
Abstract
The geographic relationships and biogeography of Apis cerana have been studied extensively, but Korean populations have not been investigated thoroughly. We sequenced the non-coding region between the tRNALeu and COII mitochondrial (mt) genes (herein named NC2) of Korean samples, along with the samples from seven Asian localities (China, Vietnam, and Thailand). Four undiscovered haplotypes were found in Korea and China, respectively. A phylogenetic analysis confirmed that Korean A. cerana belonged to the Mainland Asian group. Dominance of Japan1 haplotype in Mainland Asia including Korea suggests extensive gene flow and a common genetic origin. A newly developed non-coding region between the tRNAMet and tRNAGln mt genes (named NC1) provided nine haplotypes with twice the number of variable positions compared to those in NC2. A NC1-based phylogenetic analysis revealed the presence of two phylogenetic groups in Apis cerana from Korea suggesting two different sources.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The Asian cavity-nesting honey bee, Apis cerana, has been the subject of genetic variation and biogeographic studies in most of its temperate and tropical Asia distributional range (Deowanish et al. 1996; Smith and Hagen 1996; Sihanuntavong et al. 1999; De la Rúa et al. 2000; Smith et al. 2000, 2004; Tan et al. 2007; Takahashi et al. 2007). However, South Korean populations require additional investigation to reinforce previous findings obtained from a limited sampling (Smith and Hagen 1996; Smith et al. 2000; Takahashi et al. 2007). Those studies employed an internal spacer region located between mitochondrial (mt) tRNALeu and COII (Crozier and Crozier 1993; Cornuet et al. 1991) (herein called the second non-coding region, NC2, accordingly its position in the A. cerana mt genome). NC2 (which is 86–93-bp long in A. cerana) was firstly found in the A. mellifera mt genome (Crozier and Crozier 1993; Cornuet et al. 1991) and was the longest intergenic spacer observed in Apis species at the time of marker development. From the NC2, five haplotypes were found in South Korea (Korea4, Korea7, Korea9, Japan1, and Nepal1). The Japan1 haplotype had the highest frequency and belongs to the Mainland Asian group when incorporated into the worldwide dataset (Smith and Hagen 1996; Smith et al. 2000, 2004; Warrit et al. 2006; Takahashi et al. 2007).
Other studies employed either a restriction enzyme analysis or direct sequencing using the mitochondrial genes COI, 12S rRNA, 16S rRNA, or a nuclear ribosomal internal transcribed spacer 2 for Korean samples (Chang et al. 2000; Kim et al. 2010). Although these markers possess appropriate merits (e.g., Hajibabaei et al. 2007; Cha et al. 2007; Oh et al. 2009), their variability in A. cerana was insufficient to uncover hidden diversity. In contrast, A. cerana NC2-based biogeographic and genetic diversity studies show the utility of NC2 for illustrating A. cerana biogeographic history and diversity (Smith et al. 2000; Tan et al. 2007).
In this study, we collected A. cerana from nine Korean localities and from seven overseas localities (five from China, one from Vietnam, and one from Thailand). These samples were sequenced for NC2, genetic relationships with the preexisting worldwide haplotypes were identified, and genetic diversity of the Korean populations was compared with that of samples from other countries.
In addition, we developed an additional marker. Although NC2 is informative for the illustration of worldwide genetic diversity and biogeography of A. cerana, additional markers will be useful to expand our understanding of these aspects. The full-length A. cerana mt genome from China has been reported (Tan et al. 2011), and a genomic comparison of Apis species shows three additional spacer sequences that are longer than NC2 and unique to the A. cerana mt genome (Crozier and Crozier 1993; Tan et al. 2011; Wang et al. 2013). We assessed the potential of these three spacer sequences and found the internal spacer region located between tRNAMet and tRNAGln (termed NC1) the most useful one. The newly found NC1 was used to scrutinize the phylogenetic relationships in Korean A. cerana.
2 Materials and methods
2.1 Sampling and DNA extraction
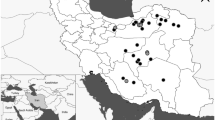
Adult workers were collected from 118 A. cerana colonies in nine Korean localities and in 66 colonies from foreign countries, such as five localities in China and one each in Vietnam and Thailand (Figure 1; Table I). The field-collected bees were preserved in 95 % ethanol until molecular experiments were performed. Total DNA was extracted from one or two legs using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA).

Map indicating the Apis cerana sampling localities. 1 Cheongsong-gun, Gyeongsangbuk-do; 2 Chungju-si, Chungcheongbuk-do; 3 Hongcheon-gun, Gangwon-do; 4 Namwon-si, Jeollabuk-do; 5 Icheon-si, Gyeonggi-do; 6 Yanggu-gun, Gangwon-do; 7 Cheongyang-gun, Chungcheongnam-do; 8 Pyeongchang-gun, Gangwon-do; 9 Cheongwon-gun, Chungcheongbuk-do; 10 Kunming, Yunnam province, China; 11 Yellow Mountain, Anhui province, China; 12 Changbai Mountains, Jiling province, China; 13 Tianshui, Gansu province, China; 14 Bei**g, China; 15 Vin Phuc, Vietnam; and 16 Chanthaburi, Thailand.
2.2 Primer design
To amplify the NC2 region (~89 bp), primers were designed at COI for the forward direction and at COII for the reverse direction from A. cerana mt genome (Tan et al. 2011) as follows: forward, 5′-CAATTTCATCTTTAGGATC-3′ and reverse, 5′-GTTCAAATAATTTCAATATTGTG-3′. Total product length was ~470 bp, but the gene regions showed no variations at all. Thus, only the NC2 region was utilized for subsequent analyses.
Sequence alignment of the three Apis mt genomes (GenBank accession numbers NC_001566 for A. mellifera, GQ162109 for A. cerana, and JX982136 for A. florea) (Crozier and Crozier 1993; Tan et al. 2011; Wang et al. 2013) shows that A. cerana has three additional unique non-coding regions located between tRNAMet and tRNAGln (231 bp), between COIII and tRNAGly (66 bp), and between tRNAPro and ND6 (50 bp), respectively (called NC1, NC3, and NC4, respectively, considering their positions in the A. cerana mt genome). Primers were designed at tRNASer and tRNAlle of the A. cerana mt genome sequence for NC1, COIII, and ND3 for NC3, and at ND4L and ND6 for NC4 (Figure 2).

Locations and primer sites of the three major non-coding regions in the Apis cerana mitochondrial genome. Primer sites of a NC1 at tRNASer and tRNAIle, b NC3 at COIII and ND3, and c NC4 at ND4L and ND6. S, E, M, Q, A, I, G, T, and P indicate tRNASer, tRNAGlu, tRNAMet, tRNAGln, tRNAAla, tRNAIle, tRNAGly, tRNAThr, and tRNAPro, respectively.
2.3 Polymerase chain reaction and sequencing
Thirty-five amplification cycles (94 °C for 1 min, 50–53 °C for 1 min, and 72 °C for 1 min) were conducted after an initial denaturation step at 94 °C for 5 min and a final extension step for 10 min at 72 °C to amplify the non-coding regions. PCR amplification was conducted using the AccuPower PCR PreMix (Bioneer, Daejeon, Korea) in a Biometra Thermal Cycler (T-gradient Thermoblock, Goettingen, Germany). Electrophoresis was carried out in 0.5× TAE buffer on 0.5 % agarose gels to confirm successful DNA amplification. The PCR product was purified using a PCR purification kit (Qiagen, Hilden, Germany). DNA sequencing was conducted using the ABI PRISM® BigDye® Terminator ver. 3.1 Cycle Sequencing Kit with an ABI 3100 Genetic Analyzer (PE Applied Biosystems, Foster City, CA, USA). All products were sequenced from both strands.
2.4 Sequence analysis and phylogenetic inference
Sequence delimitation and alignment were conducted using the CLUSTAL X ver. 1.8 program (Thompson et al. 1997). When homologous sequences from two individuals differed by ≥one nucleotide base or an insertion/deletion (indel), the sequences were considered different haplotypes. Haplotypes found in previous studies (e.g., Smith and Hagen 1996) were named identically (e.g., Japan1), but newly found haplotypes were named after one of the localities where they occurred.
The preexisting NC2 haplotypes originating from 955 individuals in 11 countries were downloaded from previous studies and GenBank for the phylogenetic analysis (Online Resource 1; Smith and Hagen 1996; Smith et al. 2000; Smith et al. 2004; Tan et al. 2007; Takahashi et al. 2007). A few haplotypes were excluded due to short sequence length, non-A. cerana haplotype, or redundancy. Eventually, 48 haplotypes were obtained and were aligned with the newly acquired haplotypes.
The phylogenetic analysis was conducted using the minimum evolution method (Rzhetsky and Nei 1992) using uncorrected p distance incorporated in PAUP* ver. 4.0b10 (Swofford 2002). Gaps were manually replaced with hypothetical nucleotides that are not found at the particular position in the alignment to prevent loss of sequence information. The analysis was performed by heuristic search using the selecting tree-bisection-reconnection option for the branch swap** algorithm, the random addition option for the starting point, one tree held at each step during stepwise addition, and with the steepest descent option not in effect. The tree is presented as a 50 % majority-rule consensus tree and was rooted using two haplotypes of A. koschevnikovi (GenBank accession numbers AB072437 and AB072438; Takahashi et al. 2002).
The median-joining algorithm (Bandelt et al. 1999) was used to further illustrate the relationships among NC2 haplotypes. Possible evolutionary pathways among haplotypes were visualized using SplitsTree 4.11.3 (Huson and Bryant 2006). This method adds to the network median vectors (consensus sequences) by starting with the minimum spanning trees combined within a single network. Such vectors can be interpreted as possibly extant unsampled sequences or extinct ancestral sequences (Bandelt et al. 1999). Because the NC1 haplotypes are only available from the current study, the NC1-based phylogenetic tree was rooted to midpoint, although other methods were the same to those used for NC2.
2.5 Genetic diversity analysis
The NC2 sequences from the current study were incorporated into available sequence information of previous Mainland Asian populations to upgrade the genetic diversity estimates of the Korean population using Arlequin ver. 3.5 (Excoffier and Lischer 2010). Gaps in sequence alignment were replaced with hypothetical nucleotides to prevent loss of sequence information as did for phylogenetic analysis.
3 Results
3.1 Sequence analysis and distribution of the NC2 haplotypes
Ten NC2 haplotypes of 96–98 bp were obtained (Japan1, Korea11, Korea12, Korea13, Korea14, IndiaB4, China6, China7, China8, and China9) (Table I). The sequence alignment of the haplotypes showed 11 variable positions, composed of six transitions (2 T⇔C and 4 G⇔A), three transversions (one each T⇔A, C⇔A, and G⇔T), and two indels. An unrooted pairwise comparison divergence in the sequences ranged from 3.093 % (three positions) to 1.031 % (one position). The highest sequence divergence was found when haplotype China6 found in a single individual in a Chinese locality (locality 11) was compared to nearly all other haplotypes, except Japan1. Among the 10 haplotypes, Japan1 and IndiaB4 were newly reported previously (Online Resource 1; Smith and Hagen 1996), but the remaining eight were found here. Korea11, Korea12, Korea13, Korea14, Japan1, and Japan1 were found in Korea. Japan1, IndiaB4, China6, China7, China8, and China9 were found in China. Only Japan1 was found in Vietnam, and Japan1 and IndiaB4 were found in Thailand.
3.2 Phylogenetic relationships
The phylogenetic analysis showed A. cerana NC2 haplotypes divided in two large groups (the Mainland Asian group and the Sundaland group) and three subgroups in the Philippine islands (Online Resource 2). The formation of a boundary between the sister group, the Mainland Asian group and the Sundaland group in the Malay Peninsula, was explained mainly by the presence of the Bilauktaung Mountain range, which hampers gene flow between Myanmar and Thailand in the Malay Peninsula (Smith et al. 2000). The Mainland Asian group was the most haplotype-rich (32 among 56 haplotypes), containing samples from diverse Asian countries, such as India, Burma, Thailand, Nepal, Russia, China, Korea, and Japan. The newly acquired haplotypes all belonged to the Mainland Asian group, reinforcing the previous findings that Korean A. cerana belongs to the Mainland Asian group.
The NC2-based median-joining network confirmed the presence of two large A. cerana groups, but the three subgroups found in the phylogenetic tree were recognized as two subgroups (Figure 3). One of the interesting features was the close relationships of one subgroup of the Philippine island group to the Mainland Asian group, rather than a close relationship to each other. The nine Mainland Asian group haplotypes found so far in Korea (Korea11, Korea12, Korea13, and Korea14 in this study; and Korea4, Korea7, Korea9, Japan1, and Nepal1 in previous studies) were scattered within the network, but six of them (Korea4, Korea7, Korea9, Korea11, Korea12, and Korea14) were located around Japan1, which has been found most frequently and widely in Mainland Asia. The Mainland Asian group network showed that a substantial number of haplotypes (15 haplotypes) was derived from the common Japan1 haplotype, presenting a star phylogeny (Figure 3).

Median-joining networks indicating the relationships among the NC2 haplotypes. Branch lengths represent the amount of character-state change occurring on that branch. Black dots represent haplotypes, and white dots indicate the hypothetical haplotypes that were not found in this study.
3.3 Nucleotide diversity
The genetic diversity of the Korean samples was compared to that of samples from other countries reported previously (Smith et al. 2000, 2004; Warrit et al. 2006; Tan et al. 2007; Takahashi et al. 2007). The frequency and distribution of the NC2 haplotypes from previous studies are presented in Online Resource 1. Haplotype and nucleotide diversities were high overall in the Philippines, India, Malaysia, Thailand, and Burma (Table II). Among these countries, the Philippines contained haplotypes from all subgroups of the Philippine island group (MindanaoP, Palawan1, Palawan2, and Cebu1, Luzon1, Luzon2, Mindanao1, Mindanao2, MindanaoL, and Negros1). The Mainland Asian group (Thai1, Japan1, ThaiN1, ThaiN2, and ThaiN3) and the Sundaland group (KoSamui1, ThaiS1, and Malay1) were found in Thailand; and the Mainland Asian group (BurmaN1, BurmaN2, BurmaN3, Japan1, and Nepal1) and the Sundaland group (ThaiS1) were found in Burma. In contrast, India and Malaysia had either the Mainland Asian group (IndiaB1, IndiaB3, IndiaB3, and IndiaB4) or the Sundaland group (Malay1, Malay2, Malay3, Malay4, Borneo1, Borneo2, and Borneo3), but diversity estimates remained substantially high. If the unincorporated, unaligned sequences from India (IndiaY1 and IndiaY2), Indonesia (SulawesiY1, SulawesiShort, SangiheY1, and SulawesiShort), and the Philippines (PhilippineShort and SangiheY1) were incorporated (Online Resource 1), the genetic diversity in these countries increased further. In contrast, haplotype and nucleotide diversities in Korea were substantially lower but slightly higher than those of China and Japan.
3.4 NC1 characterization
Sequencing of three additional non-coding regions (called NC1, NC3, and NC4, respectively; Figure 2) from a few A. cerana individuals showed that NC3 was ~66-bp long and composed mostly of A + T nucleotides (1.5 % of G + C content) with the AT repeats close to the 3′-end of tRNAGly. NC4 was ~50-bp long and composed of A + T nucleotides with several stretches of A or T nucleotides (6 % of G + C content). In contrast, NC1 was longer (~231 bp) and had rather higher G + C content (7.7 %) than the other non-coding regions. Thus, NC1, located between tRNAMet and tRNAGln, was eventually selected and sequenced in all samples. The sequence alignment showed that NC1 was composed of four non-variable regions (NV1–NV4) and three variable regions (V1–V3) (Online Resource 3), with nine variable positions consisting of five transitions (1 T⇔C and 4 G⇔A) and four indels (Online Resource 4).
Nine NC1 haplotypes (named A. cerana non-coding region 1 haplotype 01–09, ACNC101–ACNC109) were obtained; eight were found in Korean A. cerana, and four were found solely in Korea (Table III). The uncorrected sequence divergence of the haplotypes ranged from 2.597 to 0.433 % with a maximum of six as variable positions. Eleven localities had two or more NC1 haplotypes (Table III).
3.5 Phylogenetic relationships inferred from NC1
The phylogenetic relationships among the NC1 haplotypes revealed two groups (Figure 4; groups A and B). Group A contained ACNC101 and ACNC102, which were found most frequently and widely (seven among nine Korean localities, all five Chinese localities, and each Vietnam and Thailand) and ACNC109, which was found only in a Chinese locality (locality 14). In contrast, group B contained the remaining six haplotypes, which were found in seven of the nine Korean localities, excluding two Korean and Chinese localities each, which only had ACNC101 and ACNC102 (localities 1, 6, 12, and 13). Consequently, five Korean (localities 2, 4, 5, 8, and 9) and one Chinese locality (locality 10) had the haplotypes belonging to both phylogenetic groups.

Relationships among Apis cerana NC1 haplotypes. The tree was constructed with the minimum evolution method with a midpoint-rooted minimal tree and presented as an unrooted 50 % majority-rule consensus tree using PAUP* ver. 4.0b10 software. Parentheses indicate the localities where the corresponding haplotypes were found. Nodal support was obtained by 1000 replicates.
4 Discussion
4.1 NC2 haplotype distributions and relationships
Nine haplotypes are known in Korea, composed of four found new here (Table I) and five found previously (Online Resource 1) (Smith and Hagen 1996; Smith et al. 2000; Takahashi et al. 2007). Thus, extended sampling of additional localities revealed previously undetected A. cerana diversity in Korea. Furthermore, including the new haplotypes increased maximum sequence divergence from 2.062 % (two positions) to 3.093% (three positions) in Korean A. cerana. The extended A. cerana sampling in China was also fruitful in that four new haplotypes specific to China (China6, China7, China8, and China9) were added to the preexisting eight haplotypes (Tan et al. 2007). IndiaB4, which was found previously only in India (Smith and Hagen 1996) was also found in a Chinese (locality 10) and a Thailand locality (locality 16). The A. cerana samples outside Korea were limited in this study, but the detection of new haplotypes validated further sampling in these regions. Additionally, finding an extended distribution of a certain haplotype (e.g., IndiaB4) may emphasize some older relationships between bees in this region (Smith and Hagen 1996).
We also found that Japan1 was distributed most extensively with the highest frequency and accounted for 71 % of the Korean samples (Table II). A high frequency of Japan1 in Mainland Asian countries was reported previously (106 among 226 samples from six countries; Tan et al. 2007). Takahashi et al. (2007) focused on the Japanese islands and showed the dominance of Japan1 (530 among 535 individuals from 47 colonies). Nevertheless, one Korean and one Chinese locality (localities 1 and 14, respectively) possessed only non-Japan1 haplotypes specific to each locality (Table III). The current and previous studies collectively suggest that the A. cerana haplotype distribution on the Mainland Asian continent including Korea includes the widely distributed Japan1, along with a few locally limited haplotypes.
According to the simple isolation by distance model, the variance in the geographic distribution of an mtDNA lineage is proportional to its age (Neigel and Avise 1993). Thus, the most widespread haplotypes with the highest frequencies may be the oldest ones, whereas their progeny with relatively lower frequencies are expected to be found close to the areas where they arose (Watterson and Guess 1977). Such perspectives are reflected in the network as a star-like phylogeny in which widespread older haplotypes are located in the center of the network and recently derived rarer ones are stretched near the external nodes. Thus, Japan1, as the most widespread haplotype, may be the oldest one and involved in extensive gene flow, whereas the haplotypes stretched near the external nodes may be derived from Japan1 (e.g., Korea11, Korea12, Korea14) and distributed in limited areas of the Mainland Asian continent (Figure 3). A favorable post-Pleistocene climate and habitat may have allowed an increase in population size and extensive Japan1-based gene flow onto the Mainland Asian continent (Smith et al. 2000). Consequently, a substantial number of Japan1-close haplotypes, whose distributions are confined to limited regions, are found in the Mainland Asian group and allowed for the highest number of NC2 haplotypes in the Mainland Asian group (32 among 56 haplotypes; Figure 3). Japan1 may explain the similarly low level of genetic diversity in Korea, China, and Japan as has been previously pointed out (Tan et al. 2007). A connection between the Korean peninsula via the Korean offshore islands and the Japanese islands by a lower sea level during the Pleistocene may have facilitated Japan1-based gene flow from China to Korea and extended to the western boundary of Japan, although a specific cause for such a sweep requires additional study.
4.2 Utility of NC1 for inferring Korean A. cerana phylogeny
Somewhat higher variability was detected in the NC1 sequences. Nine NC1 haplotypes, ranging in size from 227 to 231 bp, provided sequence divergence of 2.597–0.433 % and six maximally variable sites. However, the 96–98-bp-long NC2 using the same individuals provided 10 haplotypes with three maximally variable sites and sequence divergence of 3.093–1.031 %. Thus, the number of haplotypes (ten vs. nine) and the maximum sequence divergence (3.093 vs. 2.597 %) were slightly higher in NC2 than those in NC1. Nevertheless, the maximum number of variable sites doubled in NC1 (from three to six), providing a higher number of populations with more than two haplotypes (from six to 11) and a higher number of haplotypes in Korean populations (from five to eight) in the NC1 sequences. Considering these aspects, NC1 can be used as another mtDNA-based analysis.
The NC1-based phylogenetic analysis revealed two groups (Figure 4; groups A and B). Previous studies also suggested that Korean A. cerana can be categorized into two genetic groups (Chang et al. 2000; Kim et al. 2010). The presence of two distinctive groups roughly concordant with the geographic regions in Korea was reported based on the HinfI enzyme digestion pattern of the mt COI amplicon: higher altitude northeastern localities in one group and lower altitude southwestern localities in the other group (Chang et al. 2000). Another study that directly sequenced the COI gene (488 bp) found two haplotypes, each of which is found solely in different localities, but the distribution pattern differed from the previous study of Chang et al. (2000). Our NC1 data were similar in that the two phylogenetic groups are present in Korea, but completely differed from both previous studies in that the two groups occurred without any regional subdivisions (Figures 1 and 4). In fact, the two most frequent haplotypes, ACNC101 and ACNC102 in group A, were widespread without any regional bias (Table III). Apis cerana may have been introduced to Korea from two different sources in India via China long ago (Choi 1989). Characteristic A. cerana beekee**, such as continuous hives in mountainous locations, may have allowed A. cerana stocks to remain as they were introduced but more likely human intervention along with natural dispersal may have facilitated mingling of random populations. Consequently, two mt lineages were identified by phylogenetic analysis, but no lineage was confined regionally. Further molecular marker with extensive geographic sampling from Korea and neighboring countries is required to understand the pathways of the two mtDNA lineages in Korea.
In summary, the NC2 region was sequenced to understand the genetic relationships and diversity of A. cerana in Korea. We found relatively low diversity but discovered unreported haplotypes from Korea and China. Japan1 dominated all haplotypes. Korean A. cerana belonged to the Mainland Asian group, as reported previously. Newly developed NC1 was informative, as it provided more Korean population haplotypes than those of NC2. The NC1 phylogenetic analysis provided two Korean A. cerana lineages, but a region-based interpretation was unlikely. We interpreted this as a result of an A. cerana introduction from two different sources and a randomized admixture of the sources.
References
Bandelt, H.J., Forster, P., Röhl, A. (1999) Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 6, 37–48
Cha, S.Y., Yoon, H.J., Lee, E.M., Yoon, M.H., Hwang, J.S., **, B.R., Han, Y.S., Kim, I. (2007) The complete nucleotide sequence and gene organization of the mitochondrial genome of the bumblebee, Bombus ignitus (Hymenoptera: Apidae). Gene 392, 206–220
Chang, Y.D., Byun, Y.W., Yeo, Y.S., Lee, M.Y. (2000) Nucleotide sequence variation in mitochondrial DNA of oriental honeybee (Apis cerana Fab.). Korean J. Apic. 15, 87–102
Choi, S.Y. (1989) Apiology. Jiphyeonsa, Seoul
Cornuet, J.M., Garnery, L., Solignac, M. (1991) Putative origin and function of the intergenic region between COI and COII of Apis mellifera L. mitochondrial DNA. Genetics 128, 393–403
Crozier, R.H., Crozier, Y.C. (1993) The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics 133, 97–117
De la Rúa, P., Simon, U.E., Tilde, A.C., Moritz, R.F.A., Fuchs, S. (2000) MtDNA variation in Apis cerana populations from the Philippines. Heredity 84, 124–130
Deowanish, S., Nakamura, J., Matsuka, M., Kimura, K. (1996) mtDNA variation among subspecies of Apis cerana using restriction fragment length polymorphism. Apidologie 27, 407–413
Excoffier, L., Lischer, H.E.L. (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567
Hajibabaei, M., Singer, G.A., Hebert, P.D., Hickey, D.A. (2007) DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends Genet. 23, 167–172
Huson, D.H., Bryant, D. (2006) Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267
Kim, H.-K., Lee, M.Y., Lee, M.L., Choi, Y.S., Hong, I.P., Kim, M.S., Lee, K.G., **, B.R. (2010) Genetic diversity of the Apis cerana in Korea by mitochondrial DNA and ITS ribosomal DNA sequence. Korean J. Apic. 25, 9–16
Neigel, J.E., Avise, J.C. (1993) Application of a random walk model to geographic distributions of animal mitochondrial DNA variation. Genetics 135, 1209–1220
Oh, H.K., Yoon, H.J., Kim, M.J., Jeong, H.U., Kim, S.R., Hwang, J.S., Bae, C.H., Kim, I. (2009) ITS2 ribosomal DNA sequence variation of the bumblebee, Bombus ardens (Hymenoptera: Apidae). Genes Genomics 31, 293–303
Rzhetsky, A., Nei, M. (1992) A simple method for estimating and testing minimum-evolution trees. Mol. Biol. Evol. 9, 945–967
Sihanuntavong, D., Sittipraneed, S., Klinbunga, S. (1999) Mitochondrial DNA diversity and population structure of the honey bee, Apis cerana, in Thailand. J. Apic. Res. 38, 211–219
Smith, D.R., Hagen, R.H. (1996) The biogeography of Apis cerana as revealed by mitochondrial DNA sequence data. J. Kansas Entomol. Soc. 69, 249–310
Smith, D.R., Villafuerte, L., Otis, G., Palmer, M.R. (2000) Biogeography of Apis cerana F. and A. nigrocincta Smith: insights from mtDNA studies. Apidologie 31, 265–279
Smith, D.R., Warrit, N., Hepburn, H.R. (2004) Apis cerana from Myanmar (Burma): unusual distribution of mitochondrial lineages. Apidologie 35, 637–644
Swofford, D.L. (2002) PAUP*. Phylogenetic analysis using Parsimony (*and other method) software ver. 4.10, Sinauer Associates. Sunderland, Massachusetts
Takahashi, J., Nakamura, J., Sasaki, M., Tingek, S., Akimoto, S. (2002) New haplotypes for the non-coding region of mitochondrial DNA in cavity nesting honey bees Apis koschevnikovi and Apis nuluensis. Apidologie 33, 13–19
Takahashi, J., Yoshida, T., Takagi, T., Akimoto, S., Wood, K.S., Deowanish, S., Hepburn, R., Nakamura, S., Matsuka, M. (2007) Geographic variation in the Japanese islands of Apis cerana japonica and in A. cerana populations bordering its geographic range. Apidologie 38, 335–340
Tan, K., Warrit, N., Smith, D.R. (2007) Mitochondrial DNA diversity of Chinese Apis cerana. Apidologie 38, 238–246
Tan, H.W., Liu, G.H., Dong, X., Lin, R.Q., Song, H.Q., Huang, S.Y., Yuan, Z.G., Zhao, G.H., Zhu, X.Q. (2011) The complete mitochondrial genome of the Asiatic cavity nesting honeybee Apis cerana (Hymenoptera: Apidae). PLoS One 6, e23008
Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., Higgins, D.G. (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 24, 173–216
Wang, A.R., Kim, M.J., Park, J.S., Choi, Y.S., Thapa, R., Lee, K.Y., Kim, I. (2013) Complete mitochondrial genome of the dwarf honeybee, Apis florea (Hymenoptera: Apidae). Mitochondrial DNA 24, 208–210
Warrit, N., Smith, D.R., Lekprayoon, C. (2006) Genetic subpopulations of Varroa mites and their Apis cerana hosts in Thailand. Apidologie 37, 19–30
Watterson, G.A., Guess, H.A. (1977) Is the most frequent allele the oldest? Theor. Popul. Biol. 7, 141–160
Acknowledgments
This study was supported by a grant from the Next-Generation BioGreen 21 Program (no. PJ009031) and the Rural Development Administration, Republic of Korea. We thank Joong Won Park for his help with sequencing at the beginning.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript editor: Marina Meixner
Variations de l’ADN mitochondrial chez les populations coréennes d’ Apis cerana (Hymenoptera: Apidae) et développement d’un autre marqueur potentiel
relation génétique / abeille asiatique / nid dans cavité / ADNmt / sequence non-codante
Variation der mitochondrialen DNA in Apis cerana (Hymenoptera: Apidae) und Entwicklung eines möglichen weiteren Markers
Genetische Verwandtschaft / asiatische höhlenbrütende Honigbiene / mitochondriale DNA / nicht-kodierende Sequenz
Joo Young Lee and Ah Rha Wang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
(DOCX 26 kb)
Online Resource 2.
(JPEG 133 kb)
Online Resource 3.
(JPEG 74 kb)
Online Resource 4.
(JPEG 191 kb)
Rights and permissions
About this article

Cite this article
Lee, J.Y., Wang, A.R., Choi, Y.S. et al. Mitochondrial DNA variations in Korean Apis cerana (Hymenoptera: Apidae) and development of another potential marker. Apidologie 47, 123–134 (2016). https://doi.org/10.1007/s13592-015-0381-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-015-0381-y