
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that currently has no cure. The aged population is growing globally, creating an urgent need for more promising therapies for this debilitating disease. Much effort has been made in recent decades, and the field is highly dynamic, with numerous trials. The main focus of these trials includes disease modification and symptomatic treatment. Some have shown beneficial outcomes, while others have shown no significant benefits. Here, we cover the outcome of recently published AD clinical trials, as well as the mechanism of action of these therapeutical agents, to re-think drug development strategies and directions for future studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Current pharmacological clinical trials for Alzheimer’s disease can be considered under two categories, namely disease-modifying therapies and symptomatic treatments. At present, no treatment has been proved sufficient to cure the disease. |
Abnormal deposition of amyloid-beta (Aβ) and hyperphosphorylation of tau are characteristics of the disease and are considered contributors to disease mechanisms; thus, much effort has been focused on inhibiting or removing these abnormalities on the basis of the amyloid hypothesis and tau hypothesis. |
Symptomatic treatment, which is based on a current understanding of the pathogenesis of the disease, may achieve short-term improvements or long-term stabilization, or slow the deterioration in one or more symptom domains. |
The inhibition of Aβ or its related proteins/enzymes alone appears insufficient to battle the disease; therefore, future studies may be more fruitful by focusing on downstream events, such as tau hyperphosphorylation and neuroinflammation, as well as mitochondrial damage in presynaptic neurons. |
Introduction
Alzheimer’s disease (AD) is pathologically characterized by the accumulation of insoluble amyloid-beta (Aβ) protein in extracellular neuritic plaques and microtubule-associated protein (MAP) tau in intracellular neurofibrillary tangles (NFTs) associated with synaptic dysfunction and neuronal loss [1]. AD is the most common cause of dementia, accounting for approximately 60–80% of cases, thus placing a heavy burden on society [2]. According to the World Alzheimer Report 2015, around 46.8 million people worldwide suffered from dementia in 2015, and this number is predicted to increase to 131.5 million by 2050.
Current pharmacological clinical trials for AD can be considered under two categories, namely disease-modifying therapies (DMTs) and symptomatic treatments [3]. Some agents have more than one mechanism of action, and in these cases, the dominant mechanism is noted and discussed. This review provides a comprehensive summary of clinical trials published in the last 5 years, as well as the mechanism of action/hypothesis behind these potential therapeutic strategies, aiming to highlight the compounds that are still under investigation and discuss the drug development directions that are still of potential therapeutic value.
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Disease-Modifying Therapies
DMTs are a collective group of therapies aimed at delaying or, ideally, reversing pathogenic mechanisms that precede symptoms and are considered to contribute significantly to the disease process. Since abnormal deposition of Aβ and hyperphosphorylation of tau are characteristics of the disease and are considered contributors to disease mechanisms, much effort has been aimed at inhibiting or removing these abnormalities.
Therapies Targeting Amyloid-Beta (Aβ) and Amyloid-Related Proteins
The abnormal deposition of Aβ has long been considered a core event in the initiation and development of AD [4]. Aβ peptides are derived from proteolytic cleavage of the N- or C-terminus of the amyloid precursor protein (APP) by β-secretases (BACE1 and BACE2) and the γ-secretase complex, respectively [4]. Two homologous proteins, presenilin 1/2 (PS1/2, encoded by PSEN1/2), are essential for γ-secretase-mediated cleavage and mutations in genes encoding APP, and PS1/2 are also found in cases of early-onset familial AD [4]. Various drugs designed to decrease Aβ or amyloid-related proteins (APP, BACE and γ-secretase) have been developed to treat AD.
Active vaccination for Aβ removal was the first DMT to be developed. The first human clinical trial started in 2000, testing AN1792 (Janssen, Pfizer) and its surface-active saponin adjuvant QS21. However, unexpected meningoencephalitis occurred in AD patients, and the phase II trial was terminated [5, 6]. Improved active vaccination strategies were then developed using vanutide cridificar (also known as ACC-001, Elan/Wyeth) that linked the Aβ short fragments to a carrier which inactivated diphtheria toxin and minimized the inflammatory responses seen in AN1792 [7]. However, despite an acceptable safety profile, no significant cognitive improvement has been observed, and the clinical trial was therefore terminated [8].
More recently, passive vaccination, using either naturally occurring autoantibodies or humanized murine monoclonal antibodies to Aβ peptide as immunogens, have been developed and assessed in clinical trials. Intravenous immunoglobulin (IVIg) is derived from the plasma of healthy donors and used as an immunogen for passive vaccination. The presence of natural anti-Aβ antibodies, which have lower avidity for single Aβ molecules but higher affinity to oligomers and fibrils, have been reported in IVIg, and very importantly, the anti-Aβ antibodies generated in response to this antigen have been shown to be safe [9]. Compared with placebo (low-dose albumin), 18-month treatment with IVIg (0.2 or 0.4 g/kg every 2 weeks) reduced plasma Aβ42 levels in patients with mild-to-moderate AD, but without cognitive or functional improvement [10]. A more recent study testing the effect on patients with very early versus entrenched mild cognitive impairment (MCI) showed that five infusions of IVIg (0.4 g/kg every 2 weeks) reduced both brain atrophy and cognitive decline, and slowed progression to AD. This beneficial effect was significant at 1 year of treatment, but not at 2 years [11]. In a small-scale trial, IVIg treatment did not alter the levels of Aβ40 or Aβ42 in cerebrospinal fluid (CSF), but rather increased high-molecular-weight (HMW) oligomers, suggesting that HMW-Aβ might be a better post-IVIg treatment biomarker of target engagement in Aβ clinical trials [12]. Apart from IVIg, humanized immunoglobulin monoclonal antibodies against Aβ have also been designed to increase the clearance of Aβ in the brain. Bapineuzumab is aimed at disaggregating deposited amyloid in plaques by binding N-terminal 5 residues of Aβ [13]. This antibody has been tested in two phase III trials on mild-to-moderate AD cases, one in carriers of the apolipoprotein E (ApoE) ε4 allele, and the other in non-carriers. Intravenous (IV) infusion with bapineuzumab significantly reduced accumulation of fibrillar Aβ; however, a decrease in phosphorylated tau in CSF was only significant in ApoE ε4 allele carriers, and neither had obvious effects on stabilizing brain volume or clinical outcomes [14–16]. Interestingly, AD patients treated with bapineuzumab had a larger ventricular volume compared with placebo, possibly due to Aβ clearance [17]. Safety and tolerability (up to 3 years) of this antibody was generally considered good [18]; however, the doses tested on ApoE ε4 allele homozygous carriers were often lower than those tested on non-carriers because of amyloid-related imaging abnormalities (ARIA) in carriers [19]. ARIAs, including cerebral edema (ARIA-E) and cerebral microhaemorrhages (ARIA-H), are abnormal findings detected in the brain of AD patients via magnetic resonance imaging (MRI), which are associated with amyloid-modifying therapies. Because of the lack of efficacy of IV administration in phase III trials, subcutaneous (s/c) injection has been evaluated in a phase II trial. Though no significant change was observed in the cerebral amyloid signal assessed by positron emission tomography (PET) imaging, fewer ARIAs were detected when compared with IV administration [20], indicating that s/c injection is safer than IV infusion. Bapineuzumab failed, but it was found at pathology that 28% of patients did not have AD, which drove the further design of solanezumab. Solanezumab (Eli Lilly and Co.) targets soluble monomeric Aβ peptides at residues 16–26, and subsequently inhibits aggregation of Aβ [21]. In the first two phase III trials (EXPEDITION and EXPEDITION2), solanezumab was tested in mild-to-moderate AD patients, with no significant reductions found in cognitive or functional decline [22]. However, reductions in these measures were detected in a subgroup of participants with mild AD [23]. To confirm this beneficial outcome, EXPEDITION3 was subsequently performed, recruiting only mild AD cases. Disappointingly, EXPEDITION3 failed to show a similar cognitive benefit [24]. One possible reason is that in 25% of mild AD patients recruited in EXPEDITION and EXPEDITION2, there was no evidence of amyloid-related changes detected with CSF biomarkers or amyloid PET scanning, suggesting a diverse underlying disease pathology. The lack of clinical efficacy of bapineuzumab and solanezumab could be explained by the following: (1) not being used early enough in the disease course; (2) not targeting the most critical Aβ species for AD pathogenesis (bapineuzumab targets all forms of Aβ, whereas solanezumab only targets monomeric Aβ); (3) the doses or durations tested in previous trials have not been sufficient.
Attempts to treat AD with novel monoclonal antibodies are still ongoing, and trials have been reported with results on their safety, tolerability, pharmacokinetics and biomarkers. MABT510A (also known as crenezumab, Genentech, Roche) is an IgG4 antibody that binds both monomeric and aggregated Aβ species to realize the clearance of Aβ [25], which was supported by the observations in the ABBY and BLAZE phase II trials, showing a significant reduction in CSF Aβ levels in crenezumab-treated patients compared with the placebo controls [26]. BIIB037 (also known as aducanumab, Biogen, Eisai) has been shown to remove Aβ plaques in AD patients by selectively reacting with soluble oligomers and insoluble fibrils [27]. However, phase III trials of these two antibodies for prodromal AD were terminated in 2019 due to failure to reach the primary and secondary endpoints. Surprisingly, a subsequent analysis of a larger data set showed that the primary endpoint was met in patients on the highest dose (10 mg/kg) of BIIB037, and the US Food and Drug Administration (FDA) recently approved it under the agency’s accelerated approval pathway after the marketing license was granted. Although the approval decision has engendered enthusiasm in the AD drug market, it has also sparked controversy from scientists due to the ambiguous results of clinical trials for the development of this medication (www.alzforum.org/therapeutics/aducanumab). AAB-003 (Janssen, Pfizer) is a modified bapineuzumab with a three-amino-acid mutation in the lower hinge region of bapineuzumab introduced to reduce Fc-receptor-mediated effector function and antibody-induced ARIA, allowing a higher dose (up to 8 mg/kg) to be used [28]. GSK933776 (GSK) is also an Fc-inactivated antibody designed to reduce ARIAs, so a maximal dosage of 6 mg/kg has been tested in a phase I trial [29]. Both AAB-003 and GSK933776 have shown good safety and tolerability, but they have not been further developed for AD treatment due to lack of significant changes in CSF biomarkers in the completed trials. BAN2401 (Biogen) specifically targets large, soluble Aβ protofibrils, and was developed based on increasing evidence that oligomers and protofibrils have higher toxicity than monomers or insoluble fibrils [30]. A phase III clinical trial of this antibody was initiated in 2019 (www.alzforum.org/therapeutics/ban2401). Donanemab (Eli Lilly and Co.) was shown in a phase II trial to slow decline on the Integrated Alzheimer’s Disease Rating Scale (iADRs) by 32% compared to placebo, which is critical for the continued development of this type of therapy ( Mintun et al. 31 Donanemab in early Alzheimer’s disease. The New England Journal of Medicine, in preview. https://doi.org/10.1056/NEJMoa2100708).
Small molecules targeting Aβ have also been designed for the treatment of AD. Aβ peptides are initially derived from APP, with APP processing correlating with disease mechanisms; therefore, APP itself has become a target for therapy. A phase II clinical trial showed that 12-month treatment with phenserine (Axonyx) in patients with mild AD decreased the Aβ42/40 ratio in the CSF and improved cognition [32], possibly by suppressing translation of APP mRNA. However, a larger-scale phase III trial failed to show similar cognitive improvement (www.alzforum.org/new/detail.asp?id=1257). APP is cleaved by β-secretase in an amyloidogenic processing pathway, with BACE1 the major β-secretase in the brain. The BACE1 inhibitor thalidomide has been tested in a phase II trial in patients with mild-to-moderate AD. Because of the poor safety and tolerability of this drug, the effective dosage for blocking BACE1 could not be achieved [33]. Although BACE2 is expressed mainly in peripheral tissues, it is also expressed to a lesser extent in the brain, and has been considered to contribute to the generation of Aβ, compensating for changes in BACE1 activity [34]. Therefore, inhibition of BACE1 alone may not be sufficient to block the formation of Aβ, and dual inhibition may be required. Such attempts have been tested in clinical trials, but none so far has been successful. AZD3293 (also known as lanabecestat, AstraZeneca) showed good tolerability and safety, even at the highest dose of 150 mg daily, with prolonged suppression of Aβ peptides in both plasma and CSF when given at most doses in a phase I trial [35]. Other BACE dual antagonists, including MK-8931 (also known as verubecestat, Merck) [36], JNJ-54861911 (also known as atabecestat, Janssen) [37], CNP520 (also known as umibecestat, Novartis) [38] and E2609 (also known as elenbecestat, Eisai/Biogen) [39], have all shown significant reductions in CSF biomarkers in both healthy subjects and AD patients, with good tolerability at multiple dosages in phase I trials. However, except for CNP520, the phase II/III trials of AZD3293, MK-8931, JNJ-5486911 and E2609 have all had early discontinuation due to lack of efficacy and/or safety concerns based on interim futility analyses. Further attempts combining the use of a BACE inhibitor (elenbecestat, Eisai/Biogen) with a monoclonal antibody (BAN2401) starting in early 2020 will be assessed in two upcoming clinical trials targeting primary and secondary prevention of the pre-symptomatic stage of AD (company press release: Eisai: Alzheimer’s clinical trials consortium selects elenbecestat and BAN2401). In addition, a novel BACE inhibitor, NB-360 (Novartis), has been identified as having potent inhibitory effects in a variety of preclinical animal models and is likely to be developed for clinical trials [40, 41]. Small molecules targeting γ-secretase, either by its modulator BMS-932481 (Bristol Myers Squibb) [42] or by its inhibitor avagacestat (Bristol Myers Squibb) [43], have been assessed in phase I and phase II clinical trials, respectively. However, due to a lack of efficacy and safety, trials for both compounds were terminated. Next, the clearance of soluble Aβ peptides requires ApoE via a protein–protein interaction, and homozygosity for the ApoE gene allele ε4 has been shown to be the strongest genetic risk factor for AD [44]. ApoE is predominantly synthesized in glial cells, and lipidated by ATP-binding cassette A1 (ABCA1) before binding and internalizing soluble Aβ for lysosomal clearance. ABCA1 production is under the control of nuclear retinoid X receptors (RXR); therefore, stimulating RXR by its agonist promotes lipidation of ApoE by increasing the expression of ApoE and ABCA1, and subsequently stimulating the clearance of Aβ1-42 from the brain [45]. Bexarotene is an RXR agonist that has been tested in patients with moderate AD with amyloid-positive PET scans. However, bexarotene only reduced brain amyloid associated with increased serum Aβ1-42 in ApoE ε4 allele non-carriers, and not in carriers. Furthermore, the drug elevated blood triglyceride levels, which is considered a cardiovascular risk factor, and hence clinical trials for this drug were also terminated [46].
There is an equilibrium between Aβ levels in plasma and in the central nervous system (CNS); therefore, reducing plasma Aβ levels can be realized via plasma exchange (PE), in which antibodies that bind to soluble Aβ in plasma may prevent its transport back into the brain and thereby facilitate a net clearance of Aβ (peripheral sink hypothesis) [47]. A phase II trial demonstrated a borderline elevated CSF level and a decreased plasma level of Aβ1-42 post-PE in association with improved cognition and memory [48]. However, behavioural and functional measurements in the PE group were worse than those in controls, probably due to psychiatric symptoms (especially anxiety) triggered by the sustained invasive procedure. This has limited further testing of this treatment in larger-scale clinical trials.
So far, though most of the drugs are safe and well tolerated at the dosages tested, the lack of clinical efficacy is a consistent problem facing clinical trials testing treatments for reducing Aβ. This could be due to the use of inappropriate biomarkers for outcome evaluations, or the interventions being started at an inappropriate stage of the disease. However, the number of failed trials also raises the question of whether Aβ alone is responsible for the pathogenesis of the disease, and thus a better understanding of disease mechanisms is urgently needed.
Therapies Targeting Tau
Neurofibrillary tangles are the other key pathological feature in AD, and it has been hypothesized that the hyperphosphorylation of tau triggers the pathogenic cascade of the disease, eventually causing neuronal cell dysfunction and death [49]. Therefore, tau protein, and especially its hyperphosphorylated form, is considered another potential therapeutic target for AD. Currently, only a few candidates have been identified to specifically suppress tau aggregation, by either immunotherapy or small molecule compounds. One possible reason for this is that tau is widely expressed in the brain, with crucial physiological function in neuronal cells, and therefore drugs can only be developed to specifically target the abnormal protein, leaving normal tau to maintain its functions.
AADvac1 (AXON), which is a tau vaccine directed against soluble extracellular forms, is a synthetic peptide derived from amino acids 294–305 of the tau sequence. A phase I trial showed that 29 out of 30 participants demonstrated positive IgG titres against tau after vaccination [50], suggesting excellent immunogenicity of this vaccine in humans, and thus warranting further clinical trials.
Small molecules have also been tested in clinical trials to assess their inhibition of tau aggregation. For instance, TRx0237 (also known as leuco-methylthioninium-bis(hydromethanesulfonate); LMTM, TauRx Therapeutics) underwent a phase III trial in 891 mild-to-moderate AD patients for a treatment period of 15 months. Unfortunately, the trial analysis was negative in reaching any clinical outcomes [51]. Meanwhile, two phase II trials for VEL015 (sodium selenite, Royal Melbourne Hospital) [52] and methylthioninium (MT, TauRx Therapeutics) [53] have been conducted in patients with mild-to-moderate AD, mainly to collect data on safety and tolerability. Further studies will depend on the results of these trials.
Nerve Growth Factor (NGF)
NGF has been demonstrated to have neuroregenerative and neuroprotective effects on cholinergic neurons in the basal forebrain in animal studies [54]. In addition, declining NGF levels have been observed in brain regions critical to cognition and memory in AD patients [55]. On the basis of these findings, a gene therapy was developed using implanted encapsulated cells sustainably releasing NGF. A 12-month trial injecting these cells into the basal forebrain resulted in reduced brain atrophy in responders [56]. A subsequent observational study over 10 years showed reduced loss of cholinergic neurons in AD patients responding to NGF (as indicated by axonal sprouting, cell hypertrophy and existence of neuronal markers) [57]. Implantation of NGF cells positively regulated cholinergic activity (choline acetyltransferase [ChAT], acetylcholinesterase [AChE] and cortical nicotinic receptor expression) and improved scores on the Mini-Mental State Examination (MMSE) [58]. These trials collectively support NGF gene therapy as a symptomatic treatment or potentially a disease-modifying therapy for AD.
Symptomatic Treatments
For patients with advanced AD, continuous loss of global function and cognition seems to be unavoidable. Symptomatic treatments may achieve short-term improvements or long-term stabilization, or slow deterioration in one or more symptom domains. The way these treatments work for symptom relief is based on a current understanding of the pathogenesis of the disease, as well as signalling pathway cross-talk during the development of the disease.
Symptomatic Treatments Based on the Cholinergic Hypothesis
Based on the findings of changes in metabolic levels of acetylcholine in post-mortem AD brains, the cholinergic hypothesis was proposed as one of the reasons for cognitive impairment and behavioural disturbance in AD patients. The cholinergic hypothesis is based on the continuous loss of cholinergic neurons in the progression of AD, leading to cortical deficiencies in cholinergic neurotransmission [59]. Therefore, the restoration of cholinergic function might be a potential symptomatic treatment to minimize the severity of cognitive loss for AD patients.
AChE is the primary enzyme responsible for the hydrolysis of acetylcholine (ACh) into choline and acetate. So far, three FDA-approved AChEIs, namely rivastigmine, donepezil and galantamine, have been tested via different administration routes (oral and transdermal patches) with different concomitant medications (memantine, solifenacin). Rivastigmine, which inhibits both AChE and butyrylcholinesterase (BuChE), should provide more benefits to patients with concurrent vascular disease, which was confirmed in a randomized clinical trial in AD patients with or without vascular risk factors [60]. However, the conclusions have been controversial, as another trial showed that 24-week treatment with a transdermal rivastigmine patch improved frontal lobe function in AD patients with mild, but not moderate, white matter hyperintensities (WMH), suggesting that less AD pathology and more vascular pathology may attenuate the response to AChEIs [61]. The transdermal patch may be able to replace oral capsules, with higher tolerability and fewer treatment-related adverse events including gastrointestinal (GI) side effects [62]. Currently, there are three approved doses for rivastigmine patches: 4.6 mg/24 h (5 cm2), 9.5 mg/24 h (10 cm2) and 13.3 mg/24 h (15 cm2). A higher dose or a combined treatment may have better clinical outcomes. For instance, in the ACTION trial, when comparing the effects of the lowest and highest doses in treating patients with severe AD for 24 weeks, the highest dose demonstrated better outcomes in the prospective and retrospective analyses [63–65]. A 6-month combined treatment with a rivastigmine patch (9.5 mg/24 h) and a cognition-focused intervention in a cohort of a population with severe AD, on the other hand, demonstrated superior efficacy compared with rivastigmine treatment alone [66]. It is noteworthy that the clinical response to rivastigmine is reduced in the presence of the K variant of butyrylcholinesterase (BCHE-K), found in 17–40% of AD patients [67]. Unlike rivastigmine, donepezil and galantamine are AChE-selective and reversible inhibitors. For donepezil, cognitive improvement correlates with its plasma concentration but not its metabolites, with higher doses most effective. However, its clinical efficacy is usually limited by the use of smaller tolerable doses (5 or 10 mg/day) that inhibit on average only 19.1% of cortical AChE activity [68]. Higher doses that achieve greater inhibition of central AChE and improve cognitive function can be used, as shown in a study of 223 Asian patients with moderate to severe AD comparing effective doses of 23 mg/day versus 10 mg/day [69]. However, sustained release of 23 mg/day was not superior to instant release of 10 mg/day in a Japanese cohort [70]. To amplify the cholinergic function of donepezil in the CNS without inducing peripheral symptoms, a peripheral anticholinergic agent (CPC-201, also known as solifenacin) was co-administered to achieve an escalating donepezil dose of 40 mg/day, which was well tolerated by 88% of patients with moderate AD in a 26-week treatment [71]. The improvement in tolerability of donepezil is critical for patients with severe AD, because withdrawal of donepezil increases the risk of nursing home placement, as demonstrated in the DOMINO-AD trial, regardless of whether the N-methyl-d-aspartate (NMDA) receptor antagonist, memantine, was co-administered [72]. In another study conducted in institutionalized patients with moderate-to-severe AD, discontinuation of cholinesterase inhibitors led to a higher occurrence of hallucinations and delusions [73]. Donepezil has also been tested on prodromal cases at a dose of 10 mg/day for 1 year, showing decreased hippocampal atrophy [74] and cortical thinning [75]. In addition to its inhibition of AChE, galantamine also positively modulates nicotinic receptors. A 2-year randomized study showed a clinical benefit of galantamine in mild-to-moderate dementia patients in terms of reducing cognitive or functional decline, mortality and nursing home placement [76]. Surprisingly, a post hoc analysis of the same study revealed that such benefits were diminished in patients additionally taking memantine [77].
The NMDA receptor (NMDAR) is a central modulator of synaptic plasticity and memory function. Pathologically, excessive NMDAR upregulation results in excitotoxicity by overloading intracellular calcium and triggering apoptosis. In AD, excessive NMDAR activation leads to increased production of pathological Aβ and hyperphosphorylated tau [78]. Memantine, a non-competitive NMDAR antagonist and an open-channel blocker for nicotinic receptors, has been approved for symptomatic treatment in AD, either as add-on therapy to AChEI in moderate-to-severe cases or as single therapy in less severe cases. On the basis of good clinical efficacy and tolerability as well, as increased medication persistence when co-prescribing memantine and donepezil, a fixed-dose combination capsule containing 28 mg memantine extended-release and 10 mg donepezil was successfully tested in two phase I studies [79]. When comparing these two drugs after a 24-week treatment period in a Chinese cohort of mild-to-moderate cases, memantine was more effective in alleviating agitation, whereas donepezil was superior in object-naming ability [80].
Several agents have been tested as concomitant drugs with AChEIs. Idalopirdine, a selective 5-hydroxytryptamine-6 receptor (5HT6R) antagonist, was tested in AChEI-treated patients with mild-to-moderate AD, but showed no cognitive benefits over a 24-week treatment in three phase III studies [81]. Cilostazol, a selective cyclic nucleotide phosphodiesterase 3 (PDE3) inhibitor and an anti-platelet drug, was tested in a case–control study as add-on therapy to AChEIs, showing a reduction in cognitive decline potentially as a consequence of different vascular targets by these two drugs (AChEIs and cilostazol dilate vessels in an endothelial-dependent and independent manner, respectively) [82]. A potent, selective α7 nicotinic acetylcholine receptor (nAChR) agonist, ABT-126, has been tested in mild-to-moderate AD patients who were taking stable doses of AChEIs; however, this drug failed to show any add-on benefits [83]. The α4β2 neuronal nicotinic receptor was selectively targeted in order to avoid the activation of α3 or muscarinic subtypes, but the partial α4β2 agonist, ABT-089, administered as an adjunctive agent to donepezil over a broad range of doses failed to show any add-on benefits [84]. The acetylcholine release agent, ST101, has demonstrated a dose–response cognitive benefit in patients with mild-to-moderate AD taking donepezil [85]. Tideglusib, the glycogen synthase kinase-3 (GSK-3) inhibitor, was evaluated in a phase II trial in AD patients treated with AChEI and/or memantine (NMDAR inhibitor [NMDARI]) for 26 weeks, but demonstrated no clinical benefits [86].
Other Symptomatic Drug Candidates to Improve Cognition
There are some other symptomatic drug candidates tested to improve cognition in AD patients. Bryostatin 1, a protein kinase C epsilon (PKCε) agonist which crosses the blood–brain barrier (BBB), showed good tolerability and improved cognitive function in nine AD patients in a phase II trial [87], although larger-scale clinical trials are required to determine efficacy. BI 409306 (Boehringer Ingelheim), a cGMP-specific selective phosphodiesterase 9A (PDE9A) antagonist, suppresses PDE9A-mediated cGMP hydrolysis, leading to reduced signal transduction. When tested in healthy volunteers, BI 409306 was increased in plasma and CSF and subsequently crossed the BBB, resulting in elevated levels of cGMP in the CSF [88, 89]. UE2343, an 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitor penetrating the brain and specifically blocking intracellular cortisol levels without interfering with the plasma level, has shown good tolerability and safety in a phase I study, and its peak concentration in CSF was nine-fold greater than its half-maximal inhibitory concentration (IC50) [90]. The clinical benefits await to be tested. Sembragiline (also known as RO4602522, RG1577 and EVT302) is a potent, selective and reversible monoamine oxidase B (MAO-B) inhibitor developed to treat AD. A PET study revealed almost complete inhibition of central MAO-B at an oral dose of 1 mg per day in AD patients; however, no cognitive benefits were detected after treatment [91].
Symptomatic Drug Candidates for Neuropsychiatric Symptoms
In a large proportion of AD patients, the neuropsychiatric symptoms (NPS) and behavioural and psychological symptoms of dementia (BPSD) are often treated with antipsychotic drugs (e.g. risperidone, olanzapine, quetiapine) [92]. Of interest, risperidone, but not galantamine, has been found to alter CSF biomarkers (total tau, p-tau, Aβ1-42 and Aβ42/40 ratio) in AD, and some antipsychotics may reduce brain atrophy, suggesting an impact on the disease processes [93, 94]. For sleep disorders, trazodone improved circadian rhythm disturbance [95], whereas low-dose risperidone improved and stabilized sleep patterns in the long term [96]. For severe AD cases with BPSD, the anticonvulsant and mood stabilizer lamotrigine has demonstrated efficacy in a clinical trial [97]. It was also recently reported that nabilone, a synthetic oral tetrahydrocannabinol analogue, may have potential for the treatment of agitation in patients with moderate-to-severe AD [98]. Selective serotonin reuptake inhibitors (SSRIs) have been approved for decades to treat depression. As mature medications, SSRIs have been tested in clinical trials to determine their efficacy in AD-related depression and other psychological symptoms. Firstly, escitalopram was well tolerated by AD dementia patients with depression; however, this drug did not show any clinical benefits in the reported studies [99, 100]. Secondly, citalopram has been shown to control agitation/aggression as well as irritability, anxiety, delusion and hallucination, and the minimal effective duration of treatment was shown to be 9 weeks [101]. However, its side effect profile, especially its impact on sleep, must be considered when determining the dosage used in AD patients [102]. The authors further suggested that those with moderate agitation but less cognitive impairment were more likely to obtain benefits from citalopram, whereas more severe cases, especially dementia patients, could be at higher risk of sleep disturbances [103]. In a secondary analysis, the benefits of citalopram were only partially (11%) due to sedation, and largely due to unknown mechanisms [104].
In addition, AChEIs and NMDARIs have also been tested in the treatment of AD-related neuropsychiatric symptoms. A higher level of acetylcholine obtained by a combined treatment with donepezil and choline alphoscerate displayed less behavioural disturbance in patients with mild-to-moderate AD as compared to treatment with donepezil alone [105]. A further analysis for this study revealed that the combined treatment was particularly effective in improving apathy [106]. Rivastigmine patch monotherapy has been demonstrated as an effective treatment in controlling agitation in mild-to-moderate AD, pointing out another indication of this cognition-improving drug. Such benefits were not observed when combined with memantine [107], although memantine monotreatment showed significant benefits in controlling agitation/aggression symptoms [108]. Memantine at a dose of 20 mg/day was also reported to improve sleep in AD patients [109]. However, memantine used in combination with antipsychotics had no benefits either in the prevention of neuropsychiatric relapse or in cognitive improvement [110].
Dextromethorphan-quinidine has been approved to treat pseudobulbar affect (PBA) in the USA and Europe. Dextromethorphan is the active component, while quinidine increases the bioavailability of dextromethorphan by slowing its oxidation. Dextromethorphan is a weak NMDARI, an σ1 receptor agonist, a serotonin and norepinephrine reuptake inhibitor, and a neuronal α3β4 receptor antagonist. Due to the observations in previous clinical trials showing the anti-agitation effect of this drug, dextromethorphan-quinidine was tested in moderate-to-severe AD patients with agitation, showing strong effects in the 10-week phase II randomized trial [111].
Symptomatic Drug Candidates for Gait Disorder
Gait disorders are the consequence of cortical lesions that affect critical motor areas, as well as cognitive deficits that disturb gait control. Impaired gait is frequently seen in patients with both AD-related and non-related dementia, which significantly elevates the risk of falls and affects future mobility [112]. A phase II trial using donepezil showed gait improvement in 43 patients with mild AD [113], which will now progress to phase III trials. In another open-label trial, the results showed that AChEIs and NMDARIs improved high-level gait control rather than gait performance itself because of the improvements in executive function [114].
Conclusions and Further Prospects
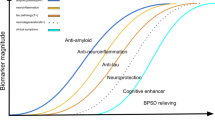
This review has provided a comprehensive summary of human pharmacological clinical trials for the pharmacological treatment of AD published between January 2015 and November 2020, including DMTs and symptomatic treatments (summarized in Fig. 1), as well as the potential mechanism of action (summarized in Fig. 2). Currently, the dominant disease-modifying strategy is the targeting of Aβ or its related proteins at various stages of AD and in people with different risk profiles, and treatments targeting tau protein are also being attempted. To date no DMT has shown strong evidence of significantly delaying or reversing the progression of the disease, although clinical trials are still ongoing with BACE inhibitors (umibecestat, elenbecestat), monoclonal antibodies against Aβ (BAN2401, aducanumab and donanemab), tau vaccines (AADvac1) and small molecule inhibitors against tau (VEL015 and methylthioninium), whereas the BACE 1/2 dual inhibitor NB-360 is still in the preclinical phase. Symptomatic treatments have been more effective in achieving short-term improvements or longer-term stabilization, with treatments targeting the cholinergic system already approved for use. Treatments still under investigation include dextromethorphan/quinidine and nabilone for agitated AD, as well as bryostatin 1 and UE2343 for cognitive impairment. The sole inhibition of Aβ or its related proteins/enzymes appears insufficient for battling the disease; therefore, future studies may be more fruitful if focusing on downstream events, such as tau hyperphosphorylation and neuroinflammation, as well as mitochondrial damage in presynaptic neurons.

Drugs tested at different clinical trial phases. Drugs in black indicates ongoing development, while those in grey indicate discontinued drugs

Mechanisms behind the drugs tested in clinical trials. Aβ peptides are derived from proteolytic cleavage of the N- or C-terminus of the amyloid precursor protein (APP) by β-secretases (BACE1 and BACE2) and γ-secretase complex [presenilins (PS1/2), nicastrin (Nct), anterior pharynx defective 1 (Aph1), presenilin enhancer 2 (PEN-2)], respectively. ApoE is lipidated by ATP-binding cassette A1 (ABCA1) before binding and internalizing soluble Aβ for lysosomal clearance. ABCA1 production is under the control of nuclear retinoid X receptors (RXR); therefore, stimulating RXR by its agonist promotes lipidation of ApoE and subsequent removal of Aβ1-42 from the brain. The oligomerization of monomeric Aβ protein is supposed to be attenuated by amyloid vaccine, autoantibodies or humanized monoclonal antibodies against Aβ, resulting in the reduced formation of insoluble fibrils and plaques in the outer space between neurons (1). The amyloid pathology is associated with tau hyperphosphorylation, which initiates the formation of neurofibrillary tangles, the other key pathological feature in AD, and eventually causes neuronal cell dysfunction and death. Tau aggregation may be suppressed by either immunotherapy (tau vaccine) or small molecule compounds (2). It has been reported that Aβ oligomers are capable of triggering neuroinflammation in a TLR-dependent manner, and anti-inflammation treatment may provide therapeutic benefits in AD (3). The self-aggregation of Aβ on neuronal membranes leads to lipid peroxidation, subsequently depolarizing the synaptic membrane and causing excessive calcium influx and mitochondrial damage, all of which affect the physiological functions of neuronal cells (4). The aggregation of Aβ also stimulates the production of reactive oxygen species (ROS) to generate toxic oxidized proteins and peroxided lipids, leading to endoplasmic reticulum (ER) stress (5). Meanwhile, the cholinergic hypothesis has pointed out a continuous loss of cholinergic neurons in AD progression, leading to cortical deficiencies in cholinergic neurotransmission. Acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) are the primary enzymes responsible for the hydrolysis of neurotransmitter acetylcholine (ACh) into choline and acetate. Thus, the main strategies for maintaining cholinergic neurotransmission are either through the inhibition of AChE and/or BuChE or through the promotion of ACh transport into the post-synaptic neurons. Finally, in AD progression, the neurotransmitter glutamate (glu) released from glutamatergic neurons induces the influx of calcium (Ca2+) into responsive neurons through NMDAR. Meanwhile, Ca2+ also enters through voltage-gated Ca2+ channels. The intracellular calcium overload results in neuronal apoptosis, which may be prevented by the use of NMDAR antagonists or calcium channel blockers. Antagonist/antibodies (in red arrow boxes) and agonists (in green arrow boxes) are listed beside targets, with ✓ (checkmark) and X (cross mark) indicating ongoing or terminated drug development, repectively
References
Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, et al. Tau, tangles, and Alzheimer’s disease. Biochim Biophys Acta. 2005;1739:216–23.
As A. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 2018;14:367–425.
Cummings J, Lee G, Ritter A, et al. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (N Y). 2020;6: e12050.
Murphy MP, LeVine H 3rd. Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis. 2010;19:311–23.
Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54.
Nicoll JA, Wilkinson D, Holmes C, et al. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–52.
Pasquier F, Sadowsky C, Holstein A, et al. Two phase 2 multiple ascending-dose studies of vanutide cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer’s disease. J Alzheimers Dis. 2016;51:1131–43.
Hull M, Sadowsky C, Arai H, et al. Long-term extensions of randomized vaccination trials of ACC-001 and QS-21 in mild to moderate Alzheimer’s disease. Curr Alzheimer Res. 2017;14:696–708.
Dodel R, Hampel H, Depboylu C, et al. Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer’s disease. Ann Neurol. 2002;52:253–6.
Relkin NR, Thomas RG, Rissman RA, et al. A phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology. 2017;88:1768–75.
Kile S, Au W, Parise C, et al. IVIG treatment of mild cognitive impairment due to Alzheimer’s disease: a randomised double-blinded exploratory study of the effect on brain atrophy, cognition and conversion to dementia. J Neurol Neurosurg Psychiatry. 2017;88:106–12.
Kasai T, Kondo M, Ishii R, et al. Abeta levels in the jugular vein and high molecular weight Abeta oligomer levels in CSF can be used as biomarkers to indicate the anti-amyloid effect of IVIg for Alzheimer’s disease. PLoS ONE. 2017;12: e0174630.
Miles LA, Crespi GA, Doughty L, et al. Bapineuzumab captures the N-terminus of the Alzheimer’s disease amyloid-beta peptide in a helical conformation. Sci Rep. 2013;3:1302.
Vandenberghe R, Rinne JO, Boada M, et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res Ther. 2016;8:18.
Liu E, Schmidt ME, Margolin R, et al. Amyloid-beta 11C-PiB-PET imaging results from 2 randomized bapineuzumab phase 3 AD trials. Neurology. 2015;85:692–700.
Russu A, Samtani MN, Xu S, et al. Biomarker exposure-response analysis in mild-to-moderate Alzheimer’s disease trials of bapineuzumab. J Alzheimers Dis. 2016;53:535–46.
Novak G, Fox N, Clegg S, et al. Changes in brain volume with bapineuzumab in mild to moderate Alzheimer’s disease. J Alzheimers Dis. 2016;49:1123–34.
Ivanoiu A, Pariente J, Booth K, et al. Long-term safety and tolerability of bapineuzumab in patients with Alzheimer’s disease in two phase 3 extension studies. Alzheimers Res Ther. 2016;8:24.
Ketter N, Brashear HR, Bogert J, et al. Central review of amyloid-related imaging abnormalities in two phase III clinical trials of bapineuzumab in mild-to-moderate Alzheimer’s disease patients. J Alzheimers Dis. 2017;57:557–73.
Brody M, Liu E, Di J, et al. A phase II, randomized, double-blind, placebo-controlled study of safety, pharmacokinetics, and biomarker results of subcutaneous bapineuzumab in patients with mild to moderate Alzheimer’s disease. J Alzheimers Dis. 2016;54:1509–19.
Zhao J, Nussinov R, Ma B. Mechanisms of recognition of amyloid-beta (Abeta) monomer, oligomer, and fibril by homologous antibodies. J Biol Chem. 2017;292:18325–43.
Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:311–21.
Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 2016;12:110–20.
Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med. 2018;378:321–30.
Salloway S, Honigberg LA, Cho W, et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimers Res Ther. 2018;10:96.
Yang T, Dang Y, Ostaszewski B, et al. Target engagement in an alzheimer trial: crenezumab lowers amyloid beta oligomers in cerebrospinal fluid. Ann Neurol. 2019;86:215–24.
Sevigny J, Chiao P, Bussiere T, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. 2016;537:50–6.
Delnomdedieu M, Duvvuri S, Li DJ, et al. First-in-human safety and long-term exposure data for AAB-003 (PF-05236812) and biomarkers after intravenous infusions of escalating doses in patients with mild to moderate Alzheimer’s disease. Alzheimers Res Ther. 2016;8:12.
Andreasen N, Simeoni M, Ostlund H, et al. First administration of the Fc-attenuated anti-beta amyloid antibody GSK933776 to patients with mild Alzheimer’s disease: a randomized, placebo-controlled study. PLoS ONE. 2015;10: e0098153.
Logovinsky V, Satlin A, Lai R, et al. Safety and tolerability of BAN2401—a clinical study in Alzheimer’s disease with a protofibril selective Abeta antibody. Alzheimers Res Ther. 2016;8:14.
Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384:1691–704.
Nordberg A, Kadir A, Andreasen N, et al. Correlations between Alzheimer’s disease cerebrospinal fluid biomarkers and cerebral glucose metabolism after 12 months of phenserine treatment. J Alzheimers Dis. 2015;47:691–704.
Decourt B, Drumm-Gurnee D, Wilson J, et al. Poor safety and tolerability hamper reaching a potentially therapeutic dose in the use of thalidomide for Alzheimer’s disease: results from a double-blind, placebo-controlled trial. Curr Alzheimer Res. 2017;14:403–11.
Dominguez D, Tournoy J, Hartmann D, et al. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005;280:30797–806.
Cebers G, Alexander RC, Haeberlein SB, et al. AZD3293: pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer’s disease. J Alzheimers Dis. 2017;55:1039–53.
Kennedy ME, Stamford AW, Chen X, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med. 2016;8: 363ra150.
Timmers M, Streffer JR, Russu A, et al. Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: randomized, double-blind, placebo-controlled study. Alzheimers Res Ther. 2018;10:85.
Neumann U, Ufer M, Jacobson LH, et al. The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol Med. 2018;10: e9316.
Yan R, Vassar R. Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014;13:319–29.
Neumann U, Machauer R, Shimshek DR. The beta-secretase (BACE) inhibitor NB-360 in preclinical models: from amyloid-beta reduction to downstream disease-relevant effects. Br J Pharmacol. 2019;176:3435–46.
Neumann U, Rueeger H, Machauer R, et al. A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-beta and neuroinflammation in APP transgenic mice. Mol Neurodegener. 2015;10:44.
Soares HD, Gasior M, Toyn JH, et al. The gamma-secretase modulator, BMS-932481, modulates abeta peptides in the plasma and cerebrospinal fluid of healthy volunteers. J Pharmacol Exp Ther. 2016;358:138–50.
Coric V, Salloway S, van Dyck CH, et al. Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial. JAMA Neurol. 2015;72:1324–33.
Liu CC, Liu CC, Kanekiyo T, et al. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Zhao J, Fu Y, Liu CC, et al. Retinoic acid isomers facilitate apolipoprotein E production and lipidation in astrocytes through the retinoid X receptor/retinoic acid receptor pathway. J Biol Chem. 2014;289:11282–92.
Cummings JL, Zhong K, Kinney JW, et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene **n moderate Alzheimer’s disease. Alzheimers Res Ther. 2016;8:4.
DeMattos RB, Bales KR, Cummins DJ, et al. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2001;98:8850–5.
Boada M, Anaya F, Ortiz P, et al. Efficacy and safety of plasma exchange with 5% albumin to modify cerebrospinal fluid and plasma amyloid-beta concentrations and cognition outcomes in Alzheimer’s disease patients: a multicenter, randomized controlled clinical trial. J Alzheimers Dis. 2017;56:129–43.
Chen GF, Xu TH, Yan Y, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017;38:1205–35.
Novak P, Schmidt R, Kontsekova E, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017;16:123–34.
Gauthier S, Feldman HH, Schneider LS, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 2016;388:2873–84.
Malpas CB, Vivash L, Genc S, et al. A phase IIa randomized control trial of VEL015 (Sodium Selenate) in mild-moderate Alzheimer’s disease. J Alzheimers Dis. 2016;54:223–32.
Wischik CM, Staff RT, Wischik DJ, et al. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis. 2015;44:705–20.
Nagahara AH, Merrill DA, Coppola G, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15:331–7.
Scott SA, Mufson EJ, Weingartner JA, et al. Nerve growth factor in Alzheimer’s disease: increased levels throughout the brain coupled with declines in nucleus basalis. J Neurosci. 1995;15:6213–21.
Ferreira D, Westman E, Eyjolfsdottir H, et al. Brain changes in Alzheimer’s disease patients with implanted encapsulated cells releasing nerve growth factor. J Alzheimers Dis. 2015;43:1059–72.
Tuszynski MH, Yang JH, Barba D, et al. Nerve growth factor gene therapy: activation of neuronal responses in Alzheimer disease. JAMA Neurol. 2015;72:1139–47.
Eyjolfsdottir H, Eriksdotter M, Linderoth B, et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: application of a second-generation encapsulated cell biodelivery device. Alzheimers Res Ther. 2016;8:30.
Francis PT, Palmer AM, Snape M, et al. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–47.
Kumar V, Anand R, Messina J, et al. An efficacy and safety analysis of Exelon in Alzheimer’s disease patients with concurrent vascular risk factors. Eur J Neurol. 2000;7:159–69.
Park KW, Kim EJ, Han HJ, et al. Efficacy and tolerability of rivastigmine patch therapy in patients with mild-to-moderate Alzheimer’s dementia associated with minimal and moderate ischemic white matter hyperintensities: a multicenter prospective open-label clinical trial. PLoS ONE. 2017;12: e0182123.
Wentrup A, Oertel WH, Dodel R. Once-daily transdermal rivastigmine in the treatment of Alzheimer’s disease. Drug Des Dev Ther. 2009;2:245–54.
Farlow MR, Sadowsky CH, Velting DM, et al. Evaluating response to high-dose 13.3 mg/24 h rivastigmine patch in patients with severe Alzheimer’s disease. CNS Neurosci Ther. 2015;21:513–9.
Isaacson RS, Ferris S, Velting DM, et al. Cognitive efficacy (SIB) of 13.3 versus 4.6 mg/24 h rivastigmine patch in severe Alzheimer’s disease. Am J Alzheimers Dis Other Dement. 2016;31:270–7.
Grossberg GT, Farlow MR, Meng X, et al. Evaluating high-dose rivastigmine patch in severe Alzheimer’s disease: analyses with concomitant memantine usage as a factor. Curr Alzheimer Res. 2015;12:53–60.
D’Onofrio G, Sancarlo D, Addante F, et al. A pilot randomized controlled trial evaluating an integrated treatment of rivastigmine transdermal patch and cognitive stimulation in patients with Alzheimer’s disease. Int J Geriatr Psychiatry. 2015;30:965–75.
Han HJ, Kwon JC, Kim JE, et al. Effect of rivastigmine or memantine add-on therapy is affected by butyrylcholinesterase genotype in patients with probable Alzheimer’s disease. Eur Neurol. 2015;73:23–8.
Bohnen NI, Kaufer DI, Hendrickson R, et al. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2005;76:315–9.
Han SH, Lee JH, Kim SY, et al. Donepezil 23 mg in Asian patients with moderate-to-severe Alzheimer’s disease. Acta Neurol Scand. 2017;135:252–6.
Homma A, Atarashi H, Kubota N, et al. Efficacy and safety of sustained release donepezil high dose versus immediate release donepezil standard dose in Japanese patients with severe Alzheimer’s disease: a randomized double-blind trial. J Alzheimers Dis. 2016;52:345–57.
Chase TN, Farlow MR, Clarence-Smith K. Donepezil plus solifenacin (CPC-201) treatment for Alzheimer’s disease. Neurotherapeutics. 2017;14:405–16.
Howard R, McShane R, Lindesay J, et al. Nursing home placement in the donepezil and memantine in moderate to severe Alzheimer’s disease (DOMINO-AD) trial: secondary and post-hoc analyses. Lancet Neurol. 2015;14:1171–81.
Herrmann N, O’Regan J, Ruthirakuhan M, et al. A randomized placebo-controlled discontinuation study of cholinesterase inhibitors in institutionalized patients with moderate to severe Alzheimer disease. J Am Med Dir Assoc. 2016;17:142–7.
Dubois B, Chupin M, Hampel H, et al. Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer’s disease. Alzheimers Dement. 2015;11:1041–9.
Cavedo E, Dubois B, Colliot O, et al. Reduced regional cortical thickness rate of change in donepezil-treated subjects with suspected prodromal Alzheimer’s disease. J Clin Psychiatry. 2016;77:e1631–8.
Hager K, Baseman AS, Nye JS, et al. Effects of galantamine in a 2-year, randomized, placebo-controlled study in Alzheimer’s disease. Neuropsychiatr Dis Treat. 2014;10:391–401.
Hager K, Baseman AS, Nye JS, et al. Effect of concomitant use of memantine on mortality and efficacy outcomes of galantamine-treated patients with Alzheimer’s disease: post-hoc analysis of a randomized placebo-controlled study. Alzheimers Res Ther. 2016;8:47.
Talantova M, Sanz-Blasco S, Zhang X, et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci USA. 2013;110:E2518–27.
Boinpally R, Chen L, Zukin SR, et al. A novel once-daily fixed-dose combination of memantine extended release and donepezil for the treatment of moderate to severe Alzheimer’s disease: two phase I studies in healthy volunteers. Clin Drug Investig. 2015;35:427–35.
Zhang N, Wei C, Du H, et al. The effect of memantine on cognitive function and behavioral and psychological symptoms in mild-to-moderate Alzheimer’s disease patients. Dement Geriatr Cogn Disord. 2015;40:85–93.
Atri A, Frolich L, Ballard C, et al. Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: three randomized clinical trials. JAMA. 2018;319:130–42.
Tai SY, Chen CH, Chien CY, et al. Cilostazol as an add-on therapy for patients with Alzheimer’s disease in Taiwan: a case control study. BMC Neurol. 2017;17:40.
Florian H, Meier A, Gauthier S, et al. Efficacy and safety of ABT-126 in subjects with mild-to-moderate Alzheimer’s disease on stable doses of acetylcholinesterase inhibitors: a randomized, double-blind placebo-controlled study. J Alzheimers Dis. 2016;51:1237–47.
Lenz RA, Pritchett YL, Berry SM, et al. Adaptive, dose-finding phase 2 trial evaluating the safety and efficacy of ABT-089 in mild to moderate Alzheimer disease. Alzheimer Dis Assoc Disord. 2015;29:192–9.
Gauthier S, Rountree S, Finn B, et al. Effects of the acetylcholine release agent ST101 with donepezil in Alzheimer’s disease: a randomized phase 2 study. J Alzheimers Dis. 2015;48:473–81.
Lovestone S, Boada M, Dubois B, et al. A phase II trial of tideglusib in Alzheimer’s disease. J Alzheimers Dis. 2015;45:75–88.
Nelson TJ, Sun MK, Lim C, et al. Bryostatin effects on cognitive function and PKCvarepsilon in Alzheimer’s disease phase IIa and expanded access trials. J Alzheimers Dis. 2017;58:521–35.
Boland K, Moschetti V, Dansirikul C, et al. A phase I, randomized, proof-of-clinical-mechanism study assessing the pharmacokinetics and pharmacodynamics of the oral PDE9A inhibitor BI 409306 in healthy male volunteers. Hum Psychopharmacol Clin Exp. 2017;32:e2569. https://doi.org/10.1002/hup.2569.
Moschetti V, Boland K, Feifel U, et al. First-in-human study assessing safety, tolerability and pharmacokinetics of BI 409306, a selective phosphodiesterase 9A inhibitor, in healthy males. Br J Clin Pharmacol. 2016;82:1315–24.
Webster SP, McBride A, Binnie M, et al. Selection and early clinical evaluation of the brain-penetrant 11beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) inhibitor UE2343 (Xanamem). Br J Pharmacol. 2017;174:396–408.
Sturm S, Forsberg A, Nave S, et al. Positron emission tomography measurement of brain MAO-B inhibition in patients with Alzheimer’s disease and elderly controls after oral administration of sembragiline. Eur J Nucl Med Mol Imaging. 2017;44:382–91.
Reeves S, McLachlan E, Bertrand J, et al. Therapeutic window of dopamine D2/3 receptor occupancy to treat psychosis in Alzheimer’s disease. Brain. 2017;140:1117–27.
Bloniecki V, Aarsland D, Blennow K, et al. Effects of risperidone and galantamine treatment on Alzheimer’s disease biomarker levels in cerebrospinal fluid. J Alzheimers Dis. 2017;57:387–93.
Lee YM, Park JM, Lee BD, et al. Gray matter volumes and treatment response of psychotic symptoms to risperidone in antipsychotic-naive Alzheimer’s disease patients. J Clin Psychiatry. 2016;77:e8-13.
Grippe TC, Goncalves BS, Louzada LL, et al. Circadian rhythm in Alzheimer disease after trazodone use. Chronobiol Int. 2015;32:1311–4.
Yin Y, Liu Y, Zhuang J, et al. Low-dose atypical antipsychotic risperidone improves the 5-year outcome in Alzheimer’s disease patients with sleep disturbances. Pharmacology. 2015;96:155–62.
Suzuki H, Gen K. Clinical efficacy of lamotrigine and changes in the dosages of concomitantly used psychotropic drugs in Alzheimer’s disease with behavioural and psychological symptoms of dementia: a preliminary open-label trial. Psychogeriatrics. 2015;15:32–7.
Herrmann N, Ruthirakuhan M, Gallagher D, et al. Randomized placebo-controlled trial of nabilone for agitation in Alzheimer’s disease. Am J Geriatr Psychiatry. 2019;27:1161–73.
An H, Choi B, Park KW, et al. The effect of escitalopram on mood and cognition in depressive Alzheimer’s disease subjects. J Alzheimers Dis. 2017;55:727–35.
Choe YM, Kim KW, Jhoo JH, et al. Multicenter, randomized, placebo-controlled, double-blind clinical trial of escitalopram on the progression-delaying effects in Alzheimer’s disease. Int J Geriatr Psychiatry. 2016;31:731–9.
Weintraub D, Drye LT, Porsteinsson AP, et al. Time to response to citalopram treatment for agitation in Alzheimer disease. Am J Geriatr Psychiatry. 2015;23:1127–33.
Leonpacher AK, Peters ME, Drye LT, et al. Effects of citalopram on neuropsychiatric symptoms in Alzheimer’s dementia: evidence from the CitAD study. Am J Psychiatry. 2016;173:473–80.
Schneider LS, Frangakis C, Drye LT, et al. Heterogeneity of treatment response to citalopram for patients with Alzheimer’s disease with aggression or agitation: the CitAD randomized clinical trial. Am J Psychiatry. 2016;173:465–72.
Newell J, Yesavage JA, Taylor JL, et al. Sedation mediates part of Citalopram’s effect on agitation in Alzheimer’s disease. J Psychiatr Res. 2016;74:17–21.
Carotenuto A, Rea R, Traini E, et al. The effect of the association between donepezil and choline alphoscerate on behavioral disturbances in Alzheimer’s disease: interim results of the ASCOMALVA trial. J Alzheimers Dis. 2017;56:805–15.
Rea R, Carotenuto A, Traini E, et al. Apathy treatment in alzheimer’s disease: interim results of the ASCOMALVA trial. J Alzheimers Dis. 2015;48:377–83.
Yoon SJ, Choi SH, Na HR, et al. Effects on agitation with rivastigmine patch monotherapy and combination therapy with memantine in mild to moderate Alzheimer’s disease: a multicenter 24-week prospective randomized open-label study (the Korean EXelon Patch and combination with mEmantine Comparative Trial study). Geriatr Gerontol Int. 2017;17:494–9.
Gauthier S, Loft H, Cummings J. Improvement in behavioural symptoms in patients with moderate to severe Alzheimer’s disease by memantine: a pooled data analysis. Int J Geriatr Psychiatry. 2008;23:537–45.
Ishikawa I, Shinno H, Ando N, et al. The effect of memantine on sleep architecture and psychiatric symptoms in patients with Alzheimer’s disease. Acta Neuropsychiatr. 2016;28:157–64.
Ballard C, Thomas A, Gerry S, et al. A double-blind randomized placebo-controlled withdrawal trial comparing memantine and antipsychotics for the long-term treatment of function and neuropsychiatric symptoms in people with Alzheimer’s disease (MAIN-AD). J Am Med Dir Assoc. 2015;16:316–22.
Cummings JL, Lyketsos CG, Peskind ER, et al. Effect of dextromethorphan-quinidine on agitation in patients with Alzheimer disease dementia: a randomized clinical trial. JAMA. 2015;314:1242–54.
Amboni M, Barone P, Hausdorff JM. Cognitive contributions to gait and falls: evidence and implications. Mov Disord. 2013;28:1520–33.
Montero-Odasso M, Muir-Hunter SW, Oteng-Amoako A, et al. Donepezil improves gait performance in older adults with mild Alzheimer’s disease: a phase II clinical trial. J Alzheimers Dis. 2015;43:193–9.
Beauchet O, Barden J, Liu-Ambrose T, et al. Anti-dementia drugs, gait performance and mental imagery of gait: a non-randomized open-label trial. Drugs Aging. 2016;33:665–73.
Acknowledgements
Funding
No funding or sponsorship was received for publication of this review. The Rapid Service Fee was funded by the authors.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors’ Contributions
Manuscript concept and design: PL, JS. Manuscript drafting: PL, JS, QC, YY, JG. Critical revision of the manuscript: DC, JG.
Disclosures
** Lin, Junyu Sun, Qi Cheng, Yue Yang, Dennis Cordato, Jianqun Gao have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lin, P., Sun, J., Cheng, Q. et al. The Development of Pharmacological Therapies for Alzheimer’s Disease. Neurol Ther 10, 609–626 (2021). https://doi.org/10.1007/s40120-021-00282-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-021-00282-z