
Abstract
BRCA1 mutations are associated with ovarian cancer. Previous studies reported that murine granulosa cell (GC) Brca1 loss caused ovarian-uterine tumors resembling serous cystadenomas, but the pathogenesis of these tumors may have been confounded by ectopic Brca1 expression and altered estrous cycling. We have used Tg.AMH.Cre conferring proven ovarian and GC-specific Cre activity to selectively target Brca1 disruption, denoted Brca1 GC−/−. Furthermore, ovary-specific Brca1 GC−/− was combined with global Trp53 haploinsufficiency (Trp53 +/−) and transgenic follicle-stimulating hormone (Tg.FSH) overexpression as a multi-hit strategy to investigate additional genetic and hormonal ovarian tumorigenesis mechanisms. However, 12-month-old Brca1 GC−/− mice had no detectable ovarian or uterine tumors. Brca1 GC−/− mice had significantly increased ovary weights, follicles exhibiting more pyknotic granulosa cells, and fewer corpora lutea with regular estrous cycling compared to controls. Isolated Brca1 GC−/− mutation lengthened the estrous cycle and proestrus stage; however, ovarian cystadenomas were not observed, even when Brca1 GC−/− was combined with Trp53 +/− and overexpressed Tg.FSH. Our Brca1 GC−/− models reveal that specific intra-follicular Brca1 loss alone, or combined with cancer-promoting genetic (Trp53 loss) and endocrine (high serum FSH) changes, was not sufficient to cause ovarian tumors. Our findings show that the ovary is remarkably resistant to oncogenesis, and support the emerging view of an extragonadal, multi-hit origin for ovarian tumorigenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ovarian cancer is diagnosed annually in nearly a quarter of a million women globally, and is responsible for over 140,000 deaths each year (WHO, cancer incidence and mortality worldwide in 2008). Five to 15 % of ovarian cancers are due to known hereditary factors, and approximately 90 % of these can be attributed to germline mutations in BRCA1 [1, 2]. Germline BRCA1 mutations confer a 40 % risk of develo** ovarian cancer by the age of 70, and an inherited predisposition to fallopian tube cancer, which may precede ovarian involvement [3–5], providing a basis for the tubal origin of ovarian cancer hypothesis [2]. Somatic BRCA1 mutations are rare, yet reduced or absent protein expression is found in 90 % of sporadic ovarian tumors [6–8]. Mutations in human tumor suppressor BRCA1/2 and TP53 genes have been strongly linked with the development of ovarian cancers. Somatic mutations in TP53 have been found in over 96 % of human high-grade serous ovarian cancers [9, 10], and TP53 protein accumulation is observed in ovarian serous carcinomas [11, 12] and BRCA1-related cancer [13], providing a clinical marker that may contribute to the cause and progression of ovarian cancers.
Mouse models of ovarian tumorigenesis have been established by targeting Brca1 disruption using transgenic (Tg) Cre-loxP-induced genomic modification. Adenoviral-mediated delivery of Tg.Cre to target murine ovarian surface epithelium (OSE) showed that local OSE disruption of both Brca1 and Trp53 resulted in ovarian tumors resembling leiomyosarcomas [14, 15], whereas inactivation of either Brca1 or Trp53 alone produced few tumors [14, 16]. Chodankar et al. utilized Tg.Cre driven via the FSHR promoter (Tg.FSHR.Cre) to direct loss of Brca1 in granulosa cells (GC) of mouse ovarian follicles [17, 18]. Female mice with Tg.FSHR.Cre-mediated Brca1 disruption developed cystic ovarian and uterine horn tumors resembling serous cystadenomas by 7–8 months of age [17, 18]. The presence of normal Brca1 alleles in these cystadenomas led to the proposal that GC-specific Brca1 disruption may influence tumor development indirectly via an undefined paracrine mediator secreted by the modified GCs [17]. However, this Brca1-modified model exhibited extraovarian (e.g., pituitary) Cre-induced Brca1 disruption and altered estrous cycling [17, 18], presenting multiple causes for the pathogenesis of tumors in this model. Non-follicular FSHR expression has also been reported in ovarian surface epithelium and tumors [19] as well as other cancers [20].
Here, we have investigated the selective role of ovarian Brca1 disruption using GC-specific Tg.Cre directed via the anti-Müllerian hormone (AMH) gene promoter [21, 22]. In addition, we combined Tg.AMH.Cre-mediated GC-specific Brca1 disruption with Trp53 haploinsufficiency (Trp53 +/−) to study effects of both ovarian cancer-associated factors. Furthermore, we combined Brca1/Trp53 disruption with our Tg model overexpressing circulating FSH activity (transgenic follicle-stimulating hormone (Tg.FSH)) and steroids [23]. It has been proposed that repetitive ovulatory trauma (“incessant ovulation hypothesis”) and high circulating concentrations of gonadotropins may contribute to ovarian cancer development or progression [24], as well as the related “excessive gonadotropin” hypothesis [25], which proposes elevated gonadotropin (and estradiol) levels as a causative factor in ovarian cancer risk. Our Tg.AMH.Cre-induced Brca1 disruption model (Brca1 GC−/−) combined with Trp53 +/− and Tg.FSH provides a unique experimental in vivo platform to determine the impact of multiple factors proposed to contribute to ovarian tumorigenesis.
Materials and Methods
Genetic Mouse Models
Age-matched Tg and non-Tg female littermates used in experiments were housed under controlled conditions (12 h light/dark cycle) with ad lib access to food and water. Mice weights and general well-being were monitored weekly over the period of 6–12 months of age. All animal procedures were approved by the Animal Care and Ethics Committee of the Northern Sydney Local Health District and performed in accordance to the National Health and Medical Research Council code of practice for care and use of animals and the NSW Animal Research Act (1985).
Brca1GC−/− and Trp53+/− Mice
To generate Brca1 GC−/− mice, Tg.AMH.Cre mice were crossbred with Brca1-floxed mice [26], Brca1 exons 5 and 13 flanked by loxP sites (STOCK Brca1 tm2Brn). Tg.AMH.Cre mice exhibit GC-specific Cre expression and activity, as well as no detectable pituitary or uterine Cre activity [21, 22]. The Tg.AMH.Cre-mediated Brca1 deletion genotype [27], Brca1 GC−/−, was combined with a global Trp53 haploinsufficiency (Trp53 +/−) model (B6.129S2-Trp53 tm1Tyj /J) [28].
Tg.FSH Mice
Our Tg.FSH mouse model, previously referred to as TgFSHH for high FSH levels [23], exhibits pituitary-independent FSH expression driven via the rat Ins2 promoter [23, 29]. Tg.FSH mice of predominantly C57BL/6 strain were crossbred with Tg.AMH.Cre mice and then combined with the Brca1-floxed and Trp53 +/− background.
Genoty**
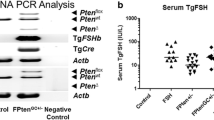
Genomic DNA was isolated from toe, tail tip, ovary, or pituitary tissue by lysis using proteinase K as previously described [30]. Genomic DNA was obtained from uterine and fallopian tube tissue sections as recommended by the manufacturer (Isolate II FFPE RNA/DNA kit, Bioline, NSW, Australia). DNA samples were used for PCR genoty** to detect floxed, Tg.Cre-mediated mutant, or wild-type mouse Brca1 [27], and the heterozygous Trp53 +/− background [28]. Mice containing Tg.AMH.Cre and Tg.FSH were detected using PCR conditions as previously described, with actin providing an internal sample control [29, 31]. Primer pairs and expected amplicon product sizes for genoty** are listed in Table 1. Designated genotype abbreviations for the different mouse groups examined are listed in Fig. 1a.

a Experimental female groups were defined by genoty** to detect the presence of specific gene mutations and transgenes, abbreviated as shown. b Whole ovary genoty** showed the presence of floxed Brca1 and the Tg.AMH.Cre-loxP-mediated Brca1 deletion. c In contrast, the pituitary, fallopian tubes, and uterus had no detectable Brca1 deletion. Genoty** wild-type animals produced the expected PCR products for normal Brac1 introns 3 and 13, respectively. Trp53 heterozygosity was shown with PCR products from both wild-type and mutated alleles. Transgenic Cre and Tg.FSH (β-subunit) screening produced the expected PCR products, and the beta-actin PCR product confirmed the presence of genomic DNA in all samples
Estrous Cycle Analysis
Estrous cycle analysis was performed just prior to sacrifice of mice at 12 months of age. Vaginal samples (in 20 μL of sterile 0.9 % saline) were obtained daily at 8:00–9:00 am over 5–12 days, and smeared onto glass slides and stained with 0.05 % Trypan Blue for microscopic classification into estrous stages [32, 33].
Serum Collection, Tissue Processing, and Hormone Analysis
Mice were examined weekly for general health (activity, body weight, fur condition), and at 12 months of age, mice were weighed and blood was collected by cardiac exsanguination, under isoflurane/oxygen/nitrous oxide or ketamine/xylazine anesthesia. Blood was allowed to clot at room temperature for 20 min before centrifugation at 5000 rpm for 5 min, and then serum was collected and stored at −80 °C until assay. Serum Tg.FSH levels were measured by species-specific (human) FSH immunoassay as described previously [34]. Ovaries and uteri were removed and weighed, and the pituitary, tail tip, or toe were removed and immediately frozen (liquid N2) for DNA or RNA (pituitary, tail, toe, one ovary), steroid assay (liquid chromatography tandem mass spectrometry, LC-MS/MS), or fixed in 4 % paraformaldehyde (ovary, uterus) overnight and transferred to 70 % ethanol for histological analysis.
Tissue Histology and Corpora Lutea Quantification
One fixed ovary from each mouse was embedded in paraffin, sectioned at 8 μm, and every 10th section stained with hematoxylin and eosin (H&E). Total corpus luteum counts were undertaken by light microscopy under a ×40 oil objective and using Stereo Investigator software (MicroBrightfield, Williston, VT). Total follicle numbers and pyknotic GC numbers were quantified as previously described [32, 35, 36]. Ovarian stroma content was quantified using ImageJ (National Institute of Health, USA), using every 10th ovarian section (10/ovary) as described [37, 38]. Uterine/fallopian tube 20-μm sections were embedded in paraffin, and tissue from 4 sections was dissected under light microscopy and used for genomic DNA isolation described above.
Intraovarian Steroid Analysis
Frozen ovaries were transferred to 5-mL glass tubes containing 300 μL of homogenization buffer (0.5 % (w/v) BSA, 5 mM EDTA in PBS, pH 7.4) and homogenized on ice for 20 s using an IKA T10 basic disperser on the highest setting. The dispersing element was gently wiped to remove any remaining tissue and rinsed in PBS between samples. Homogenized samples were centrifuged (3000 rpm, 10 min, 4 °C) and supernatants removed from insoluble debris (pellet) and transferred to fresh 1.5-mL plastic tubes, stored at −80 °C until analysis. Sample estradiol and estrone levels were measured by LC-MS/MS [39, 40]. Limits of detection (defined as lowest level detected with a coefficient variation (CV) of <20 %) were estradiol (2 pg/ovary) and estrone (1 pg/ovary).
Statistical Analysis
Statistical analysis was performed by two-way ANOVA using Brca1 (i.e., Brca1 GC−/−) and Tg.FSH status as main effects with non-significant interactions ignored and a suitable linear contrast to define the additional effects of Trp53 heterozygosity (Trp53 +/− ) (NCSS version 9). p values for each main effect, interaction, and linear contrast are stated with values below 0.05 regarded as significant. Comparisons between specific groups used one-way ANOVA as indicated. All data are presented as mean ± SEM.
Results
Genetic Mouse Models
Genoty** confirmed the presence of specific gene mutations and transgenes in experimental mice, with groups designated as shown in Fig. 1a. Whole ovary genoty** showed the presence of floxed Brca1 (545- and 620-bp PCR products for modified introns 3 and 13, respectively) and the Tg.AMH.Cre-loxP-mediated Brca1 deletion (594-bp product), as shown in Fig. 1b. In contrast, the pituitary, fallopian tubes, uterus (Fig. 1b), and toe (not shown) had no detectable Brca1 deletion, consistent with ovary-specific Cre-mediated Brca1 loss. Genoty** wild-type animals produced the expected 390- and 494-bp PCR products for normal Brca1 introns 3 and 13, respectively. Trp53 heterozygosity was shown by 450- and 650-bp PCR products corresponding to wild-type and mutated alleles, respectively (Fig. 1b). Transgenic Cre and Tg.FSH (β-subunit) screening produced the expected PCR products, and the beta-actin PCR product confirmed the presence of genomic DNA in all samples.
Tg.FSH Expression
After cross-breeding Tg.FSH × Brca1-floxed mice, serum Tg.FSH levels were 28.5 ± 3.5 IU/L (N = 4) and 53.4 ± 17.3 IU/L (N = 3) in 9- and 12-month-old Brca1 floxedfloxed females, respectively. As expected, serum Tg.FSH levels were not detectable in non-Tg control females (C, B, and BT; refer to Fig. 1). Serum Tg.FSH levels were 20.3 ± 6.4 IU/L (N = 10) in aged control Tg.FSH (F) mice, whereas significantly lower levels (p < 0.001) were measured in mice containing the Brca1 GC−/− genotype (BF 2.5 ± 0.5 IU/L; BTF 2.9 ± 0.6 IU/L).
Effect on Estrous Cycling
The ability of aged 12-month-old mice to display estrous cycling was not affected by the follicular loss of Brca1 (Brca1 GC−/− status, p = 0.57), or combined with T background (Trp53 +/− effect, p = 0.71) and/or the presence of F (Tg.FSH, p = 0.59). A similar percentage of mice (66 %) were cycling in all groups (Table 2). Overall, there was no significant Brca1 GC−/− genotype effect on cycle length or proestrus stage (Brca1 GC−/− , p = 0.06). However, estrous cycle and proestrus stage length were significantly longer in B versus control C mice (one-way ANOVA, p < 0.05), as shown in Table 2. There were no significant differences between combined preovulatory (proestrus + estrus) stage times, or between diestrus stage times for any genotype (Table 2). However, post-ovulatory (metestrus + diestrus) stage times displayed a Brca1 GC−/− genotype effect (Brca1 GC−/− status, p < 0.05), with longer combined stages in the B and BF groups (one-way ANOVA, p < 0.05) (Table 2).
Ovarian Weights, Histology, Corpora Lutea, and Follicle Numbers
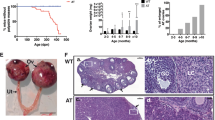
Ovary weights were significantly increased in 12-month-old females carrying the Brca1 GC−/− genotype (Brca1 GC−/−, p < 0.001; Tg.FSH, p = 0.78; Trp53 +/−, p = 0.48) (Fig. 2a). Ovarian stroma was significantly increased in Brca1 GC−/− females (Brca1 GC−/−, p < 0.001), and there were more regions of increased lipid-laden stromal cell types in B compared to control ovaries, as shown in Fig. 3. Total ovarian follicle counts were significantly affected by the B genotype (Brca1 GC−/−, p < 0.05; Trp53 +/−, p < 0.05; Tg.FSH, p = 0.90), in particular the reduced follicle numbers found in BF compared to F ovaries (p < 0.01, one-way ANOVA), shown in Fig. 2. In addition, ovaries carrying the B genotype displayed aberrant follicles that contained higher levels of pyknotic granulosa cells (Brca1 GC−/−, p < 0.001), which were not altered in equivalent follicles of F ovaries (Tg.FSH, p = 0.75) (Fig. 3b). Total numbers of corpora lutea with a normal appearance were significantly decreased in 12-month-old females bearing the B genotype (Brca1 GC−/−, p < 0.001; Tg.FSH, p = 0.79) (Fig. 2).

a Relative ovary weights were significantly increased in 12-month-old females from all Brca1 GC−/− genotypes. Total follicle counts were significantly reduced in Brca1 GC−/− (and Trp53 +/−) but not Tg.FSH-exposed mice. Total numbers of corpora lutea (CL) with a normal appearance were significantly decreased in 12-month-old Brca1 GC−/− genotype (B containing) but not Tg.FSH (F) mice. Genotype abbreviations defined in Table 1 shown in bar graph by white, C; light gray, B; dark gray, BT; white hatch, F; light gray hatch, BF; dark gray hatch; BTF. Data shown as mean ± SEM, N = 7–12 per genotype. Asterisk indicates significant difference via one-way ANOVA compared to C and F mice. b Analysis of ovarian histology showed that no macroscopic tumors were present in 12-month-old females of any genotypes examined (×4 magnification). Number sign indicates the corpus luteum (CL), circumflex accent indicates hemorrhagic cyst

a Ovarian stroma was significantly increased in B (Brca1 GC−/−) genotype-exposed females. In addition, there were more regions of increased lipid-laden stromal cell types in Brca1 GC−/− but not Tg.FSH-exposed ovaries. Representative images of C and BF ovary sections depict the increased proportion of stroma found in Brca1 GC−/− genotype mice (×4 magnification with × 20 magnification inset). N = 7–12 per genotype. b The ovaries from Brca1 GC−/− females displayed aberrant follicles which contained higher levels of pyknotic granulosa cells compared to equivalent control and Tg.FSH females. Representative images of follicles from C and BT genotype females demonstrating pyknotic granulosa cells found in Brca1 GC−/− genotype mice. (×20 magnification) N = 7–12 per genotype
Analysis of ovarian histology showed that no macroscopic tumors were present in 12-month-old females of any genotypes examined (Fig. 2b). Likewise, there were no detectable differences in uterine histology from 12-month-old females from any model examined (Fig. 4).

Analysis of uteri histology of H&E-stained sections showed that no macroscopic tumors were present in 12-month-old females of any genotypes examined (×4 magnification)
Effect on Intraovarian Estradiol Content
Intraovarian estrone content was not significantly reduced in B genotype females (Brca1 GC−/−, p = 0.29; Tg.FSH, p = 0.07; Trp53 +/−, p = 0.49), although there was a significant interaction between B and F genotypes (Tg.FSH/Brca1 GC−/−, p < 0.05). Tg.FSH significantly increased (p < 0.05) intraovarian estradiol content, which was fourfold higher in F compared to control ovaries (Fig. 5), whereas B and T genotypes had no effect (Brca1 GC−/−, p = 0.17; Trp53 +/−, p = 0.90).

Analysis of intraovarian estrone (E1) and estradiol (E2) content showed no significant difference in the E1 level in Brca1 GC−/− (B genotype) ovaries. Ovarian E2 content was not affected by the B and T genotypes, whereas the E2 level was significantly higher in the F group females. Data shown as mean ± SEM, N = 5 per genotype
Discussion
We established a series of unique customized mouse models to determine the specific role of localized Brca1 loss in ovarian tumorigenesis. Proven ovary-specific Cre activity provided by Tg.AMH.Cre [21] was used to selectively target GC Brca1 disruption, Brca1 GC−/−. In addition, we combined Brca1 GC−/− with genetic (Trp53 loss) and endocrine changes (high FSH) considered to be promoters of ovarian cancer. Our findings show that follicular Brca1 loss alone or combined with Trp53 +/− and Tg.FSH expression did not cause ovarian or uterine tumors, suggesting that the intraovarian environment itself appears remarkably resistant to these genetic and hormonal mechanisms in oncogenesis.
Aged Brca1 GC−/− females exhibited larger relative ovarian weights compared to age-matched control females, which largely reflected an increase in the proportion of ovarian stroma featuring more areas of enlarged lipid-filled cells. Similar foam-like cells were reported in a separate mouse model exhibiting abnormal follicle maturation and few corpora lutea [41]. The Brca1 GC−/− ovaries displayed more follicles with a higher level of degenerating GCs, as well as fewer corpora lutea. BRCA1 has been implicated in a wide variety of cellular processes, including the regulation of genome integrity [42], DNA damage recognition and repair [43], cell cycle checkpoint control [44, 45], and apoptosis [46–48]. Disruption of theses pathways may contribute to the higher levels of pyknotic GCs in the follicles of Brca1 GC−/− ovaries, and possibly explain the reduced number of corpora lutea.
Despite the relative increase in size, Brca1 GC−/− ovaries did not exhibit detectable tumors at 12 months of age, which contrasts with the benign cystadenomas reported in <8-month-old mice with Tg.FSHR.Cre-induced Brca1 inactivation, noting similar use of the C57BL/6 background [17]. The distinct phenotype of Brca1 disruption when induced by the different Cre mouse lines (Tg.AMH.Cre in the current work compared to Tg.FSHR.Cre previously) may reflect different tissue expression levels or patterns of each Tg.Cre activity. An established LacZ-reporter system to detect Cre activity showed that Tg.AMH.Cre provides strong Cre activity in GCs alone (70–100 % of GC in most preantral-antral follicles), and no detectable extraovarian Cre activity in relevant tissues including brain, pituitary, and uterus [21]. Furthermore, AMH.Cre-targeted disruption (of the androgen receptor) produced a phenotype in female mice as young as 3 months of age [21]. It is possible AMH.Cre may provide low Cre activity relative to FSHR.Cre, and reduced penetrance may lead to a longer latency in tumor formation. However, the published Tg.FSHR.Cre-induced LacZ-reporter expression appears very weak or absent in GCs of many follicles [17]. Furthermore, ectopic expression of Tg.FSHR.Cre was reported in the pituitary [18], which does not normally express FSHR [49]. The uncertain significance of Brca1 loss upon overall pituitary function and possibly other sites of ectopic Tg.FSHR promoter expression may limit interpretation of findings with that model.
The relatively normal ovarian phenotype in TgAMH.Cre-driven mutant Brca1 mice suggests that the follicular environment is resistant to Brca1-associated tumorigenesis. Moreover, the lack of ovarian or uterine tumors in Brca1 GC−/− females does not support the proposed existence of an ovary-derived paracrine factor caused by follicular Brca1 disruption, which was predicted to induce uterine tumors in the Tg.FSHR.Cre-induced mutant Brca1 model [17]. Extraovarian rather than GC-driven defects may have caused the cystic tumors in Tg.FSHR.Cre/mutant Brca1 females [17]. These epithelial-like cystadenomas in the ovaries and uterine horns each possessed normal rather than mutant Brca1 alleles, suggesting that the ovarian tumors were secondary to uterine-derived tumors [17]. Precancerous changes are commonly seen in the fallopian tubes of women who are asymptomatic carriers of BRCA1 mutations [4, 5, 50]. Moreover, inactivation of Brca1 in mouse OSE induced preneoplastic epithelial changes similar to those observed in the ovaries of human BRCA1 mutation carriers [16]. It is possible that inactivation of BRCA1 in both epithelial and non-epithelial cells of the ovary maybe necessary for a cooperative effect that leads to ovarian carcinogenesis. Such findings are consistent with the recent “extraovarian origin hypothesis” proposing that ovarian serous tumors arise from the implantation of extraovarian tissue, in particular epithelium (benign or malignant) from the fallopian tube, from which the ovarian cancer arises secondarily [51, 52]. Proposed tumor origins include Mullerian duct derivatives surrounding the ovary [53], and many ovarian tumors are composed of cell types and cystic structures not present in normal ovarian tissue [53]. In the current model, Tg.AMH.Cre was not expressed in uterine, fallopian, or pituitary tissues, and no extraovarian Brca1 disruption was detected.
Our current findings support the proposal that granulosa cell-specific BRCA1 loss alone may be insufficient for ovarian tumorigenesis. Past research showed that disruption of BRCA1 caused a longer doubling time for ovarian and breast cancer cells [44, 45, 47, 48], and Brca1 downregulation resulted in growth arrest [54]. Furthermore, while most tumors in individuals with germline BRCA1 mutations show compound heterozygosity (with germline mutant retained) at the BRCA1 locus, consistent with a tumor suppressor role for BRCA1, some tumors retain the wild-type BRCA1 allele [55–57]. It was postulated that BRCA1 haploinsufficiency results in a destabilized genome that is more sensitive to DNA damage and other gene mutations. For instance, in the absence of the caretaker function of BRCA1, other repair pathways, such as that involving TP53, may be activated, resulting in slowed proliferation as damage is repaired [16]. Features of Brca1 disruption such as longer cellular doubling time or increased apoptosis can be overcome by loss of Trp53 [47, 48].Targeted disruption of both Brca1 and Trp53 in OSE and mammary tumor models have indicated that Trp53 may play a significant role in both Brca1-associated growth control and tumorigenesis [15, 16, 48, 58–61]. Thus, diminished BRCA1 activity may create a genetically unstable environment, the impact of which is not wholly evident until further genetic alterations take place, in a multi-hit mechanism of oncogenesis.
On this basis, we combined Trp53 haploinsufficiency with the Brca1 GC−/− background to provide a second genetic defect in a multi-hit approach. TP53 dysfunction is commonly associated with ovarian carcinoma, particularly in tumors associated with a germline BRCA1 mutation [62–64]. However, in the present study, females carrying the combined Brca1 GC−/− /Trp53 +/− mutations had no detectable ovarian or uterine tumors. Therefore, global Trp53 haploinsufficiency in all cells of the ovary, including GC and OSE, had no additive or combined effect with follicular Brca1 disruption in our model. It is likely that extraovarian and/or additional genetic mutations combine to initiate mutant BRCA1-associated tumorigenesis and ovarian cancer. Recent multi-hit murine models targeting conditional Brca1/Trp53/Pten disruption in the fallopian tube epithelial cells produced high-grade serous ovarian tumors [64], and targeting Brca1/Trp53/Rb inactivation in OSE produced ovarian carcinoma [65], highlighting the role of combined genetic defects and the possible extraovarian origin of certain ovarian cancers.
Elevated circulating FSH activity alone did not induce ovarian tumors in 1-year-old mice. FSH activates pathways associated with cell proliferation, growth, and oncogenesis dose-dependently in normal and malignant human OSE cells [19, 65], in particular during early stages of oncogenesis. Although the present findings show that elevated FSH alone is insufficient to initiate ovarian tumors, we cannot rule out a role for FSH activity in promoting the progression of existing ovarian cancers, in particular of an OSE origin in a multi-hit mechanism. Unexpectedly, serum Tg.FSH levels were lower in Brca1 GC−/− females relative to Brca1 floxed or Brca1 Wt/Wt females. Tg.AMH.Cre alone had no impact on Tg.FSH levels, and the mechanism of the lower Tg.FSH levels in Brca1 GC−/− females remains unclear, but might contribute to underestimating the impact of high circulating FSH in our multi-hit strategy. However, we previously showed that equivalent levels (2–3 IU/L) of Tg.FSH in a lower-expressing TgFSH mouse line increased ovulation [32], suggesting this level may still provide enhanced functional FSH activity. Females with combined Brca1 GC−/− /Trp53 +/− mutations and Tg.FSH expression also had no detectable ovarian or uterine tumors.
Our mouse model was based on conditional inactivation of Brca1 in GCs, which are known regulators of the estrous cycle. Therefore, we compared estrous cycling between all Brca1 GC−/− and control groups and found minimal differences between Brca1 cohorts; although the cycling data observed was highly variable within the old female groups. The current findings support the increased duration of the proestrus phase of the estrous cycle reported in the earlier Tg.FSHR.Cre/mutant Brca1 model [17, 18]. However, the Brca1 mutation had no marked impact on ability to cycle or the estrous stages in all groups examined (i.e., Brca1 with Tpr53 loss and/or high FSH), and the present cycling data did not show an increase in the preovulatory (proestrus + estrus) phase, described in Tg.FSHR.Cre/mutant Brca1 females [18]. Altered estrous cycling in Tg.FSHR.Cre/mutant Brca1 mice was proposed to reflect increased estradiol activity. Circulating estradiol levels were elevated in Tg.FSHR.Cre/Brca1 females after the administration of pregnant mare serum gonadotrophins, although direct analysis of ovarian steroid levels was not determined [18]. In the current study, intraovarian estrone and estradiol content remained normal in all Brca1 GC−/− females, whereas elevated estradiol levels were found in Tg.FSH ovaries. Previous work showed that reduced expression of BRCA1 in a human granulosa cell line led to upregulated aromatase (the rate-limiting enzyme in estradiol biosynthesis) expression [66]. In our model, it is possible that localized estradiol production may be higher in some Brca1 GC−/− follicles in order to compensate for the presence of follicles exhibiting more degenerating (pyknotic) GCs found in Brca1 GC−/− ovaries. However, direct analysis of ovarian steroids shows that the GC-specific Brca1 mutation does not alter overall ovarian estradiol content, which is consistent with no major impact upon the distinct stages of the estrous cycle.
In summary, our current findings indicate that the intraovarian environment is resistant to tumorigenic changes by combining up to three recognized tumor promoting strategies, namely the associated genetic loss of Brca1 and Trp53, and addition of elevated FSH. Follicular Brca1 modification alone or combined with genetic (Trp53 loss) and endocrine changes (high serum FSH) was not sufficient to cause ovarian or uterine tumors. Our findings indicate that extraovarian rather than ovarian GC-driven defects were likely responsible for the ovarian-uterine cystic tumors reported in the earlier Brca1 mutant model, and support an emerging view of an extragonadal origin for apparently ovarian cancers.
References
Berchuck A, Heron KA, Carney ME et al (1998) Frequency of germline and somatic BRCA1 mutations in ovarian cancer. Clin Cancer Res 4:2433–2437
Pal T, Permuth-Wey J, Betts JA et al (2005) BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104:2807–2816
Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL (2002) Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst 94:1365–1372
Leeper K, Garcia R, Swisher E, Goff B, Greer B, Paley P (2002) Pathologic findings in prophylactic oophorectomy specimens in high-risk women. Gynecol Oncol 87:52–56
Piek JMJ, van Diest PJ, Zweemer RP et al (2001) Dysplastic changes in prophylactically removed fallopian tubes of women predisposed to develo** ovarian cancer. J Pathol 195:451–456
Geisler JP, Hatterman-Zogg MA, Rathe JA, Buller RE (2002) Frequency of BRCA1 dysfunction in ovarian cancer. J Natl Cancer Inst 94:61–67
Russell PA, Pharoah PDP, De Foy K et al (2000) Frequent loss of BRCA1 mRNA and protein expression in sporadic ovarian cancers. Int J Cancer 87:317–321
Chan KYK, Ozçelik H, Cheung ANY, Ngan HYS, Khoo US (2002) Epigenetic factors controlling the BRCA1 and BRCA2 genes in sporadic ovarian cancer. Cancer Res 62:4151–4156
Schuijer M, Berns EM (2003) TP53 and ovarian cancer. Hum Mutat 21:285–291
The Cancer Genome Atlas Research N (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474:609–615
Kupryjańczyk J, Thor AD, Beauchamp R et al (1993) p53 gene mutations and protein accumulation in human ovarian cancer. Proc Natl Acad Sci U S A 90:4961–4965
Bosari S, Viale G, Radaelli U, Bossi P, Bonoldi E, Coggi G (1993) p53 accumulation in ovarian carcinomas and its prognostic implications. Hum Pathol 24:1175–1179
Zweemer RP, Shaw PA, Verheijen RM et al (1999) Accumulation of p53 protein is frequent in ovarian cancers associated with BRCA1 and BRCA2 germline mutations. J Clin Pathol 52:372–375
Quinn BA, Brake T, Hua X et al (2009) Induction of ovarian leiomyosarcomas in mice by conditional inactivation of Brca1 and p53. PLoS One 4:e8404
Clark-Knowles KV, Senterman MK, Collins O, Vanderhyden BC (2009) Conditional inactivation of Brca1, p53 and Rb in mouse ovaries results in the development of leiomyosarcomas. PLoS One 4:e8534
Clark-Knowles KV, Garson K, Jonkers J, Vanderhyden BC (2007) Conditional inactivation of Brca1 in the mouse ovarian surface epithelium results in an increase in preneoplastic changes. Exp Cell Res 313:133–145
Chodankar R, Kwang S, Sangiorgi F et al (2005) Cell-nonautonomous induction of ovarian and uterine serous cystadenomas in mice lacking a functional Brca1 in ovarian granulosa cells. Curr Biol 15:561–565
Hong H, Yen H-Y, Brockmeyer A et al (2010) Changes in the mouse estrus cycle in response to Brca1 inactivation suggest a potential link between risk factors for familial and sporadic ovarian cancer. Cancer Res 70:221–228
Parrott JA, Doraiswamy V, Kim G, Mosher R, Skinner MK (2001) Expression and actions of both the follicle stimulating hormone receptor and the luteinizing hormone receptor in normal ovarian surface epithelium and ovarian cancer. Mol Cell Endocrinol 172:213–222
Radu A, Pichon C, Camparo P et al (2010) Expression of follicle-stimulating hormone receptor in tumor blood vessels. N Engl J Med 363:1621–1630
Walters KA, Middleton LJ, Joseph SR et al (2012) Targeted loss of androgen receptor signaling in murine granulosa cells of preantral and antral follicles causes female subfertility. Biol Reprod 87(151):1–11
Lécureuil C, Fontaine I, Crepieux P, Guillou F (2002) Sertoli and granulosa cell-specific Cre recombinase activity in transgenic mice. Genesis 33:114–118
Allan CM, Kalak R, Dunstan CR et al (2010) Follicle-stimulating hormone increases bone mass in female mice. Proc Natl Acad Sci U S A 107:22629–22634
Fathalla MF (1971) Incessant ovulation? A factor in ovarian neoplasia? Lancet 298:163
Cramer D, Welch WR (1983) Determinants of ovarian cancer rsik II. Inferences regarding pathogenesis. J Natl Cancer Inst 71:717–721
Lim P, Robson M, Spaliviero J et al (2009) Sertoli cell androgen receptor DNA binding domain is essential for the completion of spermatogenesis. Endocrinology 150:4755–4765
Liu X, Holstege H, van der Gulden H et al (2007) Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A 104:12111–12116
Jacks T, Remington L, Williams BO et al (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4:1–7
Allan CM, Haywood M, Swaraj S et al (2001) A novel transgenic model to characterize the specific effects of follicle-stimulating hormone on gonadal physiology in the absence of luteinizing hormone actions. Endocrinology 142:2213–2220
Singh J, O’Neill C, Handelsman DJ (1995) Induction of spermatogenesis by androgens in gonadotropin-deficient (hpg) mice. Endocrinology 136:5311–5321
Schwenk F, Baron U, Rajewsky K (1995) A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23:5080–5081
McTavish KJ, Jimenez M, Walters KA et al (2007) Rising follicle-stimulating hormone levels with age accelerate female reproductive failure. Endocrinology 148:4432–4439
Mettus RV, Rane SG (2003) Characterization of the abnormal pancreatic development, reduced growth and infertility in Cdk4 mutant mice. Oncogene 22:8413–8421
Jimenez M, Spaliviero JA, Grootenhuis AJ, Verhagen J, Allan CM, Handelsman DJ (2005) Validation of an ultrasensitive and specific immunofluorometric assay for mouse follicle-stimulating hormone. Biol Reprod 72:78–85
Desmeules P, Devine PJ (2006) Characterizing the ovotoxicity of cyclophosphamide metabolites on cultured mouse ovaries. Toxicol Sci 90:500–509
Braw RH, Tsafriri A (1980) Effect of PMSG on follicular atresia in the immature rat ovary. J Reprod Fertil 59:267–272
Labiche A, Heutte N, Herlin P, Chasle J, Gauduchon P, Elie N (2010) Stromal compartment as a survival prognostic factor in advanced ovarian carcinoma. Int J Gynecol Cancer 20:28–33
Elie N, Labiche A, Michels J-J, Herlin P (2011) Control of low-resolution scanning of ovarian tumor stromal compartment. Image Anal Stereol 24:85–93
McNamara KM, Harwood DT, Simanainen U, Walters KA, Jimenez M, Handelsman DJ (2010) Measurement of sex steroids in murine blood and reproductive tissues by liquid chromatography-tandem mass spectrometry. J Steroid Biochem Mol Biol 121:611–618
Harwood DT, Handelsman DJ (2009) Development and validation of a sensitive liquid chromatography–tandem mass spectrometry assay to simultaneously measure androgens and estrogens in serum without derivatization. Clin Chim Acta 409:78–84
Cheng G, Weihua Z, Mäkinen S et al (2002) A role for the androgen receptor in follicular atresia of estrogen receptor beta knockout mouse ovary. Biol Reprod 66:77–84
Yu V (2000) Caretaker Brca1: kee** the genome in the straight and narrow. Breast Cancer Res 2:82–85
Yoshida K, Miki Y (2004) Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci 95:866–871
Larson JS, Tonkinson JL, Lai MT (1997) A BRCA1 mutant alters G2-M cell cycle control in human mammary epithelial cells. Cancer Res 57:3351–3355
Yan Y, Spieker RS, Kim M, Stoeger SM, Cowan KH (2005) BRCA1-mediated G2/M cell cycle arrest requires ERK1/2 kinase activation. Oncogene 24:3285–3296
Shao N, Chai YL, Shyam E, Reddy P, Rao VN (1996) Induction of apoptosis by the tumor suppressor protein BRCA1. Oncogene 13:1–7
Deng C-X (2006) BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 34:1416–1426
Xu X, Wagner KU, Larson D et al (1999) Conditional mutation of Brca 1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet 22:37–43
Dierich A, Sairam MR, Monaco L et al (1998) Impairing follicle-stimulating hormone (FSH) signaling in vivo: targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc Natl Acad Sci U S A 95:13612–13617
Colgan TJ, Murphy J, Cole DEC, Narod S, Rosen B (2001) Occult carcinoma in prophylactic oophorectomy specimens: prevalence and association with BRCA germline mutation status. Am J Surg Pathol 25:1283–1289
Kurman RJ, Shih I-M (2010) The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol 34:433–443
Kurman RJ, Shih I-M (2011) Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—shifting the paradigm. Hum Pathol 42:918–931
Dubeau L (2008) The cell of origin of ovarian epithelial tumours. Lancet Oncol 9:1191–1197
Ouchi M, Fujiuchi N, Sasai K et al (2004) BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M transition. J Biol Chem 279:19643–19648
George J, Alsop K, Etemadmoghadam D et al (2013) Nonequivalent gene expression and copy number alterations in high-grade serous ovarian cancers with BRCA1 and BRCA2 mutations. Clin Cancer Res 19:3474–3484
Esteller M, Silva JM, Dominguez G et al (2000) Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 92:564–569
Cornelis R, Neuhausen S, Arason A et al (1995) Allele loss rate at 17q12-q21 in breast and ovarian tumors from 52 germ line BRCA1-mutation carriers. Genes Chromosom Cancer 13:203–210
Cressman VL, Backlund DC, Hicks EM, Gowen LC, Godfrey V, Koller BH (1999) Mammary tumor formation in p53- and BRCA1-deficient mice. Cell Growth Differ Mol Biol J Am Assoc Cancer Res 10:1–10
Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A (2001) Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 29:418–425
Banerji S, Cibulskis K, Rangel-Escareno C et al (2012) Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486:405–409
Flesken-Nikitin A, Choi K-C, Eng JP, Shmidt EN, Nikitin AY (2003) Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res 63:3459–3463
Miki Y, Swensen J, Shattuck-Eidens D et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66–71
Szabova L, Yin C, Bupp S et al (2012) Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res 72:4141–4153
Perets R, Wyant GA, Muto KW et al (2013) Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in brca;Tp53;pten models. Cancer Cell 24:751–765
Zheng W, Lu JJ, Luo F et al (2000) Ovarian epithelial tumor growth promotion by follicle-stimulating hormone and inhibition of the effect by luteinizing hormone. Gynecol Oncol 76:80–88
Hu Y, Ghosh S, Amleh A et al (2005) Modulation of aromatase expression by BRCA1: a possible link to tissue-specific tumor suppression. Oncogene 24:8343–8348
Acknowledgments
We thank Mamdouh Khalil and the staff of the ANZAC and Kolling Institute animal facilities.
Conflict of Interest
The authors declare that they have no competing interests.
Funding
This work was supported by the National Health and Medical Research Council (Australia) Project Grant (APP1008160) (CMA, DJH, KAW, VMH), Cancer council NSW project grant (VMH), and Cancer Institute NSW Fellowship (VMH).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Upton, D.H., Fuller, E.S., Colvin, E.K. et al. Granulosa Cell-Specific Brca1 Loss Alone or Combined with Trp53 Haploinsufficiency and Transgenic FSH Expression Fails to Induce Ovarian Tumors. HORM CANC 6, 142–152 (2015). https://doi.org/10.1007/s12672-015-0222-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-015-0222-5