacogenomic data also suggest that genetic mutations within these transporters can contribute to alterations in the pharmacokinetics and responses of different drugs [62]. For example, genetic variants in genes encoding for the OCT2 and MATE transporters can influence the pharmacokinetics of metformin. The single nucleotide polymorphism 808 G>T (rs316019) is a common missense variant (~ 10–15% frequency [63, 64]) of the SLC22A2 gene that can influence the disposition of metformin among individuals from different ethnic backgrounds (Korean, Caucasian or African American populations) [65,66,67]. Similarly, the single nucleotide polymorphism 922-158 G>A (rs2289669) in the SLC47A1 gene encoding MATE1 has been shown to influence the glucose-lowering effects of metformin [68,69,70]. Current evidence suggests that the pharmacokinetic variability with known genotypes of OCT/MATE transporters is relatively small compared with genetic polymorphisms observed with drug-metabolising cytochrome P450 enzymes CYP2C9 and CYP2D6 [62]. However, genoty** of functional and common variants of OCTs can be considered [62].
Additionally, various disease conditions can influence baseline biomarker levels. While the influence of disease on the biomarkers NMN and creatinine has already been comprehensively reviewed in detail [31], only limited data have been reported for other biomarkers.
For the biomarker m1A, various solid tumour types, including colon, ovarian and cervical, are known to increase m1A baseline levels [71]. Urinary m1A excretion in patients with metastatic colorectal cancer also follows a circadian rhythm, with large between-patient and within-patient variations in urinary excretion observed in patients with other tumour types [71]. Renal impairment can also cause a false-positive increase in m1A levels [72].
For the biomarker PDA, several intrinsic and extrinsic factors, and various diseases can potentially influence changes in baseline levels. PDA is the major catabolite of vitamin B6 (pyridoxine) metabolism, and therefore increased levels are seen with extrinsic vitamin B supplementation [73]. Higher plasma levels of the metabolites pyridoxal and PDA are also seen in users of oral contraceptives [74]. Of the intrinsic factors, individual differences in the synthesis and metabolism of vitamin B6, particularly its conversion to pyridoxal and subsequently to PDA, illustrate the complex dynamics of this biomarker [75]. Several diseases can also influence biomarker levels. For example, patients with colorectal malignancy show decreased PDA levels in plasma [75, 76], while patients with rheumatoid arthritis show no alteration in plasma levels but a decrease in urine levels [77]. Renal impairment is associated with increased PDA levels [78], and decreased levels are seen in individuals with hepatic impairment, particularly those with non-alcohol-related liver disease [79]. Individuals with attention-deficit hyperactivity disorder show low levels of PDA, with a commonly used treatment for this disorder, methylphenidate, modifying PDA levels [80].
Therefore, when assessing renal transporter-mediated DDI using endogenous biomarkers, it is crucial to consider these multi-faceted factors that can influence biomarker baseline levels, as they can have a moderate-to-significant impact on the interpretation and reliability of DDI assessments. At present, our understanding about the impact of various underlying diseases on the biomarkers described above (in the context of drug development) is not fully known. Incorporating these findings into future pharmacokinetic modelling to assess renal transporter-mediated drug interactions will enhance our understanding and interpretation of biomarker level changes within a representative target patient population, notably within the oncology therapeutic area. This strategy is particularly pertinent because of the logistical complexities inherent in conducting clinical drug development DDI studies in oncology patients.
6 Using Pharmacokinetic Modelling to Integrate Endogenous Biomarkers in the Assessment of Renal Transporter-Mediated DDIs
Because of the recognised challenge in translating in vitro results to in vivo studies, physiologically-based pharmacokinetic (PBPK) models have been developed to support investigation of renal transporter-mediated DDIs during drug development [34]. Regulatory agencies encourage the use of PBPK models to guide the drug development process, using in vitro results to estimate the magnitude of the in vivo interaction [81].
In one example, PBPK models of the endogenous OCT2 and MATE1 substrates creatinine and NMN were developed to predict kinetic biomarker changes during administration of various OCT2 and MATE1 perpetrator drugs (trimethoprim, pyrimethamine and cimetidine) [34]. The model for NMN was enhanced to incorporate circadian rhythm factors, thereby accounting for the daily variations in plasma NMN levels. The developed models accurately described and predicted observed plasma concentration–time profiles and urinary excretion of both biomarkers, and the models were coupled to the previously built and evaluated perpetrator models for each perpetrator [34]. In the PBPK modelling of NMN, the authors proposed that inhibitors of MATE impair the synthesis of NMN in vivo, resulting in decreased plasma levels of NMN [34]. This hypothesis is supported by data from healthy volunteers that show a reduction in plasma NMN concentrations following co-administration with a MATE inhibitor [21]. This finding appears to differ from PBPK models developed for inhibition of coproporphyrin I, an endogenous biomarker for OATP1B, where neither circadian effects nor inhibition of coproporphyrin I (CP I) synthesis were observed [82, 83].
Population pharmacokinetic models have also been used to support quantification of PDA and HVA as endogenous biomarkers of OAT1/3 [47]. Simulations based on these models suggest that the circadian rhythm has no prominent effect on PDA and HVA plasma concentrations. The simulations also confirmed the sensitivity and robustness of using plasma PDA data to identify weak, moderate and strong OAT1/3 inhibitors using an adequately powered clinical study [47].
In a similar way, PBPK models have been developed to evaluate endogenous biomarkers. Tan and colleagues developed a PBPK model of PDA in healthy volunteers, and then incorporated a mechanistic kidney model to consider OAT1/3-mediated renal secretion [48]. The model successfully predicted the PDA plasma concentrations, AUC and CLR in healthy volunteers (HVs) at baseline and following single or multiple doses of probenecid. Simulations in patients with severe chronic kidney disease (CKD) successfully predicted the increase in PDA exposure relative to HVs. In another PBPK model developed for creatinine, the way in which renal transporters and passive permeability contribute to the disposition of creatinine was evaluated [84]. This model has been utilised to predict creatinine–drug interactions [45]. Furthermore, they extended the application of this PBPK model to patients with CKD, successfully simulating creatinine–drug interactions in this patient population [85].
The goal of using a modelling approach for NME evaluation is to incorporate in vivo data [82] for each endogenous biomarker, including m1A, NMN, and PDA, in phase I studies (single or multiple rising dose), along with in vitro data, to build respective PBPK models. This approach integrates PBPK modelling using a probe drug such as metformin or furosemide or other relevant concomitant medications. The aim is to predict the DDI of the NME with probes (e.g. metformin) or relevant concomitant medications such as methotrexate. This approach has been successfully applied in phase I studies to evaluate the use of the endogenous biomarker CP I to support prediction of DDIs with statins involving hepatic OATP1B [82, 83, 86]. Similar approaches for renal transporter-mediated DDIs are expected to be developed in the future to streamline the assessment of DDIs in drug development [8]. While most PBPK models have been developed to evaluate the NME as a perpetrator, consideration can be given in the future with regard to the development of models when the NME is a victim and a perpetrator.
7 Integrating Endogenous Biomarkers Early in Drug Development to Streamline DDI Assessments
To streamline DDI assessment in the early phases of drug development, while avoiding unnecessary clinical studies, we propose an endogenous biomarker-informed approach, as shown in Fig. 2B. Like the current decision tree, an early robust in vitro DDI characterisation is required. For NMEs that show renal transporter inhibition with projected ratios of Cmax,u/IC50 above the regulatory cut-off levels, the impact of the investigational drug on biomarker CLR can be assessed in a phase I single-rising or multiple-rising dose study. The latest FDA draft Guidance for Industry (January 2020) recommends the use of serum/plasma creatinine levels as an early index of OCT2, and MATE 1/2-K inhibition [2]. Urine levels of endogenous biomarkers are preferred over plasma levels in view of their value in providing a direct analysis of kidney function. While blood levels can still be useful, CLR is a more relevant pharmacokinetic parameter than Cmax,u. Where possible, baseline values of each biomarker should be taken over a predefined time interval (e.g. 8–24 hours) prior to administration of the NME.
If there is no significant change in AUC or CLR (i.e. within the 80–125% bioequivalence criteria), the DDI risk is minimal, and the NME can proceed to subsequent PoCP (phase II) studies without any restrictions. In the event of a substantial change in the endogenous biomarker levels (i.e. increased AUC for OAT1/3-related biomarkers or decreased CLR for OCT2- and MATE1/2-K-related biomarkers), the observed change stands as a potential parameter for consideration, in conjunction with the drug label specifications of concomitant medications. This information provides potential to inform adjustments in the dosage of affected concomitant medication (e.g. metformin), thereby guiding the strategic design of later PoCP (phase II) trials. For example, it may be prudent to explore the feasibility of administering a reduced dosage of metformin during these clinical studies [87].
In the future, data from the effect of an NME on renal endogenous biomarkers in phase I studies are likely to be combined with PBPK modelling of the affected concomitant medications (e.g. metformin) to forecast the effect of the NME on concomitant medications (see Sect. 6), and thereby guide the dosing of concomitant medications in clinical trial participants. If the NME proves to be effective and safe in phase IIb/III studies, a dedicated DDI study with a probe substrate (e.g. metformin) may be conducted for the final regulatory submission. Another approach is to seek guidance from the regulatory agency to ascertain whether the predicted effects of the NMN on concomitant medications, derived through modelling approaches, may suffice for incorporation into the proposed drug label in the final submission.
8 Conclusions
Monitoring plasma or urine levels of endogenous biomarker levels in early-phase clinical trials is an attractive and cost-effective means of assessing transporter-mediated DDI potential [29, 31, 32, 58]. Measuring biomarkers serves to complement the DDI investigation by expanding insights into potential drug interactions mediated by renal transporters. Consequently, they aid in estimating DDI risks in early-stage in vivo studies and supporting study planning and prioritisation [88].
A lack of change in the systemic exposure of sensitive and selective endogenous probes may avoid costly clinical DDI studies prompted by high false-positive predictions. This is particularly valuable for NMEs for which in vitro assays show borderline transporter-mediated DDIs. Significant increases in plasma concentrations or a decrease in CLR of endogenous biomarkers may lead to dedicated clinical DDI studies with drug probes. However, if drug–endogenous biomarker interactions can be reliably extrapolated to DDIs, such endogenous biomarkers could replace drug probes to define the likelihood of DDIs and minimise adverse events.
Further clinical research is required to fully characterise endogenous biomarkers, particularly m1A, NMN and PDA, as well as new emerging biomarkers. Further clinical evaluations will provide greater understanding about the selectivity, sensitivity and specificity of these endogenous biomarkers. In addition, the circadian effect and the impact of disease, nutrients and other intrinsic factors that may alter the exposure of these biomarkers remain to be defined for many of the biomarkers. This knowledge will further inform the design of clinical trials to take full advantage of these biomarkers.
The development of more robust PBPK modelling will assist in understanding the mechanisms of synthesis, degradation and interaction of these biomarkers with common perpetrators. By utilising data from early clinical studies on the interaction of NMEs with a relevant “cocktail” of endogenous biomarkers, the DDI potential of NMEs towards renal biomarkers can be fully characterised, with priority then given to the most sensitive or rate-limiting step of DDI risk. By doing so, the drug development process can be accelerated, thereby reducing costs, and minimising unnecessary exposure of NMEs to clinical trial participants.
In summary, the goal for the use of endogenous biomarkers in the early phase of clinical development is to reduce the need for future DDI studies. While this approach is described in current regulatory guidance, it has not become firmly established. In practice, compounds nearing approval may still require a stand-alone DDI study (e.g. with metformin) to demonstrate their inherent DDI safety.
References
Yoshida K, Zhao P, Zhang L, Abernethy DR, Rekic D, Reynolds KS, et al. In vitro-in vivo extrapolation of metabolism- and transporter-mediated drug-drug interactions: overview of basic prediction methods. J Pharm Sci. 2017;106(9):2209–13.
US Food and Drug Administration. In vitro drug interaction studies: cytochrome P450 enzyme- and transporter-mediated drug interactions. Guidance for industry. January 2020:1-46. Available from: https://www.fda.gov/media/134582/download. Accessed 25 May 2024.
Saad AAA, Zhang F, Mohammed EAH, Wu X. Clinical aspects of drug-drug interaction and drug nephrotoxicity at renal organic cation transporters 2 (OCT2) and multidrug and toxin exclusion 1, and 2-K (MATE1/MATE2-K). Biol Pharm Bull. 2022;45(4):382–93.
Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, International Transporter Consortium, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–36.
Zhang L, Liu Q, Huang SM, Lionberger R. Transporters in regulatory science: notable contributions from Dr. Giacomini in the past two decades. Drug Metab Dispos. 2022;50(9):1211–7.
Burt HJ, Neuhoff S, Almond L, Gaohua L, Harwood MD, Jamei M, et al. Metformin and cimetidine: physiologically based pharmacokinetic modelling to investigate transporter mediated drug-drug interactions. Eur J Pharm Sci. 2016;88:70–82.
Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2012;22(11):820–7.
Miyake T, Mizuno T, Takehara I, Mochizuki T, Kimura M, Matsuki S, et al. Elucidation of N (1)-methyladenosine as a potential surrogate biomarker for drug interaction studies involving renal organic cation transporters. Drug Metab Dispos. 2019;47(11):1270–80.
Ivanyuk A, Livio F, Biollaz J, Buclin T. Renal drug transporters and drug interactions. Clin Pharmacokinet. 2017;56(8):825–92.
Lepist EI, Ray AS. Renal drug-drug interactions: what we have learned and where we are going. Expert Opin Drug Metab Toxicol. 2012;8(4):433–48.
European Medicines Agency. ICH guideline M12 on drug interaction studies, Step 2b (draft), July 21, 2022. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-m12-drug-interaction-studies-step-2b_en.pdf. Accessed 25 May 2024.
Motohashi H, Inui K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 2013;15(2):581–8.
Gessner A, Konig J, Fromm MF. Clinical aspects of transporter-mediated drug-drug interactions. Clin Pharmacol Ther. 2019;105(6):1386–94.
Maideen NMP, Jumale A, Balasubramaniam R. Drug interactions of metformin involving drug transporter proteins. Adv Pharm Bull. 2017;7(4):501–5.
Feng B, Varma MV. Evaluation and quantitative prediction of renal transporter-mediated drug-drug interactions. J Clin Pharmacol. 2016;56(Suppl. 7):S110–21.
Pfizer. Methotrexate tablets, for oral use. Prescribing information, revised: 08/2020. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/040054s015,s016,s017.pdf. Accessed 8 Aug 2023.
Shen H, Holenarsipur VK, Mariappan TT, Drexler DM, Cantone JL, Rajanna P, et al. Evidence for the validity of pyridoxic acid (PDA) as a plasma-based endogenous probe for OAT1 and OAT3 function in healthy subjects. J Pharmacol Exp Ther. 2019;368(1):136–45.
Iwaki M, Shimada H, Irino Y, Take M, Egashira S. Inhibition of methotrexate uptake via organic anion transporters OAT1 and OAT3 by glucuronides of nonsteroidal anti-inflammatory drugs. Biol Pharm Bull. 2017;40(6):926–31.
Pfizer. Furosemide injection (furosemide), for intravenous or intramuscular use. Prescribing information, revised: 2/2023. Available from: https://labeling.pfizer.com/ShowLabeling.aspx?id=4630. Accessed 10 Sep 2023.
Łapczuk-Romańska J, Droździk M, Oswald S, Droździk M. Kidney drug transporters in pharmacotherapy. Int J Mol Sci. 2023;24(3):2856.
Miyake T, Kimoto E, Luo L, Mathialagan S, Horlbogen LM, Ramanathan R, et al. Identification of appropriate endogenous biomarker for risk assessment of multidrug and toxin extrusion protein-mediated drug-drug interactions in healthy volunteers. Clin Pharmacol Ther. 2021;109(2):507–16.
Chu X, Liao M, Shen H, Yoshida K, Zur AA, Arya V, et al. Clinical probes and endogenous biomarkers as substrates for transporter drug-drug interaction evaluation: perspectives from the International Transporter Consortium. Clin Pharmacol Ther. 2018;104(5):836–64.
Tornio A, Filppula AM, Niemi M, Backman JT. Clinical studies on drug-drug interactions involving metabolism and transport: methodology, pitfalls, and interpretation. Clin Pharmacol Ther. 2019;105(6):1345–61.
European Medicines Agency. Guidance on the investigation of drug interactions. Revision 1, 2015. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf. Accessed 13 Jun 2023.
Arya V, Reynolds KS, Yang X. Using endogenous biomarkers to derisk assessment of transporter-mediated drug-drug interactions: a scientific perspective. J Clin Pharmacol. 2022;62(12):1501–6.
Mathialagan S, Feng B, Rodrigues AD, Varma MVS. Drug-drug interactions involving renal OCT2/MATE transporters: clinical risk assessment may require endogenous biomarker-informed approach. Clin Pharmacol Ther. 2021;110(4):855–9.
US Food and Drug Administration. Clinical drug interaction studies: cytochrome P450 enzyme- and transporter-mediated drug interactions. Guidance for industry. January 2020:1-27. Available from: https://www.fda.gov/media/134581/download. Accessed 25 May 2024.
Dong Z, Yang X, Arya V, Zhang L. Comparing various in vitro prediction criteria to assess the potential of a new molecular entity (NME) to inhibit organic anion transporter 1 and 3 (OAT1 and OAT3) in vivo (abstract PII-075). Clin Pharmacol Ther. 2016;99:S94–5.
Willemin ME, Van Der Made TK, Pijpers I, Dillen L, Kunze A, Jonkers S, et al. Clinical investigation on endogenous biomarkers to predict strong OAT-mediated drug-drug interactions. Clin Pharmacokinet. 2021;60(9):1187–99.
Gessner A, Müller F, Wenisch P, Heinrich MR, König J, Stopfer P, et al. A metabolomic analysis of sensitivity and specificity of 23 previously proposed biomarkers for renal transporter-mediated drug-drug interactions. Clin Pharmacol Ther. 2023;114(5):1058–72.
Müller F, Sharma A, Konig J, Fromm MF. Biomarkers for in vivo assessment of transporter function. Pharmacol Rev. 2018;70(2):246–77.
Rodrigues AD, Taskar KS, Kusuhara H, Sugiyama Y. Endogenous probes for drug transporters: balancing vision with reality. Clin Pharmacol Ther. 2018;103(3):434–48.
Li Y, Talebi Z, Chen X, Sparreboom A, Hu S. Endogenous biomarkers for SLC transporter-mediated drug-drug interaction evaluation. Molecules. 2021;26(18):5500.
Türk D, Müller F, Fromm MF, Selzer D, Dallmann R, Lehr T. Renal transporter-mediated drug-biomarker interactions of the endogenous substrates creatinine and N(1)-methylnicotinamide: a PBPK modeling approach. Clin Pharmacol Ther. 2022;112(3):687–98.
Mathialagan S, Rodrigues AD, Feng B. Evaluation of renal transporter inhibition using creatinine as a substrate in vitro to assess the clinical risk of elevated serum creatinine. J Pharm Sci. 2017;106(9):2535–41.
Ito S, Kusuhara H, Kumagai Y, Moriyama Y, Inoue K, Kondo T, et al. N-methylnicotinamide is an endogenous probe for evaluation of drug-drug interactions involving multidrug and toxin extrusions (MATE1 and MATE2-K). Clin Pharmacol Ther. 2012;92(5):635–41.
Müller F, Pontones CA, Renner B, Mieth M, Hoier E, Auge D, et al. N(1)-methylnicotinamide as an endogenous probe for drug interactions by renal cation transporters: studies on the metformin-trimethoprim interaction. Eur J Clin Pharmacol. 2015;71(1):85–94.
Luo L, Kay J, Zhang J, Holliman CL, Rodrigues AD, Dowty M, et al. LC-MS/MS assay for N(1)-methylnicotinamide in humans, an endogenous probe for renal transporters. Bioanalysis. 2018;10(9):673–89.
Zheng Q, Yu X, Zhang Q, He Y, Guo W. Genetic characteristics and prognostic implications of m1A regulators in pancreatic cancer. Biosci Rep. 2021;41(4):1–13.
Tschuppert Y, Buclin T, Rothuizen LE, Decosterd LA, Galleyrand J, Gaud C, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol. 2007;64(6):785–91.
Vourvahis M, Byon W, Chang C, Le V, Diehl A, Graham D, et al. Evaluation of the effect of abrocitinib on drug transporters by integrated use of probe drugs and endogenous biomarkers. Clin Pharmacol Ther. 2022;112(3):665–75.
Müller F, Hohl K, Keller S, Schmidt-Gerets S, Deutsch B, Schuler-Metz A, et al. N(1)-methylnicotinamide as biomarker for MATE-mediated renal drug-drug interactions: Impact of cimetidine, rifampin, verapamil, and probenecid. Clin Pharmacol Ther. 2023;113(5):1070–9.
Choi H, Huang F, Flack M. The effect of BI 730357 (retinoic acid-related orphan receptor gamma t antagonist bevurogant) on the pharmacokinetics of a transporter probe cocktail, including digoxin, furosemide, metformin and rosuvastatin: an open-label, non-randomized, two-period fixed-sequence trial in healthy subjects. Clin Pharmacol Drug Dev. 2024;13(2):197–207.
Kusuhara H, Ito S, Kumagai Y, Jiang M, Shiroshita T, Moriyama Y, et al. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther. 2011;89(6):837–44.
Scotcher D, Arya V, Yang X, Zhao P, Zhang L, Huang SM, et al. Mechanistic models as framework for understanding biomarker disposition: prediction of creatinine-drug interactions. CPT Pharmacometrics Syst Pharmacol. 2020;9(5):282–93.
Shen H, Nelson DM, Oliveira RV, Zhang Y, McNaney CA, Gu X, et al. Discovery and validation of pyridoxic acid and homovanillic acid as novel endogenous plasma biomarkers of organic anion transporter (OAT) 1 and OAT3 in cynomolgus monkeys. Drug Metab Dispos. 2018;46(2):178–88.
Ahmad A, Ogungbenro K, Kunze A, Jacobs F, Snoeys J, Rostami-Hodjegan A, et al. Population pharmacokinetic modeling and simulation to support qualification of pyridoxic acid as endogenous biomarker of OAT1/3 renal transporters. CPT Pharmacometrics Syst Pharmacol. 2021;10(5):467–77.
Tan SPF, Willemin ME, Snoeys J, Shen H, Rostami-Hodjegan A, Scotcher D, Galetin A. Development of 4-pyridoxic acid PBPK model to support biomarker-informed evaluation of OAT1/3 inhibition and effect of chronic kidney disease. Clin Pharmacol Ther. 2023;114(6):1243–53.
Tsuruya Y, Kato K, Sano Y, Imamura Y, Maeda K, Kumagai Y, et al. Investigation of endogenous compounds applicable to drug-drug interaction studies involving the renal organic anion transporters, OAT1 and OAT3, in humans. Drug Metab Dispos. 2016;44(12):1925–33.
Takehara I, Terashima H, Nakayama T, Yoshikado T, Yoshida M, Furihata K, et al. Investigation of glycochenodeoxycholate sulfate and chenodeoxycholate glucuronide as surrogate endogenous probes for drug interaction studies of OATP1B1 and OATP1B3 in healthy Japanese volunteers. Pharm Res. 2017;34(8):1601–14.
Imamura Y, Tsuruya Y, Damme K, Heer D, Kumagai Y, Maeda K, et al. 6beta-hydroxycortisol is an endogenous probe for evaluation of drug-drug interactions involving a multispecific renal organic anion transporter, OAT3/SLC22A8, in healthy subjects. Drug Metab Dispos. 2014;42(4):685–94.
Liu R, Hao J, Zhao X, Lai Y. Characterization of elimination pathways and the feasibility of endogenous metabolites as biomarkers of organic anion transporter 1/3 (OAT1/3) inhibition in cynomolgus monkeys. Drug Metab Dispos. 2023;51(7):844–50.
Rodrigues AD. Reimagining the framework supporting the static analysis of transporter drug interaction risk; Integrated use of biomarkers to generate pan-transporter inhibition signatures. Clin Pharmacol Ther. 2023;113(5):986–1002.
Ma Y, Zhang M, Yang J, Zhu L, Dai J, Wu X. Characterization of the renal tubular transport of creatinine by activity-based protein profiling and transport kinetics. Eur J Pharm Sci. 2023;180: 106342.
Urakami Y, Kimura N, Okuda M, Inui K. Creatinine transport by basolateral organic cation transporter hOCT2 in the human kidney. Pharm Res. 2004;21(6):976–81.
Stopfer P, Giessmann T, Hohl K, Hutzel S, Schmidt S, Gansser D, et al. Optimization of a drug transporter probe cocktail: potential screening tool for transporter-mediated drug-drug interactions. Br J Clin Pharmacol. 2018;84(9):1941–9.
Dutta S, Shah RB, Singhal S, Dutta SB, Bansal S, Sinha S, Haque M. Metformin: a review of potential mechanism and therapeutic utility beyond diabetes. Drug Des Devel Ther. 2023;17:1907–32.
Chu X, Chan GH, Evers R. Identification of endogenous biomarkers to predict the propensity of drug candidates to cause hepatic or renal transporter-mediated drug-drug interactions. J Pharm Sci. 2017;106(9):2357–67.
Mori D, Kimoto E, Rago B, Kondo Y, King-Ahmad A, Ramanathan R, et al. Dose-dependent inhibition of OATP1B by rifampicin in healthy volunteers: comprehensive evaluation of candidate biomarkers and OATP1B probe drugs. Clin Pharmacol Ther. 2020;107(4):1004–13.
Nigam AK, Li JG, Lall K, Shi D, Bush KT, Bhatnagar V, et al. Unique metabolite preferences of the drug transporters OAT1 and OAT3 analyzed by machine learning. J Biol Chem. 2020;295(7):1829–42.
Sato H, Marutani R, Takaoka R, Mori-Fegan D, Wang X, Maeda K, et al. Model-based meta-analysis of ethnic differences and their variabilities in clearance of oral drugs classified by clearance mechanism. CPT Pharmacometrics Syst Pharmacol. 2023;12(8):1132–42.
Varma MVS. Genetic variation in organic cation transporters and considerations in drug development. Expert Opin Drug Metab Toxicol. 2023;19(3):149–64.
Lai Y, Varma M, Feng B, Stephens JC, Kimoto E, El-Kattan A, et al. Impact of drug transporter pharmacogenomics on pharmacokinetic and pharmacodynamic variability: considerations for drug development. Expert Opin Drug Metab Toxicol. 2012;8(6):723–43.
Zazuli Z, Duin N, Jansen K, Vijverberg SJH, Maitland-van der Zee AH, Masereeuw R. The impact of genetic polymorphisms in organic cation transporters on renal drug disposition. Int J Mol Sci. 2020;21(18):6627.
Song IS, Shin HJ, Shim EJ, Jung IS, Kim WY, Shon JH, Shin JG. Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther. 2008;84(5):559–62.
Chen Y, Li S, Brown C, Cheatham S, Castro RA, Leabman MK, et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genom. 2009;19(7):497–504.
Naja K, El Shamieh S, Fakhoury R. rs622342A>C in SLC22A1 is associated with metformin pharmacokinetics and glycemic response. Drug Metab Pharmacokinet. 2020;35(1):160–4.
Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Interaction between polymorphisms in the OCT1 and MATE1 transporter and metformin response. Pharmacogenet Genomics. 2010;20(1):38–44.
Tkáč I, Klimčáková L, Javorský M, Fabianová M, Schroner Z, Hermanová H, et al. Pharmacogenomic association between a variant in SLC47A1 gene and therapeutic response to metformin in type 2 diabetes. Diabetes Obes Metab. 2013;15(2):189–91.
He R, Zhang D, Lu W, Zheng T, Wan L, Liu F, Jia W. SLC47A1 gene rs2289669 G>A variants enhance the glucose-lowering effect of metformin via delaying its excretion in Chinese type 2 diabetes patients. Diabetes Res Clin Pract. 2015;109(1):57–63.
Dulong S, Huang Q, Innominato PF, Karaboue A, Bouchahda M, Pruvost A, et al. Circadian and chemotherapy-related changes in urinary modified nucleosides excretion in patients with metastatic colorectal cancer. Sci Rep. 2021;11(1):24015.
Honda I, Itoh K, Mizugaki M, Sasaki T. Creatinine at the evaluation of urinary 1-methyladenosine and pseudouridine excretion. Tohoku J Exp Med. 1999;188(2):133–8.
Driskell JA, Giraud DW, Mitmesser SH. Vitamin B-6 intakes and plasma B-6 vitamer concentrations of men and women, 19–50 years of age. Int J Vitam Nutr Res. 2000;70(5):221–5.
Rios-Avila L, Coats B, Chi YY, Midttun Ø, Ueland PM, Stacpoole PW, et al. Metabolite profile analysis reveals association of vitamin B-6 with metabolites related to one-carbon metabolism and tryptophan catabolism but not with biomarkers of inflammation in oral contraceptive users and reveals the effects of oral contraceptives on these processes. J Nutr. 2015;145(1):87–95.
Xu L, Fang Y-J, Che M-M, Abulimiti A, Huang C-Y, Zhang C-X. Association of serum pyridoxal-5’-phosphate, pyridoxal, and PAr with colorectal cancer risk: a large-scale case-control study. Nutrients. 2022;14(12):2389.
Gylling B, Myte R, Schneede J, Hallmans G, Häggström J, Johansson I, et al. Vitamin B-6 and colorectal cancer risk: a prospective population-based study using 3 distinct plasma markers of vitamin B-6 status. Am J Clin Nutr. 2017;105(4):897–904.
Chiang EP, Smith DE, Selhub J, Dallal G, Wang YC, Roubenoff R. Inflammation causes tissue-specific depletion of vitamin B6. Arthritis Res Ther. 2005;7(6):R1254–62.
Coburn SP, Reynolds RD, Mahuren JD, Schaltenbrand WE, Wang Y, Ericson KL, et al. Elevated plasma 4-pyridoxic acid in renal insufficiency. Am J Clin Nutr. 2002;75(1):57–64.
Labadarios D, Rossouw JE, McConnell JB, Davis M, Williams R. Vitamin B6 deficiency in chronic liver disease: evidence for increased degradation of pyridoxal-5′-phosphate. Gut. 1977;18(1):23–7.
Dolina S, Margalit D, Malitsky S, Rabinkov A. Attention-deficit hyperactivity disorder (ADHD) as a pyridoxine-dependent condition: urinary diagnostic biomarkers. Med Hypotheses. 2014;82(1):111–6.
Taskar KS, Pilla Reddy V, Burt H, Posada MM, Varma M, Zheng M, et al. Physiologically-based pharmacokinetic models for evaluating membrane transporter mediated drug-drug interactions: current capabilities, case studies, future opportunities, and recommendations. Clin Pharmacol Ther. 2020;107(5):1082–115.
Yoshikado T, Toshimoto K, Maeda K, Kusuhara H, Kimoto E, Rodrigues AD, et al. PBPK modeling of coproporphyrin I as an endogenous biomarker for drug interactions involving inhibition of hepatic OATP1B1 and OATP1B3. CPT Pharmacometrics Syst Pharmacol. 2018;7(11):739–47.
Kimoto E, Costales C, West MA, Bi YA, Vourvahis M, David Rodrigues A, et al. Biomarker-informed model-based risk assessment of organic anion transporting polypeptide 1B mediated drug-drug interactions. Clin Pharmacol Ther. 2022;111(2):404–15.
Scotcher D, Arya V, Yang X, Zhao P, Zhang L, Huang SM, et al. A novel physiologically based model of creatinine renal disposition to integrate current knowledge of systems parameters and clinical observations. CPT Pharmacomet Syst Pharmacol. 2020;9(6):310–21.
Takita H, Scotcher D, Chinnadurai R, Kalra PA, Galetin A. Physiologically-based pharmacokinetic modelling of creatinine-drug interactions in the chronic kidney disease population. CPT Pharmacometrics Syst Pharmacol. 2020;9(12):695–706.
Mochizuki T, Aoki Y, Yoshikado T, Yoshida K, Lai Y, Hirabayashi H, et al. Physiologically-based pharmacokinetic model-based translation of OATP1B-mediated drug-drug interactions from coproporphyrin I to probe drugs. Clin Transl Sci. 2022;15(6):1519–31.
Zamek-Gliszczynski MJ, Chu X, Cook JA, Custodio JM, Galetin A, Giacomini KM, et al. ITC commentary on metformin clinical drug-drug interaction study design that enables an efficacy- and safety-based dose adjustment decision. Clin Pharmacol Ther. 2018;104(5):781–4.
Shen H. A pharmaceutical industry perspective on transporter and CYP-mediated drug-drug interactions: kidney transporter biomarkers. Bioanalysis. 2018;10(9):625–31.
Acknowledgements
We thank Dr. Ashish Sharma (Boehringer Ingelheim, USA) and Dr. Peter Stopfer (Boehringer Ingelheim, Germany) for their review of the manuscript and useful comments.
Author information
Authors and Affiliations
Contributions
Concept and Design: Fenglei Huang conceived the initial idea for the article. Literature Search: HeeJae Choi and Shilpa Madari performed the literature search and data analysis. Drafting and Revision: Hee Jae Choi, Shilpa Madari, and Fenglei Huang drafted the manuscript. All three authors listed above critically revised the work for important intellectual content.
Corresponding author
Ethics declarations
Funding
This study was funded by Boehringer Ingelheim Pharmaceuticals. No external funds were used in the preparation of this article.
Conflicts of Interest/Competing Interests
Hee Jae Choi, Shilpa Madari and Fenglei Huang are full time employees of Boehringer Ingelheim.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
The original contributions presented are included in the article/Electronic Supplementary Material; further inquiries can be directed to the corresponding author.
Code Availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Choi, H.J., Madari, S. & Huang, F. Utilising Endogenous Biomarkers in Drug Development to Streamline the Assessment of Drug–Drug Interactions Mediated by Renal Transporters: A Pharmaceutical Industry Perspective. Clin Pharmacokinet (2024). https://doi.org/10.1007/s40262-024-01385-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s40262-024-01385-0