
Abstract
Spinocerebellar ataxia 19 (SCA19) represents a rare autosomal dominant genetic disorder resulting in progressive ataxia and cerebellar atrophy. SCA19 is caused by variants in the KCND3 gene, which encodes a voltage-gated potassium channel subunit essential for cerebellar Purkinje cell function. We describe six cases from Chile and Mexico, representing the largest report on SCA19 in Latin America. These cases encompass a range of clinical presentations, highlighting the phenotypic variability within SCA19 from an early-onset, severe disease to a late-onset, slowly progressive condition with normal lifespan. While some patients present with pure ataxia, others also show cognitive impairment, dystonia, and other neurological symptoms. The correlations between specific KCND3 variants and phenotypic outcomes are complex and warrant further investigation. As the genomic landscape of spinocerebellar ataxias evolves, comprehensive genetic testing is becoming pivotal in improving diagnostic accuracy. This study contributes to a better understanding of the clinical spectrum of SCA19, laying the groundwork for further genotype-phenotype correlations and functional studies to elucidate the underlying pathophysiology.


Similar content being viewed by others

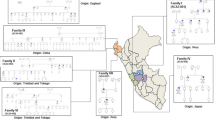
Data Availability
Not applicable
References
Baviera-Muñoz R, et al. Diagnostic efficacy of genetic studies in a series of hereditary cerebellar ataxias in Eastern Spain. Neuro Gen. 2022;8(6):e200038. https://doi.org/10.1212/NXG.0000000000200038.
Clatot J, et al. Inter-regulation of Kv4.3 and voltage-gated sodium channels underlies predisposition to cardiac and neuronal channelopathies. Int J Mol Sci. 2020;21(14):5057. https://doi.org/10.3390/ijms21145057.
Coutelier M, et al. Genetic landscape remodelling in spinocerebellar ataxias: the influence of next-generation sequencing. J Neurol. 2015;262:2382–95. https://doi.org/10.1007/s00415-015-7725-4.
Duarri A, et al. Spinocerebellar ataxia type 19/22 mutations alter heterocomplex Kv4.3 channel function and gating in a dominant manner. Cellul Mol Life Sci: CMLS. 2015;72(17):3387–99. https://doi.org/10.1007/s00018-015-1894-2.
Hsiao C-T, et al. Novel SCA19/22-associated KCND3 mutations disrupt human K V 4.3 protein biosynthesis and channel gating. Hum Mutat. 2019;40 https://doi.org/10.1002/humu.23865.
Huin V, et al. Expanding the phenotype of SCA19/22: Parkinsonism, cognitive impairment and epilepsy. Parkinsonism Relat Disord. 2017;45 https://doi.org/10.1016/j.parkreldis.2017.09.014.
Klockgether T, et al. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019;5(1):24. https://doi.org/10.1038/s41572-019-0074-3.
Kurihara M, et al. Novel de novo KCND3 mutation in a Japanese patient with intellectual disability, cerebellar ataxia, myoclonus, and dystonia. Cerebellum (London, England). 2018;17(2):237–42. https://doi.org/10.1007/s12311-017-0883-4.
Li M, et al. Rare KCND3 loss-of-function mutation associated with the SCA19/22. Front Mol Neurosci. 2022;15:919199. https://doi.org/10.3389/fnmol.2022.919199.
Pollini L, et al. KCND3-related neurological disorders: from old to emerging clinical phenotypes. Int J Mol Sci. 2020;21(16):5802. https://doi.org/10.3390/ijms21165802.
Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Gen Med: official J Am College Med Gen. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Schuermans N, et al. Exome sequencing and multigene panel testing in 1,411 patients with adult-onset neurologic disorders. Neurol Gen. 2023;9(3):e200071. https://doi.org/10.1212/NXG.0000000000200071.
Srivastava S, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Gen Med: official J Am College Med Gen. 2019;21(11):2413–21. https://doi.org/10.1038/s41436-019-0554-6.
Sullivan R, et al. Spinocerebellar ataxia: an update. J Neurol. 2019;266(2):533–44. https://doi.org/10.1007/s00415-018-9076-4.
Teive HAG, et al. The geographic diversity of spinocerebellar ataxias (SCAs) in the Americas: a systematic review. Mov Disord Clin Pract. 2019;6(7):531–40. https://doi.org/10.1002/mdc3.12822.
Zanni G, et al. Novel KCND3 variant underlying nonprogressive congenital ataxia or SCA19/22 disrupt KV4.3 protein expression and K+ currents with variable effects on channel properties. Int J Mol Sci. 2021;22(9):4986. https://doi.org/10.3390/ijms22094986.
Acknowledgements
We thank Professor Laura Jardim for fostering the contact between the clinicians from Chile and Mexico and for initial insights regarding this report.
Funding
This work was supported in part by a Grant “Fondo SAVAL para investigación en la Universidad de Chile” awarded to DAJ.
Author information
Authors and Affiliations
Contributions
DAJ, FM, MLA, JMF, DDOM, and MM performed the clinical assessment of cases.
MLB, DV, and MM conceived the study and analyzed the data.
DAJ and FM drafted the initial version of the manuscript under the supervision of MLB and MM.
All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethical Approval
This study was performed according to the principles of the Declaration of Helsinki. All participants provided informed consent for genetic studies and publication of their clinical information.
Consent for Publication
Participants provided signed informed consent for publication.
Competing Interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Avila-Jaque, D., Martin, F., Bustamante, M.L. et al. The Phenotypic Spectrum of Spinocerebellar Ataxia Type 19 in a Series of Latin American Patients. Cerebellum (2024). https://doi.org/10.1007/s12311-023-01654-x
Accepted:
Published:
DOI: https://doi.org/10.1007/s12311-023-01654-x