
Abstract
Single-cell RNA-seq (scRNA-seq) is a revolutionary technology that allows for the genomic investigation of individual cells in a population, allowing for the discovery of unusual cells associated with cancer and metastasis. ScRNA-seq has been used to discover different types of cancers with poor prognosis and medication resistance such as lung cancer, breast cancer, ovarian cancer, and gastric cancer. Besides, scRNA-seq is a promising method that helps us comprehend the biological features and dynamics of cell development, as well as other disorders. This review gives a concise summary of current scRNA-seq technology. We also explain the main technological steps involved in implementing the technology. We highlight the present applications of scRNA-seq in cancer research, including tumor heterogeneity analysis in lung cancer, breast cancer, and ovarian cancer. In addition, this review elucidates potential applications of scRNA-seq in lineage tracing, personalized medicine, illness prediction, and disease diagnosis, which reveals that scRNA-seq facilitates these events by producing genetic variations on the single-cell level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is a genetic illness that remains a major health risk for humans. As it develops, tumor cells acquire and accumulate oncogenic somatic mutations and non-genetic changes, which may promote unchecked cell growth, invasion, and resistance to treatment. The diagnosis, discovery of new drugs, design of clinical trials, and selection of therapy strategies all rely on an in-depth understanding of the molecular pathways driving carcinogenesis. Cancer genomes and transcriptomes, tumor development, intra-tumor heterogeneity, and treatment resistance are just a few of the areas where our knowledge of cancer biology has been bolstered by next-generation sequencing (NGS) in the last decade. Despite significant progress, most standard studies and datasets are based on bulk samples, which average the molecular features of cells in a tumor sample. The microenvironment of a tumor includes a wide variety of immunological, endothelial, and stromal cells, and is increasingly seen as an integral part of the tumor itself. Evidence is mounting suggesting cells in the tumor's microenvironment, in addition to cancer cells themselves, influence the tumor's biology and clinical behavior. This finding shows that bulk tumor profiling may hide cellular diversity, making it difficult to investigate the unique molecular pathways by which various tumor cell types may contribute to carcinogenesis. Therefore, many cancer researchers have used the newly available single-cell sequencing technologies to comprehend tumors at the level of individual cells to deal with the cell heterogeneity in tumors.
The use of single-cell RNA sequencing (scRNA-seq) is a contemporary NGS approach that facilitates the identification of variations in genetic and protein expression among individual cells. This technology facilitates the acquisition of genetic data from microorganisms at the individual cellular level, thereby enhancing comprehension of the microenvironment. scRNA-seq has emerged as an influential innovation due to the advent of high-throughput sequencing techniques and the development of microfluidics, allowing researchers to obtain high-quality single-cell samples and expose the individuality of every single cell [65]. Traditionally, scientists have analyzed pooled populations of cells in tissues and organs because of the limitation of techniques to thoroughly analyze each cell’s uniqueness [75]. The comparison of bulk-RNA seq and scRNA-seq has been illuminated in Fig. 1.

Comparison of working procedures of a typical single-cell RNA sequencing experiment and traditional bulk RNA analyses. The strengths and weaknesses of each method are also listed. FFPE formalin-fixed paraffin-embedded
Recent technical developments have enabled far better resolution gene expression analysis than previously achievable. The transcriptome at the single-cell level was characterized using scRNA-seq in 2009 [117]. Various techniques enhance our knowledge of the biological information in single cells, including analyzing the genomic DNA sequence, mRNA expression, protein expression, and cellular metabolites. Technological breakthroughs in cell isolation, high-throughput sequencing methods, and data analysis software have made the scRNA-seq application a reality. All these technological advancements indicate that scRNA-seq is becoming an increasingly powerful tool for evaluating developmental, normal, and pathological processes [78].
As a component of NGS, scRNA-seq has a role in exploring cell heterogeneity and uncovering novel genetic traits associated with clinical outcomes [101]. The profiled cellular programs in tumor samples have integrated genetic, epigenetic, and environmental cues, significantly improving our understanding of cancer heterogeneity [11], the cancer microenvironment [107], cancer metastasis [4], and cancer evolution in response to therapy [45]. Human hematopoietic development follows a similar pattern. For ethical reasons, it is difficult to acquire information on the lineage potential and hierarchical connections among early human hematopoietic progenitors [52, 56]. However, scRNA-seq can help to realize fate map** and lineage tracing [127]. The molecular processes and sequence of events by which the ultimate identities of cells are determined during embryogenesis or remodeling may be inferred from the changes in cell states as tissues and organs develop [134]. For example, it may help to understand and control the fates of cells in vivo, anticipate where cancers and other diseases arise, and even recreate the differentiation process of cells [118]. scRNA-seq may be further utilized in inferring disease progression and prognosis in clinical trials [50], and single-cell genomics may reveal significant changes in the percentage and gene expression of cell types during injury and healing [21]. Furthermore, comparing the single-cell transcriptome between novel cell subsets under pathological states and normal cells facilitates the discovery of correlations between gene expression and phenotypes [87].
In general, the popularization of the scRNA-seq approach has been aided by the open sharing of methods, commercialization of technology, and widespread use in biomedical and clinical research. The primary single-cell sequencing technologies and applications, including tumor progression and lineage analysis, are outlined in this overview.
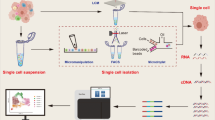
Conventional sequencing involves obscured signals from small cell populations. scRNA-seq is a set of techniques for untargeted measurement of novel genomic, transcriptomic, and proteomic information in individual cells [45]. In scRNA-seq, a single cell's DNA and RNA expression patterns can be determined by NGS, providing a better understanding of how a single cell functions and interacts with its environment. scRNA-seq technologies share common procedures, including isolating a single cell, RNA extraction, reverse transcription, preamplification, and data analysis [124]. The common workflow of scRNA-seq is illustrated in Fig. 2. Data analysis and the advent of cell separation have made it possible to create maps of individual cell types and states at the single-cell level [71].

Pictures of single-cell RNA sequencing (scRNA-seq) experiments. A typical scRNA-seq workflow shares most of the following single-cell isolation by micromanipulation, laser-capture microdissection (LCM) microfluidics-activated cell sorting (FACS), (2) cell lysis, (3) reverse transcription of mRNA to cDNA, (4) cDNA amplification and library preparation, (5) sequencing
The objective of our review is to provide a comprehensive overview of the advancements made in single-cell RNA sequencing (scRNA-seq) technology, encompassing the latest developments in emerging scRNA-seq methodologies. We conducted a comparative analysis of various scRNA-seq techniques to ascertain their respective strengths and limitations. Furthermore, our emphasis was primarily on the utilization of scRNA-seq in the identification of tumor heterogeneity and the characterization of the tumor microenvironment. Moreover, drawing from our independent investigation, our aim is to compile and condense the practical implementation of scRNA-seq in the context of tailored medical treatment, linkage map**, and prognostication of illnesses.
Single-Cell RNA Sequencing Technologies
Isolation of Single Cells (Capturing Single Cells)
The entire procedure of single-cell RNA sequencing starts with the isolation of single cells from tissues. Single cells can be separated mechanically and enzymatically into different subpopulations without affecting their viability [101]. Micromanipulation is one of the traditional methods of single-cell isolation, and individual cells are isolated from microorganisms, solid histological tissue, or embryonic stem cells with a micropipette or forceps and microscope [48]. However, due to the difficulty of its operation, the labor involved, and the high potential for mechanical damage to targeted cells, this microscopic control system is rarely used [147].
Laser capture microdissection (LCM) focuses a laser beam on cells of interest and attaches them to membranes [38]. LCM is a powerful technique for isolating cell populations or regions of interest inside a tissue. The specified region is excised by a laser using direct viewing and can then be processed for a number of subsequent investigations. From DNA and RNA sequencing through mass spectrometry, this technology has been widely employed in the research of liver disorders. However, LCM has significant limitations. Due to the cost of the microdissection apparatus, this is an expensive technology. The yield and purity of DNA, RNA, and proteins extracted from improperly preserved and processed tissue samples may not be suitable for further analysis. The absence of a coverslip on the tissue slide may impede the accurate identification of the area to be microdissected. To preserve the integrity of the molecule of interest, optimum tissue preservation, precise tissue sectioning, and staining method optimization is necessary [99].
Initially labelingsing fluorescent monoclonal antibodies, the fluorescence-activated cell sorting (FACS) method primarily relies on flow cytometry. Negative selection may also be used to undetained populations [110]. Target cells are identified and separated using this method's analysis of their molecular markers and physical traits (such as cell size and fluorescence scattering) [55]. Nonetheless, FACS systems continue to be rather restricted in several applications. Because cells must be suspended, tissues must be dissociated, resulting in the loss of cellular activities, cell–cell connections, and tissue architecture [59]. Subpopulations with comparable marker expressions are difficult to distinguish, and the overlap of emission spectra amongst fluorochromes may enhance background noise, making low-intensity samples unidentifiable. Furthermore, as established by trypan blue exclusion and necrosis/apoptosis experiments for Chinese Hamster Ovary (CHO) cells and a human monocytic cell line (THP1), FACS sorting may have non-negligible impacts on cell survival [90].
The smallest sample volume for FACS systems is several hundred microliters to milliliters [55]. This is because lengthy tube sections often result in huge dead volumes, making it impossible to utilize uncommon samples, particularly when the full sample has to be evaluated [76]. Finally, because FACS systems generally have a complex system made up of non-disposable components, the sterile operation is challenging to achieve. However, such limits are not always present or significant, depending on the instrument, process parameters (such as speed, laser type, etc.), cell type, and application [47].
A microfluidic chip with microchannels that tightly control liquid flow can be employed during cell suspension preparation to achieve low sample consumption with microfluidic methods [97]. In the past few years, microfluidic procedures have gained popularity and are gradually replacing FACS and other previously used techniques for isolating cells.
Single-cell methods generally have their own unique advantages. The micromanipulator ensures the accurate collection of cells and minimizes cell damage. LCM collects spatial information and captures single cells from solid samples. FACS can focus on cells of interest. Microfluidic methods require a low volume of reagents, which makes them cost-effective. Understanding these single-cell isolation methods helps in choosing appropriate techniques for various cells from diverse targeted bulk tissues.
Following separation, identifying single-cell sequencing data is the most difficult aspect of this approach. The plate micro-reaction system and combinatorial index technology in single-cell barcode technology solve this problem and increase single-cell detection throughput by 100-fold. The plate-like micro-reaction system, for example, contains single cells, functional beads, and reverse transcription. Oligonucleotides such as primers, cell barcodes, and unique molecular identifiers (UMI), as well as 5' to 3' poly (dT), are used to modify the surface of functional beads. The UMIs target each molecule in a cell, unlike primers, polymeric (dT) fragments, and cellular barcodes, which are unique in each micro-reaction. A UMI can mark the DNA genome, transcriptome, immunological profile, and proteome [117].
Microsphere-based technologies like Drop-seq, Seq-Well, and inDrop are used to barcode specific molecules in single cells. After barcoding targeted molecules, a critical next step is the pre-amplification of transcripts by reverse transcription. Therefore, all RNA sequences are converted to cDNA for further amplification and sequencing. Finally, the sequencing results are integrated and analyzed by computers at different levels.
Specific Protocols for Single-Cell RNA Sequencing
The first whole-genome amplification (WGA) technique developed was PCR-based amplification using Degenerate Oligonucleotide-Primed PCR (DOP-PCR) for single-cell analysis. This method is biased in amplification and PCR efficiency variations and has high error rates due to the thermostable polymerase and degenerate oligonucleotides used for primers [16]. The polymerase used is Φ29, with primary properties of excellent processivity and strand displacement activity. Since these enhancements improve genome coverage, there are fewer false-positive results. A proportionally more significant number of first-increased loci are overrepresented following exponential amplification,this overrepresentation is amplified further by greater fold amplification. Certain overrepresentation areas may have a systematic or stochastic bias [148]. Φ29 polymerase activity generates a tiny amount of chimeric sequence side products, which may be decreased by endonuclease treatment, allowing for the physical separation of amplicons through debranching [16].
PicoPLEX and multiple annealing and loo**-based amplification cycles (MALBAC) integrate the advantages of the first two methods mentioned above. Both MDA and MALBAC can successfully amplify genomes from single cells. However, in both procedures, preventing the amplification of extraneous contaminating DNA is difficult when performed in microlitre reaction volumes in a tube. Additionally, a prior investigation discovered that MDA provides significantly greater genome coverage than MALBAC (72%) [143]. scRNA-seq analyses in TNBC have indicated subclonal heterogeneity and severe disease stages. Five distinct cell clusters were detected by scRNA-seq in untreated early TNBC tumors. Patients with TNBC have a high incidence of somatic mutations, frequent TP53 mutations (83%), and complex aneuploid rearrangements (80%), resulting in considerable intra-tumor heterogeneity [5].
On the basis of single-nucleus genome sequencing, the mutation rate in ER+ tumors is comparable to that in normal cells, and TNBCs exhibit sustained intra-tumoral variety [142]. According to marker genes and clustering, immune cells, and endothelial cells exhibit unique gene expression profiles. For instance, the majority of tumor epithelial cells exhibit luminal and luminal progenitor markers, though a minority express myoepithelial cell markers such as ACTA2 and TAGLN [19]. In addition, macrophages are more prevalent in CD45-unselected malignancies and CD45-selected tumors [1, 83]. In addition, the current research provides a genetic analysis of the luminal progenitors and hormone-sensing luminal cells, the two main subpopulations of the mammary luminal compartments. Overexpression of p53 and several activated p53 target genes is shown to be present in luminal progenitor cells, which are influenced by HGF/Met signaling in the mammary epithelium, as shown by the current research. Growing data indicate that p53 regulates stem/progenitor cell self-renewal, differentiation, and plasticity in both embryonic and adult tissues [60, 114]. In the absence of p53, luminal progenitors were amplified and their proliferative and self-renewal capacities were activated in vivo. Nonetheless, it had no effect on their inherent identity. Furthermore, luminal progenitors missing p53 and stimulated in vitro by HGF were unable to establish basal-specific features, demonstrating that p53 governs the adaptive nature of luminal progenitors after Met activation[20, 122]. Previous research has shown that the lack of p53 in the mammary epithelium facilitates the process of self-renewal and symmetric division of stem cells. The aforementioned process might explain why luminal progenitors lacking p53 have a restricted ability to undertake a luminal-to-basal transition after Met activation (64). Furthermore, another research found LGR5 activation in myoepithelial cells of regenerating mammary ducts[18]. In summary, luminal, luminal progenitor markers, and myoepithelial cell markers are significant in composing the genetic heterogeneity of the oncogenic pathway of breast cancer.
Intra-tumoral heterogeneity plays a critical role in the development of TNBC and contributes to the disease's resistance to treatment, metastasis, and poor prognosis. Indeed, a worse prognosis is expected for TNBC patients in whom even a fraction of breast cancer cells is not eradicated [5]. The whole single-cell exomes of 20 TNBC patients receiving neoadjuvant chemotherapy (NAC) were sequenced in a prior study. Genotypes associated with resistance to chemotherapy in TNBC were found to be both prevalent and adaptively selected by NAC [64]. CDH1 target profiles may provide a link between E-cadherin loss and the mesenchymal phenotype acquired by tumor cells in response to NAC [80]. In certain cases, resistance genes are present in a minute proportion of primed tumor cells.
Below, we highlight some important clinical implications of the scRNA-seq findings regarding tumor heterogeneity. Patients with TNBC can benefit from chemotherapy if the tumor mass contains certain genotypes, which can be diagnosed by detecting chemoresistant clones prior to initiating NAC treatment [84]. Such subgroups of TNBC may have prognostic relevance beyond histological approaches for determining clonal extinguishment or persistence as a result of genetic clones. Hence, it is important to consider all treatment options, including potential alternatives, before choosing one[31]. Chemoresistant phenotypes can be overcome, for instance, by employing therapeutic techniques such as targeting epithelial-mesenchymal transition (EMT) signaling or decreasing hypoxia with HIF-1 inhibitors [84]. Signaling pathways related to HIF-α are depicted in Fig. 4b.
Using scRNA-seq technology, a group of researchers developed a method for identifying and analyzing single cancer cells in the operation of micro-metastasis of breast cancer in human patient-derived xenograft (PDX) models[40]. These assays contributed to the comprehension of the change in gene expression that occurs during micrometastasis. Furthermore, researchers have identified mitochondrial oxidative phosphorylation (OXPHOS) as a crucial process in metastatic seeding [15]. In the preponderance of cases (e.g., breast tumors), this transcription factor does not promote clonal growth at the primary site, preventing positive selection. In contrast, RB1 loss and other oncogenic alterations that enhance OXPHOS (see previous subsection) promote both primary tumor growth and metastasis and therefore may be selected for during clonal evolution. This theory is further supported by the fact that slow-growing tumors, such as invasive lobular carcinoma (ILC), are highly metastatic. Moreover, inhibitors of the PI3K pathway inhibit tumor growth but reprogram mitochondrial trafficking, OXPHOS, and motility[81]. Contrary to the well-known stimulatory effect of hypoxia on tumor invasion, increased invasion as a result of elevated OXPHOS may manifest. However, tumor cells with elevated OXPHOS may be better able to adapt to hypoxia and migrate away from hypoxic regions [81]. In addition, another study has demonstrated the significance of OXPHOS in the development of malignancy. Tumor growth requires active mitochondrial function and oxidative phosphorylation (OXPHOS). Recent research indicates that the absence of the retinoblastoma (RB1) tumor suppressor increases mitochondrial protein translation (MPT) and OXPHOS in breast cancer [144]. Increased OXPHOS can increase anabolic metabolism and cell proliferation, as well as cancer stemness and metastasis. STAT3, FER/FER-T, and CHCHD2 are also involved in OXPHOS in mitochondria. Scientists postulate that RB1 loss is a prototypical oncogenic change that promotes OXPHOS, that aggressive tumors acquire lethal combinations of oncogenes and tumor suppressors that stimulate anabolism as opposed to OXPHOS, and that targeting both metabolic pathways would be therapeutic [69].
Oxidative phosphorylation may aid in metastasis [37]. In addition, this technology can be used for regulating cell fate, predicting tumor origin, and remodeling cell differentiation in vitro. In recent years, the development of high-throughput scRNA-seq technology has provided a complete transcriptional map of adult tissues and embryos based on millions of individual cells [127].
Currently, single-cell genomics methods allow for an objective diagnosis of cell identity by collecting thousands of gene expression measures while maintaining the cellular resolution required for proper lineage remodeling [63]. In addition to RNA capture, genomic technologies are currently used to measure the transcriptome, epigenome, and proteome. Advances in computer technology have enabled these data to be visualized and interpreted, resulting in new insight into biological processes and the identification of new cell types. Especially relevant to developmental biologists, single-cell data can infer the differentiation routes of cells [63]. Consequently, these approaches can help in identifying transcription factors associated with particular branches of differentiation.
In recent years, a large number of analyses of the single-cell transcriptomes of develo** tissues and organs, including the inner ear, have suggested that scRNA-seq may be used to identify slight transcriptome differences between various cell types [30]. At E16.5-E18.5 d of mouse embryo development, epithelial cells isolated from the airways begin to differentiate towards the distal airway apex. In addition, they mainly differentiate into type I alveolar epithelial cells (AT1), which have a role in gas exchange, and type II alveolar epithelial cells (AT2), which produce surfactants. Quake et al. analyzed 80 single cells in the lung tissue of an E18.5 d mouse and found five cell clusters, four of which were composed of AT1, AT2, ciliated epithelium, and Clara cells. The fifth subgroup not only expressed AT1 and AT2 marker genes but also showed that progenitor cells with the characteristics of AT1 and AT2 may exist [123], which indicated that single-cell-resolution transcriptome sequencing may effectively promote the study of developmental biology [85]. By using the single-cell-binding RNA-seq method, the mechanism of mouse organogenesis has also been studied at single-cell resolution, providing a new holistic view of animal development. The above findings may also constitute a critical step towards understanding pleiotropic developmental disorders at the biological level and allow for a detailed study of the subtle effects of genes and regulatory sequences during development [14].
Owing to genome-scale single-cell analysis, our understanding has shifted over the past few years from an animation of discontinuous changes to a dynamic state driven by data [63]. These features not only predict the differentiation dynamics of thousands of genes but also infer new cell transitions and end states, interactions with the cell cycle, and the ability to "cycle between cell states." To draw an authentic lineage, the blood relationship of a cell needs to be understood. Therefore, future fate maps can provide powerful tools for tracing and reconstructing lineage relationships [125].
Single-Cell RNA Sequencing and Disease Prediction
scRNA-seq can be used to identify novel cell types, analyze stem cell differentiation and single-cell trajectory construction, and compare healthy and disease-related tissues at a single-cell level. Recent advances in cardiovascular research demonstrate the importance of these applications, as evidenced by the generation of cell atlases for mammalian cardiovascular development and stem cell differentiation. To develop effective patient-specific therapeutic strategies, single-cell omics can be used in cardiovascular precision medicine to characterize responses of individual cells to drugs or environmental stimuli. Single-cell transcriptomics has also revealed cell subpopulations in adult mouse hearts. scRNA-seq of non-cardiomyocyte cells from undamaged adult mouse hearts showed an extensive intercellular communication network involving endothelial cells, cardiac fibroblasts, and immune cells.
Recent developments in scRNA-seq enable cell types and their significance in health and illness to be separated from complex tissues and host compartments[51]. This approach has altered our capability to understand the immune system in unprecedented detail, particularly with regards to processes like hematopoiesis, carcinogenesis, the lymph node compartment, and responses to microbial ligands [42, 102]. The ever-growing amount of data has given rise to analytical techniques like computational deconvolution procedures that forecast the precise compositions of cell types from large-scale gene expression data. These algorithms, however, are reliant on preexisting knowledge or particular datasets that are part of the experimental systems[100, 109].
Cardiac Homeostasis and Disease
scRNA-seq analysis of adult mouse hearts indicated a substantial intercellular communication network. Analysis of adult myocardium at homeostasis and after ischaemic injury using SORT sequences has revealed various known cell types. Additionally, Gladkar et al. suggested that ischemia–reperfusion induces cell subsets of many different cell types and that Ckap4 is elevated in activated fibroblasts [44]. To identify gene modules of hypertrophic cardiomyocytes, the Smart-seq2 platform has been employed to conduct scRNA-seq of adult cardiomyocytes at the single-cell level [96]. Moreover, droplet-based scRNA-seq was performed on endothelial cells to explore the endothelial heterogeneity of neovascularization after myocardial infarction [105], and plasmalemma vesicle-associated protein (Plvap) was identified as a novel marker of cardiac neovascularization. The combined data show that the intact single-cell properties of cardiac ECs can help to block new angiogenesis within the myocardium. This property may help to identify new therapeutic targets for heart diseases [105].scRNA-seq has also been used to evaluate gene expression in cardiomyocytes isolated from human embryos [22]. The developmental pathway of the human heart has been mapped using single-cell tagged reverse transcription sequencing with approximately 4,000 cardiac cells from 18 human embryos. Gene expression in both cardiomyocytes and fibroblasts was found to be gradually altered throughout development. Using human and mouse scRNA-seq data, cardiomyocytes, endothelial cells, fibroblasts, and epicardial cells were compared and showed the highest transcriptional similarity in cells from human and mouse cardiomyocytes [22]. The results demonstrate higher expression of THY1 in human fibroblasts,expression of CFB and ITLN1 was lower in mouse cardiomyocytes. However, Icam2 was only expressed in the endothelial cells of mice, whereas Rnf213 was expressed exclusively in the mouse epicardium, showing different gene expression patterns. Extensive work is underway to develop a single-scale atlas of the adult heart [112]. In general, scRNA-seq plays a vital role in understanding cellular variation and in identifying critical transcriptional processes associated with cardiac development and disease.
Coronary Vessel and Disease
The formation of coronary arteries is a dynamic and regular development process, and scRNA-seq technology plays a crucial role in explaining these dynamic cellular transformations. For example, combining scRNA-seq with genetically tracked genes in mice revealed that coronary arteries derive from the pre-artery, as they develop through a specialized population of veins [116]. scRNA-seq has also been used to characterize the cellular status and fate in significant vascular lesions. Cells such as vascular endothelial cells, vascular smooth muscle cells (VSMCs), and immune cells all display different plasticity and sensitivity to extracellular signals [79, 113]. Although VSMCs are terminally differentiated cells, they are highly plastic, and the use of single-cell analysis helps to delimit their ability to differentiate into fibroblast-like cell types. Additionally, single-cell analysis can help in determining the differentiation status of VSMCs in atherosclerosis [141].
Challenges and Future Perspectives
The goal of scRNA-seq is to bring genomic studies to the cellular level, providing a new perspective on our understanding of genetics. These tools open up a new field of research to analyze the role of individual cells in ecosystems and biological biology. In recent years, there has been significant progress in obtaining high-quality single-cell data, enabling the discovery of new biological phenomena that conventional bulk genome queries cannot detect. Figure 1 illustrates the difference between traditional bulk RNA sequencing and single-cell RNA sequencing. The latter is a powerful tool to explore the complexity of cancer and its tumor environment to lead to personalized therapy. It also offers novel perspectives to investigate more ways of diagnosing and treating common diseases. In addition, scRNA-seq has been used to track cell development and stem cell research. However, this technology still faces several challenges.
Cell integrity and viability are critical for subsequent single-cell analysis, requiring that individual cells be separated quickly and accurately, with minimal damage to the cells. Therefore, to obtain high-quality scRNA-seq data, there are four major technical problems that need to be overcome: physical isolation of single cells; gene amplification of a single cell to obtain sufficient substances for further analysis; economical and efficient genome analysis queries to identify variants that validate research hypotheses; and analysis of introduced errors and biases. To maximize the quality of single-cell data and ensure that the signal is not affected by technical noise, each variable must be carefully considered when conducting single-cell experiments. The high cost of scRNA-seq is also a non-negligible problem. Although existing detection systems have brought the cost of sequencing each cell type to an acceptable level, the overall cost is still prohibitive because thousands of cells may need to be analyzed. Reducing the cost of sequencing will drive the implementation of scRNA-seq in oncology and other fields. Temporal and spatial measurement of the molecular profile by using in situ sequencing and real-time sequencing, as well as in vivo analysis of the DNA and RNA from single cells, have been developed, but these methods need enhanced sensitivity, coverage, accessibility, and cost reduction [93]. In addition, in short-read RNA-seq technology, errors and biases are mainly generated during the preparation of sequence libraries and assembly of short reads. These methods have difficulty in accurately identifying multiple different subtypes of a specific gene. Read coverage and sequencing depth must also be increased to overcome insufficient read length. Long-read RNA-seq technology overcomes the shortcomings of template expansion, reduces the false-positive rate, and can identify longer non-annotated transcripts, thus addressing the limitations of traditional short-read methods [32, 111]. However, this method is associated with problems of reduced throughput, high cost, and high sequence error, especially insertion and deletion. To reduce random errors, PacBio circular consensus sequencing (CCS) was developed, which can repeatedly read out a molecule over multiple cycles, increasing the depth of the sequence. However, this method also reduces the identification of specific isomers. In addition, the sensitivity of long-read sequencing to identify differentially expressed genes is lower than that of short-read sequencing [119]. Accordingly, a more complete and precise analysis can be obtained by hybridization of long-read and short-read sequencing [115].
The integration of artificial intelligence (AI) is increasingly being recognized as a pivotal resource in life science and healthcare research. Despite being a nascent field, research on artificial intelligence is transforming our comprehension and outlook on the scientific domain. According to recent estimates provided by the European Commission, AI-based medical startups receive approximately 13% of global venture capital investments, which amounts to €5 billion [7]. This dedication demonstrates the interest in the potential of artificial intelligence to improve healthcare. Precision medicine is a cutting-edge approach to treating disease. The generation of genomic Big Data (i.e., Big Data derived from genome sequencing), the gathering of clinical data, and the development of bioinformatics over the past ten years have made it possible to pinpoint the genetic factors underlying the onset and progression of diseases and to support clinical patient management. Personalised therapeutic treatments are still scarce despite the high expectations [43]. The inadequacy of AI infrastructure and models to support the ongoing production of large-scale genomic data represents a significant failure. Hence, the challenge lies in comprehending the heterogeneous information encompassed within this data [94, 121].
The high-throughput profiling of all RNA species generated by cells is known as transcriptomics. Transcriptomics has shown rapid growth in recent years among genetic Big Data [136]. RNA sequencing, also known as RNA-seq, is a technique that enables the characterization of dynamic biological processes that are currently active in a population of cells or in individual cells. The evaluation of the intricacy of these profiles has the potential to facilitate the identification of novel biomarkers and therapeutic targets. RNA-seq screenings are increasingly being integrated into precision medicine trials [120], AI mining of these data is thus required to determine novel clinical targets.
The increasing demand for artificial intelligence (AI) in the field of precision oncology will necessitate the presence of medical professionals and specialists who possess the ability to effectively interpret outcomes and make informed decisions regarding precision therapeutic interventions. Additionally, these individuals will play an active role in the formulation and implementation of learning strategies. Given these circumstances, a highly accurate oncology system powered by artificial intelligence will be readily accessible as needed.
In conclusion, the development of scRNA-seq and its continuous methodological improvement have led to important medical discoveries. scRNA-seq technology has been widely used in many aspects, including early mammalian embryology, tissue and organ development, the immune system, cancer, microorganism, infectious disease, and stem cell research. High-throughput scRNA-seq techniques not only reveal cellular heterogeneity during disease progression and in the immune microenvironment but also contribute to further studies on disease turnover, thus guiding early clinical diagnosis, targeted treatment, curative monitoring, and prognostic evaluation of diseases. It is expected that scRNA-seq will soon achieve 100% coverage and accuracy. Due to the rapid development of multi-omics scRNA-seq technology, single-cell genome, transcriptome, epigenome, and proteome analyses are expected to be performed simultaneously. In addition, spatially resolved transcription techniques can determine the spatial organization of cells in tissues, revolutionizing the study of tissue function and disease pathogenesis [3]. In fact, spatially resolved transcriptomic research has become a new field. Moreover, the integration of scRNA-seq and AI will become a trend in NGS. scRNA-seq will become an indispensable technology to help in more easily treating diseases and exploring the life sciences (Fig. 5).

Illustrations of single-cell RNA sequencing applications in different fields. (1) tumor heterogeneity, (2) tumor microenvironment, (3) lineage tracing, (4) personalized therapy, and (5) disease prediction
References
Adams, S., Gray, R. J., Demaria, S., Goldstein, L., Perez, E. A., Shulman, L. N., Martino, S., Wang, M., Jones, V. E., Saphner, T. J., Wolff, A. C., Wood, W. C., Davidson, N. E., Sledge, G. W., Sparano, J. A., & Badve, S. S. (2014). Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. Journal of Clinical Oncology, 32(27), 2959–2966. https://doi.org/10.1200/JCO.2013.55.0491
Andor, N., Lau, B. T., Catalanotti, C., Sathe, A., Kubit, M., Chen, J., Blaj, C., Cherry, A., Bangs, C. D., Grimes, S. M., Suarez, C. J., & Ji, H. P. (2020). Joint single cell DNA-seq and RNA-seq of gastric cancer cell lines reveals rules of in vitro evolution. NAR Genomics and Bioinformatics. https://doi.org/10.1093/nargab/lqaa016
Antipov, D., Korobeynikov, A., McLean, J. S., & Pevzner, P. A. (2016). HybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics, 32(7), 1009–1015. https://doi.org/10.1093/bioinformatics/btv688
Arvanitis, C. D., Ferraro, G. B., & Jain, R. K. (2020). The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nature Reviews Cancer, 20(1), 26–41. https://doi.org/10.1038/s41568-019-0205-x
Baslan, T., & Hicks, J. (2017). Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nature Reviews Cancer, 17(9), 557–569. https://doi.org/10.1038/nrc.2017.58
Bass, A. J., Thorsson, V., Shmulevich, I., Reynolds, S. M., Miller, M., Bernard, B., Hinoue, T., Laird, P. W., Curtis, C., Shen, H., Weisenberger, D. J., Schultz, N., Shen, R., Weinhold, N., Kelsen, D. P., Bowlby, R., Chu, A., Kasaian, K., Mungall, A. J., … Liu, J. (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature, 513(7517), 202–209. https://doi.org/10.1038/nature13480
Bhinder, B., Gilvary, C., Madhukar, N. S., & Elemento, O. (2021). Artificial intelligence in cancer research and precision medicine. Cancer Discovery, 11(4), 900–915. https://doi.org/10.1158/2159-8290.CD-21-0090
Blagodatskikh, K. A., Kramarov, V. M., Barsova, E. V., Garkovenko, A. V., Shcherbo, D. S., Shelenkov, A. A., Ustinova, V. V., Tokarenko, M. R., Baker, S. C., Kramarova, T. V., & Ignatov, K. B. (2017). Improved DOP-PCR (iDOP-PCR): A robust and simple WGA method for efficient amplification of low copy number genomic DNA. PLoS ONE. https://doi.org/10.1371/journal.pone.0184507
Bowtell, D. D., Böhm, S., Ahmed, A. A., Aspuria, P. J., Bast, R. C., Beral, V., Berek, J. S., Birrer, M. J., Blagden, S., Bookman, M. A., Brenton, J. D., Chiappinelli, K. B., Martins, F. C., Coukos, G., Drapkin, R., Edmondson, R., Fotopoulou, C., Gabra, H., Galon, J., … Balkwill, F. R. (2015). Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nature Reviews Cancer 15(11), 668–679. https://doi.org/10.1038/nrc4019
Buenrostro, J. D., Wu, B., Litzenburger, U. M., Ruff, D., Gonzales, M. L., Snyder, M. P., Chang, H. Y., & Greenleaf, W. J. (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature, 523(7561), 486–490. https://doi.org/10.1038/nature14590
Caiado, F., Silva-Santos, B., & Norell, H. (2016). Intra-tumour heterogeneity – going beyond genetics. FEBS Journal. https://doi.org/10.1111/febs.13705
Calon, A., Lonardo, E., Berenguer-Llergo, A., Espinet, E., Hernando-Momblona, X., Iglesias, M., Sevillano, M., Palomo-Ponce, S., Tauriello, D. V. F., Byrom, D., Cortina, C., Morral, C., Barceló, C., Tosi, S., Riera, A., Attolini, C. S. O., Rossell, D., Sancho, E., & Batlle, E. (2015). Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nature Genetics, 47(4), 320–329. https://doi.org/10.1038/ng.3225
Cao, J., Packer, J. S., Ramani, V., Cusanovich, D. A., Huynh, C., Daza, R., Qiu, X., Lee, C., Furlan, S. N., Steemers, F. J., Adey, A., Waterston, R. H., Trapnell, C., & Shendure, J. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science, 357(6352), 661–667. https://doi.org/10.1126/science.aam8940
Cao, J., Spielmann, M., Qiu, X., Huang, X., Ibrahim, D. M., Hill, A. J., Zhang, F., Mundlos, S., Christiansen, L., Steemers, F. J., Trapnell, C., & Shendure, J. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature, 566(7745), 496–502. https://doi.org/10.1038/s41586-019-0969-x
Chen, D., Wang, Y., Manakkat Vijay, G. K., Fu, S., Nash, C. W., Xu, D., He, D., Salomonis, N., Singh, H., & Xu, H. (2021). Coupled analysis of transcriptome and BCR mutations reveals role of OXPHOS in affinity maturation. Nature Immunology, 22(7), 904–913. https://doi.org/10.1038/s41590-021-00936-y
Chen, M., Song, P., Zou, D., Hu, X., Zhao, S., Gao, S., & Ling, F. (2014). Comparison of multiple displacement amplification (MDA) and multiple annealing and loo**-based amplification cycles (MALBAC) in single-cell sequencing. PLoS ONE. https://doi.org/10.1371/journal.pone.0114520
Chen, W., Zheng, R., Baade, P. D., Zhang, S., Zeng, H., Bray, F., Jemal, A., Yu, X. Q., & He, J. (2016). Cancer statistics in China, 2015. CA: A Cancer Journal for Clinicians, 66(2), 115–132. https://doi.org/10.3322/caac.21338
Chiche, A., Di-Cicco, A., Sesma-Sanz, L., Bresson, L., De La Grange, P., Glukhova, M. A., Faraldo, M. M., & Deugnier, M. A. (2019). P53 controls the plasticity of mammary luminal progenitor cells downstream of Met signaling. Breast Cancer Research. https://doi.org/10.1186/s13058-019-1101-8
Chung, W., Eum, H. H., Lee, H. O., Lee, K. M., Lee, H. B., Kim, K. T., Ryu, H. S., Kim, S., Lee, J. E., Park, Y. H., Kan, Z., Han, W., & Park, W. Y. (2017). Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nature Communications. https://doi.org/10.1038/ncomms15081
Cicalese, A., Bonizzi, G., Pasi, C. E., Faretta, M., Ronzoni, S., Giulini, B., Brisken, C., Minucci, S., Di Fiore, P. P., & Pelicci, P. G. (2009). The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell, 138(6), 1083–1095. https://doi.org/10.1016/j.cell.2009.06.048
Conway, B. R., O’Sullivan, E. D., Cairns, C., O’Sullivan, J., Simpson, D. J., Salzano, A., Connor, K., Ding, P., Humphries, D., Stewart, K., Teenan, O., Pius, R., Henderson, N. C., Bénézech, C., Ramachandran, P., Ferenbach, D., Hughes, J., Chandra, T., & Denby, L. (2020). Kidney single-cell atlas reveals myeloid heterogeneity in progression and regression of kidney disease. Journal of the American Society of Nephrology, 31(12), 2833–2854. https://doi.org/10.1681/ASN.2020060806
Cui, Y., Zheng, Y., Liu, X., Yan, L., Fan, X., Yong, J., Hu, Y., Dong, J., Li, Q., Wu, X., Gao, S., Li, J., Wen, L., Qiao, J., & Tang, F. (2019). Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Reports, 26(7), 1934-1950.e5. https://doi.org/10.1016/j.celrep.2019.01.079
Cusanovich, D. A., Daza, R., Adey, A., Pliner, H. A., Christiansen, L., Gunderson, K. L., Steemers, F. J., Trapnell, C., & Shendure, J. (2015). Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science, 348(6237), 910–914. https://doi.org/10.1126/science.aab1601
Dagogo-Jack, I., & Shaw, A. T. (2018). Tumour heterogeneity and resistance to cancer therapies. Nature Reviews Clinical Oncology, 15(2), 81–94. https://doi.org/10.1038/nrclinonc.2017.166
Davis, R. T., Blake, K., Ma, D., Gabra, M. B. I., Hernandez, G. A., Phung, A. T., Yang, Y., Maurer, D., Lefebvre, A. E. Y. T., Alshetaiwi, H., **ao, Z., Liu, J., Locasale, J. W., Digman, M. A., Mjolsness, E., Kong, M., Werb, Z., & Lawson, D. A. (2020). Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nature Cell Biology, 22(3), 310–320. https://doi.org/10.1038/s41556-020-0477-0
Ding, S., Chen, X., & Shen, K. (2020). Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Communications, 40(8), 329–344. https://doi.org/10.1002/cac2.12078
Dominguez, C., Tsang, K. Y., & Palena, C. (2016). Short-term EGFR blockade enhances immune-mediated cytotoxicity of EGFR mutant lung cancer cells: Rationale for combination therapies. Cell Death and Disease. https://doi.org/10.1038/cddis.2016.297
Du, X., Cheng, Z., Wang, Y. H., Guo, Z. H., Zhang, S. Q., Hu, J. K., & Zhou, Z. G. (2014). Role of Notch signaling pathway in gastric cancer: A meta-analysis of the literature. World Journal of Gastroenterology, 27, 9191–9199. https://doi.org/10.3748/wjg.v20.i27.9191
Duan, Z., Foster, R., Bell, D. A., Mahoney, J., Wolak, K., Vaidya, A., Hampel, C., Lee, H., & van Seiden, M. (2006). Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clinical Cancer Research, 12(17), 5055–5063. https://doi.org/10.1158/1078-0432.CCR-06-0861
Durruthy-Durruthy, R., Gottlieb, A., Hartman, B. H., Waldhaus, J., Laske, R. D., Altman, R., & Heller, S. (2014). Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell, 157(4), 964–978. https://doi.org/10.1016/j.cell.2014.03.036
Elion, D. L., & Cook, R. S. (2018). Genetic and phenotypic diversification of heterogeneous tumor populations. Trends in Molecular Medicine, 24(8), 655–656. https://doi.org/10.1016/j.molmed.2018.06.003
Engström, P. G., Steijger, T., Sipos, B., Grant, G. R., Kahles, A., Rätsch, G., Goldman, N., Hubbard, T. J., Harrow, J., Guigó, R., Bertone, P., Alioto, T., Behr, J., Bohnert, R., Campagna, D., Davis, C. A., Dobin, A., Gingeras, T. R., Jean, G., … Zeller, G. (2013). Systematic evaluation of spliced alignment programs for RNA-seq data. Nature Methods, 10(12), 1185–1191. https://doi.org/10.1038/nmeth.2722
Fan, H. C., Fu, G. K., & Fodor, S. P. A. (2015). Combinatorial labeling of single cells for gene expression cytometry. Science. https://doi.org/10.1126/science.1258367
Ferlay, J., Soerjomataram, I., Dikshit, R., Eser, S., Mathers, C., Rebelo, M., Parkin, D. M., Forman, D., & Bray, F. (2015). Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. International Journal of Cancer, 136(5), E359–E386. https://doi.org/10.1002/ijc.29210
Ferlay, J., Steliarova-Foucher, E., Lortet-Tieulent, J., Rosso, S., Coebergh, J. W. W., Comber, H., Forman, D., & Bray, F. (2013). Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. European Journal of Cancer, 49(6), 1374–1403. https://doi.org/10.1016/j.ejca.2012.12.027
Finotello, F., & Eduati, F. (2018). Multi-omics profiling of the tumor microenvironment: Paving the way to precision immuno-oncology. Frontiers in Oncology. https://doi.org/10.3389/fonc.2018.00430
Fletcher, R. B., Das, D., & Ngai, J. (2018). Creating Lineage Trajectory Maps Via Integration of Single-Cell RNA-Sequencing and Lineage Tracing: Integrating transgenic lineage tracing and single-cell RNA-sequencing is a robust approach for map** developmental lineage trajectories and cell fate changes. BioEssays. https://doi.org/10.1002/bies.201800056
Foley, J. W., Zhu, C., Jolivet, P., Zhu, S. X., Lu, P., Meaney, M. J., & West, R. B. (2019). Gene expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. Genome Research, 29(11), 1816–1825. https://doi.org/10.1101/gr.234807.118
Gainor, J. F., Shaw, A. T., van Sequist, L., Fu, X., Azzoli, C. G., Piotrowska, Z., Huynh, T. G., Zhao, L., Fulton, L., Schultz, K. R., Howe, E., Farago, A. F., Sullivan, R. J., Stone, J. R., Digumarthy, S., Moran, T., Hata, A. N., Yagi, Y., Yeap, B. Y., … Mino-Kenudson, M. (2016). EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: A retrospective analysis. Clinical Cancer Research, 22(18), 4585–4593. https://doi.org/10.1158/1078-0432.CCR-15-3101
Garcia-Recio, S., Thennavan, A., East, M. P., Parker, J. S., Cejalvo, J. M., Garay, J. P., Hollern, D. P., He, X., Mott, K. R., Galván, P., Fan, C., Selitsky, S. R., Coffey, A. R., Marron, D., Brasó-Maristany, F., Burgués, O., Albanell, J., Rojo, F., Lluch, A., … Perou, C. M. (2020). FGFR4 regulates tumor subtype differentiation in luminal breast cancer and metastatic disease. Journal of Clinical Investigation, 130(9), 4871–4887. https://doi.org/10.1172/JCI130323
Gierahn, T. M., Wadsworth, M. H., Hughes, T. K., Bryson, B. D., Butler, A., Satija, R., Fortune, S., Christopher Love, J., & Shalek, A. K. (2017). Seq-Well: Portable, low-cost RNA sequencing of single cells at high throughput. Nature Methods, 14(4), 395–398. https://doi.org/10.1038/nmeth.4179
Giladi, A., Paul, F., Herzog, Y., Lubling, Y., Weiner, A., Yofe, I., Jaitin, D., Cabezas-Wallscheid, N., Dress, R., Ginhoux, F., Trumpp, A., Tanay, A., & Amit, I. (2018). Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nature Cell Biology, 20(7), 836–846. https://doi.org/10.1038/s41556-018-0121-4
Giudice, M. D., Peirone, S., Perrone, S., Priante, F., Varese, F., Tirtei, E., Fagioli, F., & Cereda, M. (2021). Artificial intelligence in bulk and single-cell RNA-sequencing data to foster precision oncology. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms22094563
Gladka, M. M., Molenaar, B., de Ruiter, H., van der Elst, S., Tsui, H., Versteeg, D., Lacraz, G. P. A., Huibers, M. M. H., van Oudenaarden, A., & van Rooij, E. (2018). Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation, 138(2), 166–180. https://doi.org/10.1161/CIRCULATIONAHA.117.030742
Gohil, S. H., Iorgulescu, J. B., Braun, D. A., Keskin, D. B., & Livak, K. J. (2021). Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nature Reviews Clinical Oncology, 18(4), 244–256. https://doi.org/10.1038/s41571-020-00449-x
Govindan, R., Page, N., Morgensztern, D., Read, W., Tierney, R., Vlahiotis, A., Spitznagel, E. L., & Piccirillo, J. (2006). Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: Analysis of the surveillance, epidemiologic, and end results database. Journal of Clinical Oncology, 24(28), 4539–4544. https://doi.org/10.1200/JCO.2005.04.4859
Gross, A., Schoendube, J., Zimmermann, S., Steeb, M., Zengerle, R., & Koltay, P. (2015). Technologies for single-cell isolation. International Journal of Molecular Sciences, 16(8), 16897–16919. https://doi.org/10.3390/ijms160816897
Guo, F., Li, L., Li, J., Wu, X., Hu, B., Zhu, P., Wen, L., & Tang, F. (2017). Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Research, 27(8), 967–988. https://doi.org/10.1038/cr.2017.82
Guo, H., Zhu, P., Wu, X., Li, X., Wen, L., & Tang, F. (2013). Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Research, 23(12), 2126–2135. https://doi.org/10.1101/gr.161679.113
Heinrich, S., Craig, A. J., Ma, L., Heinrich, B., Greten, T. F., & Wang, X. W. (2021). Understanding tumour cell heterogeneity and its implication for immunotherapy in liver cancer using single-cell analysis. Journal of Hepatology, 74(3), 700–715. https://doi.org/10.1016/j.jhep.2020.11.036
Hen-Avivi, S., & Avraham, R. (2018). Immune cell type ‘fingerprints’ at the basis of outcome diversity of human infection. Current Opinion in Microbiology, 42, 31–39. https://doi.org/10.1016/j.mib.2017.09.012
Höfer, T., & Rodewald, H. R. (2018). Differentiation-based model of hematopoietic stem cell functions and lineage pathways. Blood, 132(11), 1106–1113. https://doi.org/10.1182/blood-2018-03-791517
Huang, A., Garraway, L. A., Ashworth, A., & Weber, B. (2020). Synthetic lethality as an engine for cancer drug target discovery. Nature Reviews Drug Discovery, 19(1), 23–38. https://doi.org/10.1038/s41573-019-0046-z
Huang, L., Ma, F., Chapman, A., Lu, S., & **e, X. S. (2015). Single-cell whole-genome amplification and sequencing: Methodology and applications. Annual Review of Genomics and Human Genetics, 16, 79–102. https://doi.org/10.1146/annurev-genom-090413-025352
Hwang, B., Lee, J. H., & Bang, D. (2018). Single-cell RNA sequencing technologies and bioinformatics pipelines. Experimental and Molecular Medicine. https://doi.org/10.1038/s12276-018-0071-8
Iacono, G., Massoni-Badosa, R., & Heyn, H. (2019). Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biology. https://doi.org/10.1186/s13059-019-1713-4
Iorgulescu, J. B., Braun, D., Oliveira, G., Keskin, D. B., & Wu, C. J. (2018). Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Medicine, 10(1), 87. https://doi.org/10.1186/s13073-018-0598-2
Izar, B., Tirosh, I., Stover, E. H., Wakiro, I., Cuoco, M. S., Alter, I., Rodman, C., Leeson, R., Su, M. J., Shah, P., Iwanicki, M., Walker, S. R., Kanodia, A., Melms, J. C., Mei, S., Lin, J. R., Porter, C. B. M., Slyper, M., Waldman, J., … Regev, A. (2020). A single-cell landscape of high-grade serous ovarian cancer. Nature Medicine, 26(8), 1271–1279. https://doi.org/10.1038/s41591-020-0926-0
Jahan-Tigh, R. R., Ryan, C., Obermoser, G., & Schwarzenberger, K. (2012). Flow cytometry. Journal of Investigative Dermatology, 132(10), 1–6. https://doi.org/10.1038/jid.2012.282
Jain, A. K., & Barton, M. C. (2018). P53: Emerging roles in stem cells, development and beyond. Development (Cambridge). https://doi.org/10.1242/dev.158360
Jaitin, D. A., Adlung, L., Thaiss, C. A., Weiner, A., Li, B., Descamps, H., Lundgren, P., Bleriot, C., Liu, Z., Deczkowska, A., Keren-Shaul, H., David, E., Zmora, N., Eldar, S. M., Lubezky, N., Shibolet, O., Hill, D. A., Lazar, M. A., Colonna, M., … Amit, I. (2019). Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell, 178(3), 686–698.e14. https://doi.org/10.1016/j.cell.2019.05.054
Jordan, E. J., Kim, H. R., Arcila, M. E., Barron, D., Chakravarty, D., Gao, J. J., Chang, M. T., Ni, A., Kundra, R., Jonsson, P., Jayakumaran, G., Gao, S. P., Johnsen, H. C., Hanrahan, A. J., Zehir, A., Rekhtman, N., Ginsberg, M. S., Li, B. T., Yu, H. A., … Riely, G. J. (2017). Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discovery, 7(6), 596–609. https://doi.org/10.1158/2159-8290.CD-16-1337
Kester, L., & van Oudenaarden, A. (2018). Single-cell transcriptomics meets lineage tracing. Cell Stem Cell, 23(2), 166–179. https://doi.org/10.1016/j.stem.2018.04.014
Kim, C., Gao, R., Sei, E., Brandt, R., Hartman, J., Hatschek, T., Crosetto, N., Foukakis, T., & Navin, N. E. (2018). Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell, 173(4), 879-893.e13. https://doi.org/10.1016/j.cell.2018.03.041
Kiselev, V. Y., Andrews, T. S., & Hemberg, M. (2019). Challenges in unsupervised clustering of single-cell RNA-seq data. Nature Reviews Genetics, 20(5), 273–282. https://doi.org/10.1038/s41576-018-0088-9
Klein, A. M., Mazutis, L., Akartuna, I., Tallapragada, N., Veres, A., Li, V., Peshkin, L., Weitz, D. A., & Kirschner, M. W. (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell, 161(5), 1187–1201. https://doi.org/10.1016/j.cell.2015.04.044
Kohno, T., Ichikawa, H., Totoki, Y., Yasuda, K., Hiramoto, M., Nammo, T., Sakamoto, H., Tsuta, K., Furuta, K., Shimada, Y., Iwakawa, R., Ogiwara, H., Oike, T., Enari, M., Schetter, A. J., Okayama, H., Haugen, A., Skaug, V., Chiku, S., … Shibata, T. (2012). KIF5B-RET fusions in lung adenocarcinoma. Nature Medicine, 18(3), 375–377. https://doi.org/10.1038/nm.2644
Koller, K. M., Wang, W., Schell, T. D., Cozza, E. M., Kokolus, K. M., Neves, R. I., Mackley, H. B., Pameijer, C., Leung, A., Anderson, B., Mallon, C. A., Robertson, G., & Drabick, J. J. (2016). Malignant melanoma—the cradle of anti-neoplastic immunotherapy. Critical Reviews in Oncology/Hematology, 106, 25–54. https://doi.org/10.1016/j.critrevonc.2016.04.010
Kosaisawe, N., Sparta, B., Pargett, M., Teragawa, C. K., & Albeck, J. G. (2021). Transient phases of OXPHOS inhibitor resistance reveal underlying metabolic heterogeneity in single cells. Cell Metabolism, 33(3), 649-665.e8. https://doi.org/10.1016/j.cmet.2021.01.014
Krishnamurthy, N., & Kurzrock, R. (2018). Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treatment Reviews, 62, 50–60. https://doi.org/10.1016/j.ctrv.2017.11.002
Lähnemann, D., Köster, J., Szczurek, E., McCarthy, D. J., Hicks, S. C., Robinson, M. D., Vallejos, C. A., Campbell, K. R., Beerenwinkel, N., Mahfouz, A., Pinello, L., Skums, P., Stamatakis, A., Attolini, C. S. O., Aparicio, S., Baaijens, J., Balvert, M., Barbanson, B. de, Cappuccio, A., … Schönhuth, A. (2020). Eleven grand challenges in single-cell data science. Genome Biology. https://doi.org/10.1186/s13059-020-1926-6
Laks, E., McPherson, A., Zahn, H., Lai, D., Steif, A., Brimhall, J., Biele, J., Wang, B., Masud, T., Ting, J., Grewal, D., Nielsen, C., Leung, S., Bojilova, V., Smith, M., Golovko, O., Poon, S., Eirew, P., Kabeer, F., … Shah, S. P. (2019). Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencing. Cell, 179(5), 1207–1221.e22. https://doi.org/10.1016/j.cell.2019.10.026
Lambrechts, D., Wauters, E., Boeckx, B., Aibar, S., Nittner, D., Burton, O., Bassez, A., Decaluwé, H., Pircher, A., van den Eynde, K., Weynand, B., Verbeken, E., de Leyn, P., Liston, A., Vansteenkiste, J., Carmeliet, P., Aerts, S., & Thienpont, B. (2018). Phenotype molding of stromal cells in the lung tumor microenvironment. Nature Medicine, 24(8), 1277–1289. https://doi.org/10.1038/s41591-018-0096-5
Lan, F., Demaree, B., Ahmed, N., & Abate, A. R. (2017). Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nature Biotechnology, 35(7), 640–646. https://doi.org/10.1038/nbt.3880
Lawson, D. A., Kessenbrock, K., Davis, R. T., Pervolarakis, N., & Werb, Z. (2018). Tumour heterogeneity and metastasis at single-cell resolution. Nature Cell Biology, 20(12), 1349–1360. https://doi.org/10.1038/s41556-018-0236-7
Lee, J., Hyeon, D. Y., & Hwang, D. (2020). Single-cell multiomics: Technologies and data analysis methods. Experimental and Molecular Medicine, 52(9), 1428–1442. https://doi.org/10.1038/s12276-020-0420-2
Lee, M. C. W., Lopez-Diaz, F. J., Khan, S. Y., Tariq, M. A., Dayn, Y., Vaske, C. J., Radenbaugh, A. J., Kim, H. J., Emerson, B. M., & Pourm, N. (2014). Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proceedings of the National Academy of Sciences of the United States of America, 111(44), E4726–E4735. https://doi.org/10.1073/pnas.1404656111
Li, H., Courtois, E. T., Sengupta, D., Tan, Y., Chen, K. H., Goh, J. J. L., Kong, S. L., Chua, C., Hon, L. K., Tan, W. S., Wong, M., Choi, P. J., Wee, L. J. K., Hillmer, A. M., Tan, I. B., Robson, P., & Prabhakar, S. (2017). Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nature Genetics, 49(5), 708–718. https://doi.org/10.1038/ng.3818
Libby, P., Buring, J. E., Badimon, L., Hansson, G. K., Deanfield, J., Bittencourt, M. S., Tokgözoğlu, L., & Lewis, E. F. (2019). Atherosclerosis. Nature Reviews Disease Primers. https://doi.org/10.1038/s41572-019-0106-z
Liberzon, A., Birger, C., Thorvaldsdóttir, H., Ghandi, M., Mesirov, J. P., & Tamayo, P. (2015). The molecular signatures database hallmark gene set collection. Cell Systems, 1(6), 417–425. https://doi.org/10.1016/j.cels.2015.12.004
Lin, S., Huang, C., Gunda, V., Sun, J., Chellappan, S. P., Li, Z., Izumi, V., Fang, B., Koomen, J., Singh, P. K., Hao, J., & Yang, S. (2019). Fascin controls metastatic colonization and mitochondrial oxidative phosphorylation by remodeling mitochondrial actin filaments. Cell Reports, 28(11), 2824-2836.e8. https://doi.org/10.1016/j.celrep.2019.08.011
Liu, W. Q., Zhang, H. M., Hu, D., Lu, S. J., & Sun, X. F. (2018). The performance of MALBAC and MDA methods in the identification of concurrent mutations and aneuploidy screening to diagnose beta-thalassaemia disorders at the single- and multiple-cell levels. Journal of Clinical Laboratory Analysis. https://doi.org/10.1002/jcla.22267
Loi, S., Michiels, S., Salgado, R., Sirtaine, N., Jose, V., Fumagalli, D., Kellokumpu-Lehtinen, P. L., Bono, P., Kataja, V., Desmedt, C., Piccart, M. J., Loibl, S., Denkert, C., Smyth, M. J., Joensuu, H., & Sotiriou, C. (2014). Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Annals of Oncology, 25(8), 1544–1550. https://doi.org/10.1093/annonc/mdu112
Lowery, A. J., Miller, N., McNeill, R. E., & Kerin, M. J. (2008). MicroRNAs as prognostic indicators and therapeutic targets: Potential effect on breast cancer management. Clinical Cancer Research, 14(2), 360–365. https://doi.org/10.1158/1078-0432.CCR-07-0992
Lv, B., Liu, C., Chen, Y., Qi, L., Wang, L., Ji, Y., & Xue, Z. (2019). Light-induced injury in mouse embryos revealed by single-cell RNA sequencing. Biological Research, 52(1), 48. https://doi.org/10.1186/s40659-019-0256-1
Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., Tirosh, I., Bialas, A. R., Kamitaki, N., Martersteck, E. M., Trombetta, J. J., Weitz, D. A., Sanes, J. R., Shalek, A. K., Regev, A., & McCarroll, S. A. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell, 161(5), 1202–1214. https://doi.org/10.1016/j.cell.2015.05.002
Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F., Mohammadi, S., Young, J. Z., Menon, M., He, L., Abdurrob, F., Jiang, X., Martorell, A. J., Ransohoff, R. M., Hafler, B. P., Bennett, D. A., Kellis, M., & Tsai, L. H. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature, 570(7761), 332–337. https://doi.org/10.1038/s41586-019-1195-2
Maynard, A., McCoach, C. E., Rotow, J. K., Harris, L., Haderk, F., Kerr, D. L., Yu, E. A., Schenk, E. L., Tan, W., Zee, A., Tan, M., Gui, P., Lea, T., Wu, W., Urisman, A., Jones, K., Sit, R., Kolli, P. K., Seeley, E., … Bivona, T. G. (2020). Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell, 182(5), 1232–1251.e22. https://doi.org/10.1016/j.cell.2020.07.017
Mazieres, J., Drilon, A., Lusque, A., Mhanna, L., Cortot, A. B., Mezquita, L., Thai, A. A., Mascaux, C., Couraud, S., Veillon, R., van den Heuvel, M., Neal, J., Peled, N., Früh, M., Ng, T. L., Gounant, V., Popat, S., Diebold, J., Sabari, J., … Gautschi, O. (2019). Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Annals of Oncology, 30(8), 1321–1328. https://doi.org/10.1093/annonc/mdz167
Mollet, M., Godoy-Silva, R., Berdugo, C., & Chalmers, J. J. (2008). Computer simulations of the energy dissipation rate in a fluorescence-activated cell sorter: Implications to cells. Biotechnology and Bioengineering, 100(2), 260–272. https://doi.org/10.1002/bit.21762
Nabhan, A. N., Brownfield, D. G., Harbury, P. B., Krasnow, M. A., & Desai, T. J. (2018). Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science, 359(6380), 1118–1123. https://doi.org/10.1126/science.aam6603
Navin, N. E. (2015). The first five years of single-cell cancer genomics and beyond. Genome Research, 25(10), 1499–1507. https://doi.org/10.1101/gr.191098.115
Nguyen, T. A., Yin, T. I., Reyes, D., & Urban, G. A. (2013). Microfluidic chip with integrated electrical cell-impedance sensing for monitoring single cancer cell migration in three-dimensional matrixes. Analytical Chemistry, 85(22), 11068–11076. https://doi.org/10.1021/ac402761s
Nguyen, T., Wei, Y., Nakada, Y., Chen, J. Y., Zhou, Y., Walcott, G., & Zhang, J. (2023). Analysis of cardiac single-cell RNA-sequencing data can be improved by the use of artificial-intelligence-based tools. Scientific Reports, 13(1), 6821. https://doi.org/10.1038/s41598-023-32293-1
Nilsson, M. B., Langley, R. R., & Fidler, I. J. (n.d.). Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine.
Nomura, S., Satoh, M., Fujita, T., Higo, T., Sumida, T., Ko, T., Yamaguchi, T., Tobita, T., Naito, A. T., Ito, M., Fujita, K., Harada, M., Toko, H., Kobayashi, Y., Ito, K., Takimoto, E., Akazawa, H., Morita, H., Aburatani, H., & Komuro, I. (2018). Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nature Communications. https://doi.org/10.1038/s41467-018-06639-7
Olsen, T. K., & Baryawno, N. (2018). Introduction to single-cell RNA sequencing. Current Protocols in Molecular Biology. https://doi.org/10.1002/cpmb.57
Pailler, E., Faugeroux, V., Oulhen, M., Mezquita, L., Laporte, M., Honore, A., Lecluse, Y., Queffelec, P., NgoCamus, M., Nicotra, C., Remon, J., Lacroix, L., Planchard, D., Friboulet, L., Besse, B., & Farace, F. (2019). Acquired resistance mutations to ALK inhibitors identified by single circulating tumor cell sequencing in ALK-rearranged non–small-cell lung cancer. Clinical Cancer Research, 25(22), 6671–6682. https://doi.org/10.1158/1078-0432.CCR-19-1176
Paolillo, C., Londin, E., & Fortina, P. (2019). Single-cell genomics. Clinical Chemistry, 65(8), 972–985. https://doi.org/10.1373/clinchem.2017.283895
Patel, A. P., Tirosh, I., Trombetta, J. J., Shalek, A. K., Gillespie, S. M., Wakimoto, H., Cahill, D. P., Nahed, B. V., Curry, W. T., Martuza, R. L., Louis, D. N., Rozenblatt-Rosen, O., Suvà, M. L., Regev, A., & Bernstein, B. E. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 344(6190), 1396–1401. https://doi.org/10.1126/science.1254257
Potter, S. S. (2018). Single-cell RNA sequencing for the study of development, physiology and disease. Nature Reviews Nephrology, 14(8), 479–492. https://doi.org/10.1038/s41581-018-0021-7
Puram, S. V., Tirosh, I., Parikh, A. S., Patel, A. P., Yizhak, K., Gillespie, S., Rodman, C., Luo, C. L., Mroz, E. A., Emerick, K. S., Deschler, D. G., Varvares, M. A., Mylvaganam, R., Rozenblatt-Rosen, O., Rocco, J. W., Faquin, W. C., Lin, D. T., Regev, A., & Bernstein, B. E. (2017). Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell, 171(7), 1611-1624.e24. https://doi.org/10.1016/j.cell.2017.10.044
Qian, Y., Gong, Y., Fan, Z., Luo, G., Huang, Q., Deng, S., Cheng, H., **, K., Ni, Q., Yu, X., & Liu, C. (2020). Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. Journal of Hematology and Oncology. https://doi.org/10.1186/s13045-020-00958-3
Ramsköld, D., Luo, S., Wang, Y. C., Li, R., Deng, Q., Faridani, O. R., Daniels, G. A., Khrebtukova, I., Loring, J. F., Laurent, L. C., Schroth, G. P., & Sandberg, R. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature Biotechnology, 30(8), 777–782. https://doi.org/10.1038/nbt.2282
Rohde, D., & Nahrendorf, M. (2019). Clonal and diverse: Revisiting cardiac endothelial cells after myocardial infarction. European Heart Journal, 40(30), 2521–2522. https://doi.org/10.1093/eurheartj/ehz375
Rynne-Vidal, A., Au-Yeung, C. L., Jiménez-Heffernan, J. A., Pérez-Lozano, M. L., Cremades-Jimeno, L., Bárcena, C., Cristóbal-García, I., Fernández-Chacón, C., Yeung, T. L., Mok, S. C., Sandoval, P., & López-Cabrera, M. (2017). Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. Journal of Pathology, 242(2), 140–151. https://doi.org/10.1002/path.4889
Santhakumar, C., Gane, E. J., Liu, K., & McCaughan, G. W. (2020). Current perspectives on the tumor microenvironment in hepatocellular carcinoma. In Hepatology International, 14(6), 947–957. https://doi.org/10.1007/s12072-020-10104-3
Satpathy, A. T., Granja, J. M., Yost, K. E., Qi, Y., Meschi, F., McDermott, G. P., Olsen, B. N., Mumbach, M. R., Pierce, S. E., Corces, M. R., Shah, P., Bell, J. C., Jhutty, D., Nemec, C. M., Wang, J., Wang, L., Yin, Y., Giresi, P. G., Chang, A. L. S., … Chang, H. Y. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nature Biotechnology, 37(8), 925–936. https://doi.org/10.1038/s41587-019-0206-z
Shalek, A. K., Satija, R., Adiconis, X., Gertner, R. S., Gaublomme, J. T., Raychowdhury, R., Schwartz, S., Yosef, N., Malboeuf, C., Lu, D., Trombetta, J. J., Gennert, D., Gnirke, A., Goren, A., Hacohen, N., Levin, J. Z., Park, H., & Regev, A. (2013). Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature, 498(7453), 236–240. https://doi.org/10.1038/nature12172
Shapiro, E., Biezuner, T., & Linnarsson, S. (2013). Single-cell sequencing-based technologies will revolutionize whole-organism science. Nature Reviews Genetics, 14(9), 618–630. https://doi.org/10.1038/nrg3542
Sharon, D., Tilgner, H., Grubert, F., & Snyder, M. (2013). A single-molecule long-read survey of the human transcriptome. Nature Biotechnology, 31(11), 1009–1014. https://doi.org/10.1038/nbt.2705
Skelly, D. A., Squiers, G. T., McLellan, M. A., Bolisetty, M. T., Robson, P., Rosenthal, N. A., & Pinto, A. R. (2018). Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Reports, 22(3), 600–610. https://doi.org/10.1016/j.celrep.2017.12.072
Souilhol, C., Serbanovic-Canic, J., Fragiadaki, M., Chico, T. J., Ridger, V., Roddie, H., & Evans, P. C. (2020). Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nature Reviews Cardiology, 17(1), 52–63. https://doi.org/10.1038/s41569-019-0239-5
Spike, B. T., & Wahl, G. M. (2011). P53, stem cells, and reprogramming: Tumor suppression beyond guarding the genome. Genes and Cancer, 2(4), 404–419. https://doi.org/10.1177/1947601911410224
Stark, R., Grzelak, M., & Hadfield, J. (2019). RNA sequencing: The teenage years. Nature Reviews Genetics, 20(11), 631–656. https://doi.org/10.1038/s41576-019-0150-2
Su, T., Stanley, G., Sinha, R., D’Amato, G., Das, S., Rhee, S., Chang, A. H., Poduri, A., Raftrey, B., Dinh, T. T., Roper, W. A., Li, G., Quinn, K. E., Caron, K. M., Wu, S., Miquerol, L., Butcher, E. C., Weissman, I., Quake, S., & Red-Horse, K. (2018). Single-cell analysis of early progenitor cells that build coronary arteries. Nature, 559(7714), 356–362. https://doi.org/10.1038/s41586-018-0288-7
Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., Wang, X., Bodeau, J., Tuch, B. B., Siddiqui, A., Lao, K., & Surani, M. A. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods, 6(5), 377–382. https://doi.org/10.1038/nmeth.1315
Tewary, M., Shakiba, N., & Zandstra, P. W. (2018). Stem cell bioengineering: Building from stem cell biology. Nature Reviews Genetics, 19(10), 595–614. https://doi.org/10.1038/s41576-018-0040-z
Tilgner, H., Grubert, F., Sharon, D., & Snyder, M. P. (2014). Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proceedings of the National Academy of Sciences of the United States of America, 111(27), 9869–9874. https://doi.org/10.1073/pnas.1400447111
Tirtei, E., Cereda, M., De Luna, E., Quarello, P., Asaftei, S. D., & Fagioli, F. (2020). Omic approaches to pediatric bone sarcomas. In Pediatric Blood and Cancer. https://doi.org/10.1002/pbc.28072
Topol, E. J. (2019). High-performance medicine: The convergence of human and artificial intelligence. In Nature Medicine, 25(1), 44–56. https://doi.org/10.1038/s41591-018-0300-7
Tosoni, D., Zecchini, S., Coazzoli, M., Colaluca, I., Mazzarol, G., Rubio, A., Caccia, M., Villa, E., Zilian, O., Di Fiore, P. P., & Pece, S. (2015). The Numb/p53 circuitry couples replicative self-renewal and tumor suppression in mammary epithelial cells. Journal of Cell Biology, 211(4), 845–862. https://doi.org/10.1083/jcb.201505037
Treutlein, B., Brownfield, D. G., Wu, A. R., Neff, N. F., Mantalas, G. L., Espinoza, F. H., Desai, T. J., Krasnow, M. A., & Quake, S. R. (2014). Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature, 509(7500), 371–375. https://doi.org/10.1038/nature13173
van Dijk, E. L., Jaszczyszyn, Y., Naquin, D., & Thermes, C. (2018). The third revolution in sequencing technology. Trends in Genetics, 34(9), 666–681. https://doi.org/10.1016/j.tig.2018.05.008
VanHorn, S., & Morris, S. A. (2021). Next-generation lineage tracing and fate map** to interrogate development. Developmental Cell, 56(1), 7–21. https://doi.org/10.1016/j.devcel.2020.10.021
Visscher, D. W. (2011). Genomics, histopathology, and the tumor microenvironment: New relationship or old friends re-discovered? Breast Cancer Research and Treatment, 125(3), 697–698. https://doi.org/10.1007/s10549-010-0930-z
Wagner, D. E., & Klein, A. M. (2020). Lineage tracing meets single-cell omics: Opportunities and challenges. Nature Reviews Genetics, 21(7), 410–427. https://doi.org/10.1038/s41576-020-0223-2
Wang, R., Liu, J., Li, K., Yang, G., Chen, S., Wu, J., **e, X., Ren, H., & Pang, Y. (2021). An SETD1A/Wnt/β-catenin feedback loop promotes NSCLC development. Journal of Experimental and Clinical Cancer Research. https://doi.org/10.1186/s13046-021-02119-x
Wang, Y., & Navin, N. E. (2015). Advances and applications of single-cell sequencing technologies. Molecular Cell, 58(4), 598–609. https://doi.org/10.1016/j.molcel.2015.05.005
Wang, Y., Tang, Z., Huang, H., Li, J., Wang, Z., Yu, Y., Zhang, C., Li, J., Dai, H., Wang, F., Cai, T., & Tang, N. (2018). Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proceedings of the National Academy of Sciences of the United States of America, 115(10), 2407–2412. https://doi.org/10.1073/pnas.1719474115
Wei, R., DeVilbiss, F. T., & Liu, W. (2015). Genetic polymorphism, telomere biology and non-small lung cancer risk. Journal of Genetics and Genomics, 42(10), 549–561. https://doi.org/10.1016/j.jgg.2015.08.005
Wiedmeier, J. E., Noel, P., Lin, W., von Hoff, D. D., & Han, H. (2019). Single-cell sequencing in precision medicine. Cancer Treatment and Research, 178, 237–252. https://doi.org/10.1007/978-3-030-16391-4_9
Wu, Y. H., Chang, T. H., Huang, Y. F., Huang, H. D., & Chou, C. Y. (2014). COL11A1 promotes tumor progression and predicts poor clinical outcome in ovarian cancer. British Dental Journal, 217(1), 3432–3440. https://doi.org/10.1038/onc.2013.307
Wu, Y., & Zhang, K. (2020). Tools for the analysis of high-dimensional single-cell RNA sequencing data. Nature Reviews Nephrology, 16(7), 408–421. https://doi.org/10.1038/s41581-020-0262-0
Wu, Y., & Zhou, B. P. (2010). TNF-α/NFκ-B/Snail pathway in cancer cell migration and invasion. British Journal of Cancer, 102(4), 639–644. https://doi.org/10.1038/sj.bjc.6605530
**ang, Y., Ye, Y., Zhang, Z., & Han, L. (2018). Maximizing the utility of cancer transcriptomic data. Trends in Cancer, 4(12), 823–837. https://doi.org/10.1016/j.trecan.2018.09.009
**ao, Y., & Yu, D. (2021). Tumor microenvironment as a therapeutic target in cancer. Pharmacology and Therapeutics. https://doi.org/10.1016/j.pharmthera.2020.107753
Xu, J., Gong, L., Qian, Z., Song, G., & Liu, J. (2018). ERBB4 promotes the proliferation of gastric cancer cells via the PI3K/Akt signaling pathway. Oncology Reports, 39(6), 2892–2898. https://doi.org/10.3892/or.2018.6343
Xu, X., Hou, Y., Yin, X., Bao, L., Tang, A., Song, L., Li, F., Tsang, S., Wu, K., Wu, H., He, W., Zeng, L., **ng, M., Wu, R., Jiang, H., Liu, X., Cao, D., Guo, G., Hu, X., … Wang, J. (2012). Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell, 148(5), 886–895. https://doi.org/10.1016/j.cell.2012.02.025
Yang, S., Liu, Y., Li, M. Y., Ng, C. S. H., Yang, S., & li, Wang, S., Zou, C., Dong, Y., Du, J., Long, X., Liu, L. Z., Wan, I. Y. P., Mok, T., Underwood, M. J., & Chen, G. G. (2017). FOXP3 promotes tumor growth and metastasis by activating Wnt/β-catenin signaling pathway and EMT in non-small cell lung cancer. Molecular Cancer. https://doi.org/10.1186/s12943-017-0700-1
Yao, F., Yu, P., Li, Y., Yuan, X., Li, Z., Zhang, T., Liu, F., Wang, Y., Wang, Y., Li, D., Ma, B., Shu, C., Kong, W., Zhou, B., & Wang, L. (2018). Histone variant H2A.Z is required for the maintenance of smooth muscle cell identity as revealed by single-cell transcriptomics. Circulation, 138(20), 2274–2288. https://doi.org/10.1161/CIRCULATIONAHA.117.033114
Yates, L. R., Gerstung, M., Knappskog, S., Desmedt, C., Gundem, G., van Loo, P., Aas, T., Alexandrov, L. B., Larsimont, D., Davies, H., Li, Y., Ju, Y. S., Ramakrishna, M., Haugland, H. K., Lilleng, P. K., Nik-Zainal, S., McLaren, S., Butler, A., Martin, S., … Campbell, P. J. (2015). Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nature Medicine, 21(7), 751–759. https://doi.org/10.1038/nm.3886
Yin, L., Duan, J. J., Bian, X. W., & Yu, S. C. (2020). Triple-negative breast cancer molecular subty** and treatment progress. Breast Cancer Research. https://doi.org/10.1186/s13058-020-01296-5
Zacksenhaus, E., Shrestha, M., Liu, J. C., Vorobieva, I., Chung, P. E. D., Ju, Y. J., Nir, U., & Jiang, Z. (2017). Mitochondrial OXPHOS induced by RB1 deficiency in breast cancer: Implications for anabolic metabolism, stemness, and metastasis. Trends in Cancer, 3(11), 768–779. https://doi.org/10.1016/j.trecan.2017.09.002
Zahn, H., Steif, A., Laks, E., Eirew, P., Vaninsberghe, M., Shah, S. P., Aparicio, S., & Hansen, C. L. (2017). Scalable whole-genome single-cell library preparation without preamplification. Nature Methods, 14(2), 167–173. https://doi.org/10.1038/nmeth.4140
Zhang, P., Yang, M., Zhang, Y., **ao, S., Lai, X., Tan, A., Du, S., & Li, S. (2019). Dissecting the single-cell transcriptome network underlying gastric premalignant lesions and early gastric cancer. Cell Reports, 27(6), 1934-1947.e5. https://doi.org/10.1016/j.celrep.2019.04.052
Zhang, X., Marjani, S. L., Hu, Z., Weissman, S. M., Pan, X., & Wu, S. (2016). Single-Cell sequencing for precise cancer research: Progress and prospects. Cancer Research, 76(6), 1305–1312. https://doi.org/10.1158/0008-5472.CAN-15-1907
Zheng, G. X. Y., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., Ziraldo, S. B., Wheeler, T. D., McDermott, G. P., Zhu, J., Gregory, M. T., Shuga, J., Montesclaros, L., Underwood, J. G., Masquelier, D. A., Nishimura, S. Y., Schnall-Levin, M., Wyatt, P. W., Hindson, C. M., … Bielas, J. H. (2017). Massively parallel digital transcriptional profiling of single cells. Nature Communications. https://doi.org/10.1038/ncomms14049
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, X., Zheng, Y. & Xu, K. Single-Cell RNA Sequencing: Technological Progress and Biomedical Application in Cancer Research. Mol Biotechnol 66, 1497–1519 (2024). https://doi.org/10.1007/s12033-023-00777-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-023-00777-0