
Abstract
Human polyomavirus type 1, or BK virus (BKV), is a ubiquitous pathogen belonging to the polyomaviridae family mostly known for causing BKV-associated nephropathy (BKVN) and allograft rejection in kidney transplant recipients (KTRs) following the immunosuppression regimens recommended in these patients. Reduction of the immunosuppression level and anti-viral agents are the usual approaches for BKV clearance, which have not met a desired outcome yet. There are also debating matters such as the effect of this pathogen on emerging various comorbidities and the related malignancies in the human population. In this study, a reverse vaccinology approach was implemented to design a mRNA vaccine against BKV by identifying the most antigenic proteins of this pathogen. Potential immunogenic T and B lymphocyte epitopes were predicted through various immunoinformatic tools. The final epitopes were selected according to antigenicity, toxicity, allergenicity, and cytokine inducibility scores. According to the obtained results, the designed vaccine was antigenic, neutral at the physiological pH, non-toxic, and non-allergenic with a world population coverage of 93.77%. Since the mRNA codon optimization ensures the efficient expression of the vaccine in a host cell, evaluation of different parameters showed our designed mRNA vaccine has a stable structure. Moreover, it had strong interactions with toll-like receptor 4 (TLR4) according to the molecular dynamic simulation studies. The in silico immune simulation analyses revealed an overall increase in the immune responses following repeated exposure to the designed vaccine. Based on our findings, the vaccine candidate is ready to be tested as a promising novel mRNA therapeutic vaccine against BKV.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 1971, the human polyomavirus type 1, named BK virus (BKV), was isolated from a patient’s urine sample for the first time [1]. BKV is a non-enveloped virus consisting of an icosahedral capsid with a 40-nm diameter, which encloses its circular, double-stranded DNA genome of about 5 Kb. It belongs to the polyomavirus genus of the Polyomaviridae family, which includes two other species, JC virus (JCV) and simian virus 40 (SV40). BKV is divided into four subtypes (I–IV): subtype I is the most prevalent subtype (80%), followed by subtype IV (15%), and subtypes II and III, which are rare. However, some reports show the subtype III has been detected more frequently in HIV-infected patients [2]. BKV is a prevalent virus that persistently infects about 100% of the human population by early childhood after the waning of maternal antibodies, with an IgG seroprevalence > 90% in 5–9-year-old healthy children [3,4,5,6,7,8,9]. Up to now, the exact route of BKV transmission has not been identified [10]. The foremost mode of transmission seems to be via the respiratory route, as supported by the detection of BKV DNA in the respiratory tract and tonsils of children, suggesting that the oropharynx may be the initial site of BKV infection [11,12,13]. Other proposed routes of transmission are fecal–oral, blood transfusion, and transplacental by identifying BKV in sewage, leukocytes, and fetus, respectively [12, 14,15,16,17,18]. Different studies show that it also can be transmitted via semen and through organ transplantation [19, 20, 22]. Primary BKV infection is mainly asymptomatic, and the most common symptoms, when symptoms appear, are fever and nonspecific respiratory infection or urinary tract disease [11, 20, 23, 24]. BKV goes into the circulatory system through infected tonsils, and afterward, the peripheral-blood mononuclear cells get infected and disseminate to be latent in several sites most notably in kidneys. The virus remains dormant in the uroepithelium and renal tubular epithelial cells. Intermittent reactivation with asymptomatic viruria may occur in the patients [12, 21, 25]. Moreover, clinical findings in the past decades reveal that BKV can remain latent in leukocytes, brain tissue, and lymph nodes, which remarks its notable role in the outburst of variable complications in these organs [24, 26]. Immunosuppressive therapy can reactivate the virus, so that it starts to replicate, causing a series of events, beginning with tubular cell lysis and viruria. Then, this pathogen proliferates inside the interstitium and crosses into the peritubular capillaries, causing viremia. Ultimately, tissue damage may occur following direct viral cytolytic effects and secondary inflammatory responses [12, 21, 24, 25]; the outcome is determined by the level of damage, inflammation, and fibrosis. These complicated reactions between BKV and the immune system result in different clinical pathologic manifestations of BKV. Recent studies show that BKV as well as JCV, can be an etiologic agent of progressive multifocal leukoencephalopathy (PML) [27, 28]. There are different reports regarding human neoplasia caused by BKV. Hence, the oncogenic characteristic of this pathogen should not be neglected [7]. BKV is one of the most common opportunistic pathogens causing posttransplant viral infections, leading to allograft loss in renal transplant recipients. Posttransplant viral infections are followed by potent immunosuppression regimens that are usually applied in transplant recipients with the purpose of reduction of acute allograft rejection [29, 30]. In the first posttransplant year, approximately 15% of renal transplant recipients are affected by BKV viral infection, which can cause BKV-associated nephropathy (BKVN) with limited treatment options. If unaddressed, allograft failure will be progressed [21]. There is minimal or no efficacy of anti-viral agents in clearing BKV, while causing a high incidence of side effects. Hence, reduction of immunosuppression is the mainstay of BKVN treatment [9, 21, 31,32,33], which can be a threat to renal transplant recipients. Therefore, an innovative prophylaxis strategy can exterminate most of the complications that human population is confronted by this pathogen. Vaccines have a substantial role in the eradication and prevention of viral diseases for the years [34] and can be considered as a new way of confronting BKV complications in the human population by assuring a reliable prophylactic approach. In recent years, mRNA-based vaccine technology has been appeared as a game-changing vaccine platform for therapeutics and prophylactic applications due to their high potency, rapid development capacity, low-cost manufacturing, and safe administration.
Immunoinformatics, known as computational immunology, is a branch of bioinformatics that is the interface between computational analyses and immunological data and resources. It plays a vital role in vaccine design [35, 36]. The production time and cost of vaccine development have drastically reduced by immunoinformatic approaches due to the efficient prediction of appropriate antigens, epitopes, carriers, and adjuvants needed for a vaccine [37].
In this study, we aimed to design a novel multi-epitope mRNA vaccine consist of cytotoxic T lymphocyte (CTL), helper T lymphocyte (HTL), linear B lymphocyte (LBL) epitopes derived from the BK virus antigenic proteins, combined with the highly immunogenic adjuvants and other crucial elements through the immunoinformatic, and computational strategies.
Methods
Workflow of the study
The workflow of the study is shown in Fig. 1. The analysis conducted in this study can be divided into 17 sections.

Population coverage of the selected T lymphocyte epitopes. Globally it covers 93.77% of the world’s population. The highest and lowest areas of coverage are Europe (97.54%) and Central America (8.48%) respectively
Retrieval of protein sequences
The referenced proteins of BKV (organism ID: 1,891,762) were retrieved from the UniProt proteome database (https://www.uniprot.org/proteomes/). Then, the reference sequences were downloaded and stored in the FASTA format.
Antigenic proteome selection
Antigenicity of the six proteins of the BK virus was evaluated by both VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) and ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) [38] servers. The VaxiJen server employs a novel alignment-independent method, which is based on auto cross-covariance (ACC) transformation of protein sequences into uniform vectors of principal amino acid properties. Depending on the target organisms (bacterial, viral, and tumor protein datasets), the server accuracy varies from 70 to 89% [38, 39]. The default threshold (0.4) was used. The accuracy of ANTIGENpro utilizing the combined dataset was found to be 76% based on cross-validation experiments [40].
Prediction and assessment of CTL epitopes
Both IEDB MHC-I binding tool (http://tools.iedb.org/mhci/) and RANKPEP MHC-I binding prediction tool (http://imed.med.ucm.es/Tools/rankpep.html) were used for the prediction of 9-mer CTL epitopes for 12 MHC class I supertype alleles, namely A*0101, A*0201, A*0301, A*1101, A*2301, A*2402, B*0702, B*0801, B*3501, B*4001, B*4402, and B*4403. The recommended IEDB MHC-I prediction binding tool was NetMHCpan 4.1 EL, which is trained on eluted ligand prediction method [41]. In IEDB, epitopes were selected based on their scores (high scores indicated favorable binding efficacy). RANKPEP server predicts peptide binders to MHCI molecules from protein sequence/s or sequence alignments using position specific scoring matrices (PSSMs). In addition, it predicts those MHC-I ligands whose which the C-terminal end is likely to be the result of proteasomal cleavage [42, 43]. Epitopes were selected at a 2% threshold of top-scoring peptides. According to the results of IEDB and RANKPEP servers the overlap** regions (epitopes) were selected and considered for further analysis.
Immunogenicity of the epitopes of interest was evaluated by the class I immunogenicity tool of the IEDB Analysis Resource (http://tools.iedb.org/immunogenicity/) [44]. Only epitopes that showed a positive value for immunogenicity were kept for the next stage of evaluation.
Afterward, the antigenicity of the selected CTL epitopes was evaluated by the VaxiJen v2.0 server.
The selected epitopes were evaluated for toxicity using the ToxinPred server (https://webs.iiitd.edu.in/raghava/toxinpred/algo.php), using SVM (Swiss-Prot) based method with all parameters set as default [45]. Moreover, the allergenicity of the selected epitopes was also calculated using two online tools, AllerTOP 2.0 (https://www.ddg-pharmfac.net/AllerTOP/data.html) [46] and AllegenFP v1.0 (https://ddg-pharmfac.net/AllergenFP/), with considering a priority to AllerTOP 2.0 results, since the server has a better accuracy (88.7%) than AllergenFP server (with the accuracy of 87.9%) [47, 48].
Prediction and assessment of HTL epitopes
In this study, both IEDB MHC-II binding tool server (http://tools.iedb.org/mhcii/) and RANKPEP MHC-II binding prediction tool (http://imed.med.ucm.es/Tools/rankpep.html) were used for the prediction of 15-mer HTL epitopes for eight MHC class II supertype alleles, namely DRB1*0101, DRB1*0301, DRB1*0401, DRB1*0701, DRB1*1101, DRB1*1302, DRB1*1501, and DRB5*0101. The combinatorial approach recommended by IEDB 2.22, which combines the Consensus, NN-align, SMM-align, CombLib, Sturniolo, and NetMHCIIpan methods, was chosen [49,50,51,52,53]. The host species was selected as human. Epitopes were selected based on a low adjusted rank, indicating the epitopes are good binders. RANKPEP server predicts peptide binders to MHCI molecules from protein sequence/s or sequence alignments using position-specific scoring matrices (PSSMs).
According to the results of IEDB and RANKPEP servers, the overlap** regions were selected and considered for further analysis.
Antigenicity and toxicity of the selected epitopes were evaluated by VaxiJen v2.0 and ToxinPred, respectively. Then, the antigenic and non-toxic epitopes were checked for their allergenicity through AllerTOP 2.0 and AllergenFP v1.0 with a priority for AllerTOP 2.0 results. The remaining epitopes were then assessed for their inducibility of three cytokines: (1) interferon-γ (IFN-γ) using the IFNepitope (http://crdd.osdd.net/raghava/ifnepitope/scan.php) [54] with the motif and support vector machine (SVM) hybrid approach, (2) interleukin-4 (IL-4) using IL4pred (https://webs.iiitd.edu.in/raghava/il4pred/design.php) [55] based on the motif and SVM hybrid approach with SVM threshold of 0.2, and (3) interleukin-10 (IL-10) using IL10pred (https://webs.iiitd.edu.in/raghava/il10pred/predict3.php) [56] based on the SVM prediction model with SVM threshold of − 0.3. Based on these criteria, only the epitopes that were highly antigenic, non-allergenic, non-toxic, and inducers of all the three mentioned cytokines were selected for use in our final vaccine construction.
Prediction and assessment of LBL epitopes
In this study, Bepipred linear epitope prediction 2.0 tool (http://tools.iedb.org/bcell/) was used to predict LBL epitopes, that uses the random forest algorithm with the default threshold of 0.5 [57]. The antigenicity of the projected LBL epitopes was calculated by VaxiJen v2.0 server [38] and ANTIGENpro servers. Epitopes were selected by the VaxiJen v2.0 with the threshold of 0.4. The antigenic epitopes were then checked for toxicity using ToxinPred [45] and allergenicity by using AllerTOP 2.0 [46, 47] and AllergenFP v1.0 [48] with a priority to AllerTOP 2.0 results. All the selected epitopes were finally evaluated by Igpred (https://webs.iiitd.edu.in/raghava/igpred/help.html) server for IgG, IgM, and IgA inducibility properties.
Epitope conservancy analysis
To evaluate the conservancy of the regions that were selected as the epitopes, 150 epitopes of each BKV antigenic proteins sequences representing four distinct BKV genotypes were selected randomly. Their sequences were downloaded from the NCBI data base (https://www.ncbi.nlm.nih.gov/) and then aligned using the IEDB conservancy tool (http://tools.iedb.org/conservancy/) [58].
Molecular docking studies between T lymphocyte epitopes and MHC alleles
The binding affinity of CTL and HTL epitopes with their corresponding MHC alleles was evaluated using a molecular docking simulation approach. The MHC alleles were downloaded from the RCSB PDB (https://www.rcsb.org/) [59, 60] and processed using the PyMOL software to eliminate unnecessary ligands. The 3D crystal structure of a total of 12 MHC supertype alleles of class I and II including HLA-A*0101 (PDB ID:4NQV), HLA-A*0201 (PDB ID:4UQ3), HLA-A*0301 (PDB ID:3RL2), HLA-A*1101 (PDB ID:1X7Q), HLA*A2402 (PDB ID:5HGA), HLA-B*0702 (PDB ID:5EO1), HLA-B*0801 (PDB ID:3SPV), HLA-B*3501 (PDB ID:3LKN), HLA-DRB1*0101 (PDB ID:4AH2), HLA-DRB1*0401 (PDB ID:5LAX), HLA-DRB1*1101 (PDB ID:6CPL), and HLA-DRB1*1501 (PDB ID:5V4M) were downloaded. The 3D form of epitopes was then determined using the PEP-FOLD 3.5 server (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/) [61,62,63]. The docking analyses between the epitopes and the MHC molecules were performed using the ClusPro 2.0 server (https://cluspro.bu.edu/) [64]. This web-based docking tool accomplishes rigid docking by sampling billions of conformations, energy minimization, and pairwising the root means square deviation (RMSD) of the complexes and estimates the binding energy score of the protein–protein docked complex based on the shaped complementarity. The best clusters (complexes) of epitopes and MHC alleles were selected for the construction of our vaccine based on the lowest docking energy score. In the end, PyMol was used to visualize the interaction between epitopes and MHC alleles.
Prediction of population coverage
In computational vaccine design, the population coverage study reveals the worldwide efficacy of the designed vaccine by evaluating the prevalence of human leukocyte antigen (HLA) alleles related to the epitope of interest. The combined coverage of the selected T lymphocyte epitopes with their corresponding MHC class I and II alleles as the input data were analyzed by the IEDB population coverage tool (http://tools.iedb.org/population/) [65].
Designing the mRNA vaccine construct
The desired immunogenic mRNA vaccine construct contains five main elements in its open reading frame (ORF): (1) the Kozak sequence, (2) epitopes, (3) adjuvant, (4) linkers (or spacers), and (5) stop codon. The start codon should be a part of the Kozak sequence for obvious [66, 67]; however, the sequence surrounding the stop codon may be optimized [68]. The selected epitopes that are included in the vaccine construct are antigenic, non-allergenic, non-toxic, and cytokine inducer (for HTLs only). Along with the appropriate antigenic epitopes, a potent adjuvant is critical to boost immune response [69]. In this study, 50S ribosomal protein L7/L12 was added to the vaccine construct as the adjuvant. The 50S ribosomal protein L7/L12 sequence was retrieved from the UniProt database (UniProt ID: P9WHES) [70]. The adjuvant and the LBL epitopes were linked via the EAAAK linker. Intra-LBL epitopes were combined using GPGPG linkers. Moreover, the GPGPG linker was used to conjugate LBL, HTL, and CTL epitopes. Intra-CTL epitopes were joined using AAY linkers.
Tissue plasminogen activator (tPA) secretory signal sequence (UniProt ID: P00750) and MHC I-targeting domain (MITD) (UniProt ID: Q8WV92) were added into the 5′ and 3′ regions of the ORF, respectively. The signal peptide was linked to the adjuvant by the AAY linker.
7-methyl(3-O-methyl) GpppG Cap (also known as ARCA) capped the 5′ end and then 5′ UTR from human β-globin (NCBI ID: NM 000,518.5) and 3′ UTR from rabbit β-globin gene (GenBank ID: V00882.1) flanked the 5′ end and 3′ end of the vaccine mRNA, respectively. In the 3′ end of the vaccine, a 120-nucleotide long poly(A) tail was also added.
Prediction of antigenicity, allergenicity, toxicity, and physicochemical properties of the final vaccine construct
The translated peptide form of the mRNA vaccine was used for predicting the different properties of the designed vaccine. Only adjuvant and the selected epitopes alongside with their linkers were considered for evaluation of the final vaccine; tPA and MITD sequences were excluded, as they will be cleaved while entering the secretory pathway and the MHC-I pathway, respectively. The antigenicity of the vaccine was predicted by the VaxiJen v2.0 [38] and ANTIGENpro [40] servers. To predict whether the vaccine is an allergen or not, AllerTOP 2.0 [46] and allergenFP v1.0 [48] servers were used. The toxicity of the vaccine peptide was evaluated using the ToxinPred server [45]. ProtParam (https://web.expasy.org/protparam/) server [71] was used to predict various physiochemical properties of vaccine construct such as amino acid composition, molecular weight (Mw), theoretical isoelectric point (pI), instability index (II), aliphatic index (AI), and grand average of hydropathicity (GRAVY).
Analysis of post‐translational modification
Post-translational modification (PTM) analysis, including glycosylation, phosphorylation, and acetylation of the designed vaccine construct, was carried out using NetNGlyc 1.0, NetPhos 3.0, and NetAct 1.0 servers, which are available at http://www.cbs.dtu.dk/services/. Additionally, lipid PTMs as N-terminal glycines myristoyl and GPI-modification were evaluated in the vaccine construct, by MyrPS/NMT (https://mendel.imp.ac.at/myristate/) [72] and big-PI/GPI animals servers (https://mendel.imp.ac.at/gpi/gpi_server.html) [73], respectively.
Tertiary structure modelling and validation
Several 3D models of the vaccine were generated using trRosetta server (https://yanglab.nankai.edu.cn/trRosetta/). This server uses a deep residual neural network to predict the inter-residue distance and orientation distributions of the input sequence. Then, it converts predicted distance and orientation distributions into smooth restraints to build 3D structure models-based on direct energy minimization [74,75,76]. The best model of the vaccine was chosen after validation by the SAVES server (https://saves.mbi.ucla.edu/) that included ERRAT [77], Verify-3D [78, 79], and PROCHECK (by Ramachandran Plot analysis) [80] tools as well as the ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) [81, 82]. The nonbonded atomic interactions are examined by ERRAT [77] while Verify-3D analyzes the compatibility of the 3D protein model with its amino acid sequence [83]. The geometrical and stereochemical constraints of the 3D protein model by PROCHECK in the Ramachandran Plot [80]. ProSA-web provides an easy-to-use interface to the program ProSA. It calculates an overall quality score for a specific input structure, which is displayed in the context of all known protein structures. Besides, any problematic parts of a structure are shown and highlighted in a 3D molecule viewer. If the calculated score falls outside the characteristic range of native proteins, the structure likely contains errors [81].
Molecular docking analysis of the vaccine peptide
The ClusPro 2.0 server (https://cluspro.bu.edu/) was used to evaluate the interaction between the vaccine peptide and toll-like receptor 4 (TLR4), as well as MHC class I and class II receptors [64]. The ClusPro 2.0 server can predict protein–protein interactions using the PIPER docking algorithm, which generates four sets of models using different scoring schemes (balanced, electrostatic-favored, hydrophobic-favored, and van der Waals + electrostatics). The crystal structures of TLR 4 (PDB ID: 4G8A), TLR9 (UniProt ID: Q9NR96), C-type lectin 2 (PDB ID: 3WBP), MHC class I receptor (PDB ID:1I1Y), and MHC class II receptor (PDB ID: 1KG0) were retrieved from RCSB PDB (https://www.rcsb.org/) [59, 60] and used as receptor molecules in the docking study, while the vaccine peptide was used as the ligand molecule.
The PROtein binDIng enerGY prediction (PRODIGY) tool of the HADDOCK server (https://wenmr.science.uu.nl/prodigy/) [84] was utilized to compute the binding energy of receptor-ligand interactions based on their 3D structure. Relying on intermolecular contacts and properties obtained from non-interface surfaces, the PRODIGY server can predict the binding free energy (ΔG) and dissociation constant (Kd). The FireDock server (https://bioinfo3d.cs.tau.ac.il/FireDock/) [85, 86] was used to refine and re-score the docked complexes. It ranks the complexes based on binding score and global binding energy. The interacting residues between the vaccine and TLR4 were mapped using the PDBsum server (https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html) [87].
Molecular dynamic simulation of the vaccine-TLR4 complex
The iMODS server (http://imods.chaconlab.org/) [88] was utilized for the molecular dynamic (MD) studies of the vaccine-TLR4 complex. Based on the 3D structure of the given protein, the iMODS server is used for normal mode analysis, mobility (NMA B-factors), deformability, eigenvalues, covariance matrix, and linking matrix. The vaccine-TLR4 complex with the lowest binding energy was used for dynamics simulation analysis.
Codon optimization and secondary structure prediction of the mRNA vaccine
Codon optimization of the final peptide vaccine construct is considered as a crucial step for the efficient translation of the mRNA vaccine within the host cells. The codons of the final vaccine were optimized for efficient expression in human cells by using the GenSmart Codon Optimization Tool server (https://www.genscript.com/tools/gensmart-codon-optimization).
In the next step, the rare codon analysis tool (https://www.genscript.com/tools/rare-codon-analysis) was used for the assessment of the quality of the optimized codons. Three parameters having key roles in an optimized protein expression in the host expression system were calculated: (1) Codon adaptation index (CAI), which shows the translation efficiency of the mRNA, (2) codon frequency distribution (CFD), which represents the presence of any tandem unusual codons, and (3) GC content.
The optimized DNA sequence generated by the GenSmart codon optimization tool was then converted to RNA sequence by the DNA > RNA > Protein tool (http://biomodel.uah.es/en/lab/cybertory/analysis/trans.htm).
Afterward, the RNAfold tool of ViennaRNA Package 2.0 (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) [89] was used to predict the secondary structure of the mRNA construct. The server predicts the mRNA structures thermodynamically and assigns a minimal free energy score (MFE). Hence, the MFE structure and the centroid secondary structure, as well as their minimum free energy, were evaluated for further analysis.
In silico immune simulation
The C-IMMSIM server (https://kraken.iac.rm.cnr.it/C-IMMSIM/) was used to perform an in silico immune simulation to estimate the real-life immunogenic and immune response of the designed peptide vaccine [90]. The server is an agent-based model that uses position-specific scoring matrices (PSSM) derived from machine learning techniques for predicting immune interactions. According to the most currently used vaccines, the minimum recommended interval between the first and second doses is 4 weeks [91]. Three doses of injection, each containing 1000 antigen proteins and no LPS, were administered at 1, 84, and 168 time-steps (the first dose was given at time = 0, and each time-step equals to 8 h of real life). The other parameters, including random seed, simulation volume, and simulation step were kept at 12345, 10 µl, and 1050, respectively.
Results
Retrieval of protein sequences
The proteome of BKV consists of six proteins; the protein sequences were downloaded from the Uniprot database, which are shown in Table. 1 with their corresponding accession numbers.
Antigenic proteome selection
The results obtained from VaxiJen v2.0 and ANTIGENpro servers revealed that five out of the six proteins were antigenic (Table 1), which were selected as vaccine targets for the in silico analysis of mRNA vaccine development against BKV.
Prediction and assessment of CTL epitopes
A total of 63 CTL epitopes, binding to MHC-I supertype alleles, were selected according to high-scored and overlap** epitope regions resulted from both IEDB and RANKPEP servers. Thirty-two epitopes out of 63 were predicted to be positive in terms of immunogenicity by the IEDB class I immunogenicity tool, and 18 out of these 32 epitopes were identified as antigen by the VaxiJen server. Out of these 18 epitopes, 18 were founded to be non-toxic by the ToxinPred server, and 14 were non-allergen by both AllerTOP 2.0 and AllergenFP v1.0 servers. Out of the 14 non-allergen epitopes, four were selected as final CTL epitopes based on the lowest docking energy score with their corresponding MHC alleles evaluated using ClusPro 2.0 server. The final epitopes are highlighted in red (Table S1).
Prediction and assessment of HTL epitopes
The results of overlap** peptide regions of IEDB and RANKPEP MHC-II binding tools led our epitope selection to 29 high-scored HTL epitopes for eight MHC-II supertype alleles to be used in our vaccine construct. Ten out of 29 epitopes were predicted as an antigen by the VaxiJen server. All the 10 antigenic epitopes were determined as a non-toxic by ToxinPred, and only three of them were identified as a non-allergen by both AllerTOP 2.0 and AllergenFP v1.0 servers. After considering the IFN-γ, IL-4, and IL-10 inducibility utilizing IFNepitope, IL4pred, and IL10pred servers, respectively, only one epitope met all the listed criteria as to be selected for vaccine construction (Table 2, and Table S2).
Prediction and assessment of LBL epitopes
Bepipred Linear Epitope Prediction 2.0 predicted 53 LBL epitopes of variable length ranging from six to 29 amino acids. Thirty-six of them were identified as an antigen by the VaxiJen server. Among them, 32 epitopes were non-toxic, and 15 epitopes were non-allergen. Out of the 15 epitopes, four most antigenic ones were selected; all of them were recognized as IgG-epitopes by the Igpred server (Table 2).
Epitope conservancy analysis
The conservancy of the nine final selected epitopes analyzed by the IEDB conservancy tool indicated epitopes as high conserved regions (Table 3).
Molecular docking between T lymphocyte epitopes and MHC alleles
Five out of 15 selected T lymphocyte epitopes were chosen for the vaccine construct based on the best cluster members, and the lowest weighted energy score of the complex of epitopes and MHC alleles. The selected epitopes with their corresponding MHC alleles are shown in supplementary Tables S1 and S2.
Prediction of population coverage
Population coverage of the selected T lymphocyte epitopes used in the vaccine construct was the focus of our IEDB population coverage analysis. The global population coverage of our vaccine was 93.77%. Europe, North America, and Oceania regions showed the highest rate of regional coverage among all with the percentage of 97.54%, 94.07%, and 92.69%, respectively, while Central America showed the least coverage with the percentage of 8.48% (Fig. 2).

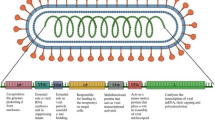
The workflow of designing the mRNA vaccine against BKV
Notably, MHC molecules are highly polymorphic, and over a thousand different human MHC alleles have been known. In this regard, MHC molecules present a vastly varying binding specificity to the cognate epitopes. Thus, the population coverage scores are different among various ethnicities.
Vaccine construction
The final construct of our mRNA vaccine contained the following part in order from the N-terminal to the C-terminal direction as shown in Fig. 3.

Schematic presentation of the designed mRNA BKV vaccine
7-methyl(3-O-methyl) GpppG (ARCA) Cap–5′UTR–Kozak sequence–tPA (Signal peptide)–AAY linker–50S ribosomal protein L7/L12 (Adjuvant)–EAAAK linker–LBL epitope–GPGPG linker–LBL epitope–GPGPG linker–LBL epitope–GPGPG linker–LBL epitope–GPGPG linker–HTL epitope–GPGPG linker–CTL epitope–AAY linker–CTL epitope–AAY linker–CTL epitope–AAY linker–CTL epitope–AAY linker–MITD sequence–Stop codon–3′UTR–Poly (A) tail.
Prediction of antigenicity, allergenicity, toxicity, and physicochemical properties of the final vaccine construct
The results of antigenicity, allergenicity, toxicity, and physiochemical properties analyses determined the vaccine to be safe and suitable for the human usage (Table 4).
VaxiJen and ANTIGENpro both predicted the vaccine is an antigen with scores of 0.5553 and 0.884519, respectively. AllerTOP 2.0 and AllergenFP v1.0 both projected the vaccine is a non-allergen. ToxinPred evaluated the vaccine is non-toxic. In the context of physicochemical properties, the vaccine peptide consists of 359 amino acids, and its molecular weight is 37.91 kDa. The pI of the vaccine was calculated to be 7.70, revealing that it is almost basic in nature; the pI of a positively charged protein is greater than 7. Among the 359 amino acid residues, 45 were negatively charged (Asp + Glu) and 46 were positively charged (Arg + Lys). The instability index (II) of 30.34 was computed for the vaccine peptide, implying that the construct would be stable after expression (II of > 40 indicates instability) [92]. The aliphatic index was calculated as 79.47, indicating the peptide construct is thermostable [93]. The GRAVY was calculated to be negative (− 0.271), which revealed the hydrophilic nature of the vaccine. The vaccine peptide has a half-life of 30 h in human reticulocytes (in vitro), > 20 h in yeast (in vivo), and > 10 h in Escherichia coli (in vivo).
Analysis of post‐translational modifications
Various PTMs were predicted for the final vaccine construct. The analysis performed by the NetNGlyc-1.0 server revealed that there was no N-glycosylation site in the vaccine construct. Prediction of phosphorylation modification carried out by the NetPhos-3.0 server displayed 19 phosphorylation sites (Ser: 8, Thr: 11, Tyr: 0) in the construct. No N-acetylation site was found by the NetAcet-1.0 server. Additionally, there was no lipid PTMs at the N-terminal glycine myristoyl, and GPI-modification predicted by MyrPS/NMT and the big-PI/GPI animals servers, respectively (Table S3).
Tertiary structure modelling and validation
The 3D model of the final vaccine was built by the trRosetta web server; five models were generated. The best model was chosen based on the higher quality of the estimated TM-score and validation results of ERRAT, Verify-3D, PROCHECK, and ProSA-web servers and are shown in Fig. 4A (Table S4). The final selected 3D model had the ERRAT value of 94.774 and the Verify 3D score of 61.28% (Fig. 4B and C). The Ramachandran plot analysis of the final model revealed that 94.2%, 5.5%, 0.3%, and 0% of residues are located in the most favored regions, additional allowed regions, generously allowed regions, and disallowed regions, respectively (Fig. 4D). Moreover, the Z-score calculated by the ProSA-web server was − 5.44 (Fig. 4E).

Tertiary structure prediction and validation of the vaccine peptide: A 3D structure of the vaccine peptide B ERRAT analysis C verify 3D analysis D Ramachandran plot analysis from PROCHECK server and E Z-score analysis from Pro-SA web
Molecular docking analysis of the vaccine peptide
Molecular docking analysis was performed to confirm the interaction between the designed vaccine peptide and its receptors. Using ClusPro 2.0 server, the peptide vaccine was docked with TLR 4 (PDB ID: 4G8A), TLR9 (UniProt ID: Q9NR96), C-type lectin 2 (PDB ID: 3WBP), MHC class I receptor (PDB:1I1Y), and MHC class II receptor (PDB: 1KG0). The docked poses with the highest cluster member were subsequently analyzed for binding affinities and dissociation constants at 37 °C utilizing the PRODIGY web server, which provided the ∆G values of the vaccine-TLR4, vaccine-TLR9, vaccine-C-type lectin 2, vaccine-MHC I receptor, and vaccine-MHC II receptor complexes as − 18.7, − 13.0, − 15.8, − 15.5, and − 12.2, respectively (Supplementary Figs S1 and S2). It can be concluded that all the interactions were energetically feasible, which is evident from the negative values of Gibbs free energy and the low dissociation constant. Moreover, the ∆G value of the vaccine-TLR4 complex was greater among other studied receptors, indicating the specificity of the docked model. The corresponding ∆G values along with the dissociation constant (Kd) values of the docked complexes are shown in Table 5.
The refinement process of the docked complexes conducted by the FireDock server revealed the global energy of the vaccine-TLR4, vaccine-MHC I receptor, and vaccine-MHC II receptor complexes were − 173.38, − 156.08, and − 201.61, respectively (Table 6). The docked complex of vaccine-TLR4 along with their interacting residues evaluated by PDBsum server are shown in Fig. 5A and B.


A Vaccine-TLR4 complex B Interacting residues between docked TLR4 (chain A) and vaccine (chain E) predicted by PDBsum C Deformability graph D B-factor graph E Eigenvalue (F) Covariance matrix (correlated residues in red, uncorrelated in white or anti-correlated in blue motions) (G) Elastic network model (darker gray regions show stiffer regions)
Molecular dynamics simulation
To determine the stability and physical motions of the atoms and molecules of the designed vaccine construct, the MD simulation study was performed by the normal mode analysis (NMA) through the iMODS server; and the vaccine-TLR4 docked complex was subjected to our MD simulation study.
Peaks in the deformability graph reveal the deformable regions of the complex (Fig. 5C). The B-factor graph indicates the relationship between NMA and PDB regions in the complex (Fig. 5D). The eigenvalue for the complex was 9.859968e−07 (Fig. 5E), and it increased gradually in each mode during the dynamics. The covariance matrix (Fig. 5F) depicts the relationship between amino acid duplets, with the correlated residues colored in red, anti-correlated residues colored in white, and non-correlated residues colored in blue, all scattered in dynamical regions. The elastic network model, which is illustrated as a connecting matrix, classifies which atom pairs are connected by springs. As seen in Fig. 5G, the higher stiff areas were found in each of the separate chains of the complex. In conclusion, all results of the iMODS analysis indicated the decrease in mobility and deformability in the coordinates of the complex structure, which signifies the stability of the vaccine-TLR4 complex interface.
Codon optimization and secondary structure prediction of the mRNA vaccine
The codons of the vaccine construct, for efficient mRNA translation in the host cells, were optimized by the GenSmart codon optimization tool. The total length of the mRNA was 1077 nucleotides. The quality of the optimized sequence was assessed via the rare codon analysis tool. The estimated CAI value of the sequence was 0.92; a CAI value of greater than 0.8 is considered for acceptable expression in the host organism (Fig. 6A). The CFD of the sequence was 0%, suggesting that there were no unusual tandem codons in the optimized sequence. Tandem rare codons can reduce translation efficiency or even disengage the translational machinery (Fig. 6B). The average GC content of the adapted sequence was 62.95%, revealing that the vaccine candidate could be expressed well in human cells. The optimal GC content percentage range for efficient expression is between 30 and 70% (Fig. 6C). The secondary structures of the mRNA along with their corresponding free energies were evaluated by the RNAfold server. The MFE secondary structure had a minimum free energy of − 434.70 kcal/mol, while the centroid secondary structure had a minimum free energy of − 389.20 kcal/mol. These results suggest that the mRNA could remain stable after manufacturing (Fig. 6D and E).

Codon optimization and secondary structure prediction of designed mRNA vaccine:(A) CAI value, (B) CFD value, (C) GC curve, (D) MFE secondary structure and (E) Centroid secondary structure of the vaccine mRNA
In silico immune simulation studies
The simulated immunological response was investigated through the C-ImmSim server.
The primary response was characterized by high IgM levels. B-cell populations and the levels of IgG1 + IgG2, IgM, and IgG + IgM antibodies were increased enormously after the second and third injections with concomitant reduction of the antigen concentration, which indicates the emergence of immunological memory and thereby proficient immunity upon subsequent exposures to the antigen (Fig. 7A and B). The TH (T helper) and TC (T cytotoxic) cell populations responded remarkably with the corresponding memory development (Fig. 7C and D). After the third injection, IgG1 levels increased, while IFN-γ concentration and TH cell population remained high throughout the exposure period. The level of cytokines after each injection increased concomitantly reflected by the escalation of IFN-γ and IL-2, which are considered as the most significant cytokines for an anti-viral immune response (Fig. 7E).

In silico simulation of immune response using vaccine as an antigen: (A) Antigen and immunoglobulins, (B) B-cell population, (C) TH (helper) cell population (D) TC (cytotoxic) cell population per state, (E) Cytokine and interleukin production
Discussion
Human polyomavirus type 1, or BKV, is a ubiquitous pathogen that causes various comorbidities and related malignancies in the human population. It is a significant risk factor for immunocompromised individuals, specifically in kidney transplant recipients, by causing renal transplant dysfunction and allograft loss [21, 24]. The efficacy of therapeutics in clinical studies is currently not as much as the desired amount, and only the limited studies have addressed immunoinformatic approaches to design vaccine targets for BKV [12, 94]. In this study, a reverse vaccinology strategy was applied to design a mRNA vaccine against BKV by identifying the most antigenic proteins of the pathogen.
Conventional vaccine approaches, such as live-attenuated, inactivated pathogens, and subunit vaccines are efficient for the prevention of different diseases; however, they do not have met the need for a completely safe and non-allergenic vaccine with a rapid development and large-scale production criteria [95, 96]. Peptide-based vaccines have been reported with lower immunogenicity indexes, especially when used alone without adjuvants [97, 98]. Plasmid DNA vaccines, known as the third generation of vaccines, evoke safety issue concerns such as activation of oncogenes due to genomic incorporation of immunizing DNA and the possible power of eliciting anti-DNA antibodies [99, 100]. Major technological innovation and research investment over the past decade demonstrate that the rapidly growing field of mRNA therapeutic agents has become a potent platform to solve many of the challenges in vaccine development for both infectious disease and cancer [101]. The use of mRNA vaccine is prior to subunit, killed and live-attenuated virus, as well as DNA-based vaccines, due to several features of that; as mRNA is a non-infectious and non-integrating platform, there is no potential risk of infection or insertional mutagenesis. Moreover, the in vivo half-life of mRNA can be regulated by various modifications and delivery methods, as it is degraded by normal cellular processes [102,103,104,105]. The immunogenicity of mRNA vaccines can also be modified and downmodulated to increase their safety profile [106]. Comparatively, low cost of production, capability of rapid development, and acceptable efficacy of vaccine could be as other beneficial aspects of mRNA vaccine. Due to their cytoplasmic expression without the need of entering into the nucleus, they possess a higher efficacy than DNA vaccines [107]. The mere alteration of a mRNA sequence can express almost every targeting protein with new indications, and utilizing the already established production process results in versatility, flexibility, time-saving, and cost-reduction manufacturing [67, 98, 108]. Though mRNA vaccines have some inherent drawbacks such as immunogenicity, instability, and delivery inefficiency, recent advances in synthesis technology and structural modifications of mRNA sequences have led to some prosperous results [109,110,111,112,113]. Considering that the first two mRNA vaccines developed by Pfizer/BioNTech (BNT162b2) and Moderna (mRNA-1273) were approved by the FDA through emergency use authorization in the USA in December 2020 [114, 115].
To our knowledge, this is the first study to design a mRNA multi-epitope BKV vaccine by identifying the most-antigenic epitopes in the entire BKV proteome.
Vaccination could provide immunological memory, which can persist for several years to several decades [116,117,118]. Stimulation of both B and T lymphocyte-mediated immunity is considered vital for any successful vaccination strategy by providing a faster and more efficient immune response in the host upon encountering the target pathogen in the future.
The cytotoxic activity of CTLs is an important part of the immune response in viral infections. Virus-infected cells degrade some of the viral proteins and present them to the CTLs in combination with MHC I molecules. The recognition of degraded parts of viral proteins, called epitopes, by CTLs leads to the killing of infected cells by releasing of cytotoxic molecules [119]. In our study, 63 CTL epitopes were identified. The highest score of binding along with overlap** regions was detected by using a variety of methods for further analysis of immunogenicity, antigenicity, allergenicity, and toxicity. Following the analysis, 14 epitopes were confirmed to be both safe and antigenic, which were suitable to be incorporated in the vaccine construct (Supplementary Table S1).
After the proteolytic cleavage of viral antigens, antigen-presenting cells display viral particles to HTLs, also known as CD4 + T cells, in combination with MHC-II molecules, which causes activation of HTLs. Afterward, the HTLs secrete a wide range of cytokines and chemokines such as IFN-γ, IL-4, and IL-10, which play vital roles in the immune response against the pathogen [120,121,122,123]. Here, 29 HTL high-scored and shared peptide regions were selected for further analyses of antigenicity, allergenicity, toxicity, and cytokine-inducing properties such as IFN-γ, IL-4, and IL-10. After evaluating the epitopes, only one epitope had all the proper criteria and was selected in the vaccine construct (Table 2).
B lymphocytes secrete specific antibodies to neutralize specific viral pathogens. Through differentiation into long-lived plasma cells and memory B lymphocytes, long-term immunological protection is provided [124, 125]. The activation of B lymphocytes happens through the binding of B cell receptors to either soluble or membrane-bound epitopes [126]. Reliable prediction of B lymphocyte epitopes via various computational tools is a crucial part of designing an efficient vaccine [127, 128]. There are two types of B cell epitopes, linear and conformational B cell epitopes [127]. In our investigation, 53 LBL epitopes were discovered for further analysis; after evaluating them for antigenicity, allergenicity, toxicity, and IgG inducibility, four LBLs were identified as suitable for using in the vaccine construct (Table 2).
According to the conservancy results of the selected epitopes among BKV strains, high protection against this viral invader was predicted (Table 3).
Molecular docking is an integral part of vaccine design studies, which is widely used to evaluate the binding affinity of CTL and HTL epitopes to their corresponding MHC alleles.
Our selected 15 T lymphocytes (14 CTLs and one HTL) epitopes had 12 corresponding MHC alleles. We subjected each of these CTL and HTL epitopes and one of their corresponding alleles to molecular docking. Among them, only four CTLs had the best cluster members and the lowest energy score, which indicates the highest binding affinity between them. Surprisingly, the only HTL epitope had the acceptable binding energy and cluster members and was confirmed to be involved in our vaccine construct (Supplementary Tables S1 and S2). All of these 5 T lymphocytes were finally selected for the vaccine construct (Table 2).
The population coverage analysis of the T lymphocyte epitopes upon the distribution of MHC alleles around the globe revealed a decent worldwide coverage of 93.77%. Furthermore, a higher degree of coverage was predicted for countries that were reported as the most affected regions by BKV (Fig. 2).
In our study, a potential multi-epitope mRNA vaccine model against the BKV, including the T- and B-cell epitopes was constructed. The TLR-4 agonist, 50S ribosomal protein L7/L12 protein from Mycobacterium tuberculosis, was used as an adjuvant to enhance the immune responses. 50S ribosomal protein L7/L12 induces MyD88 and TRIF signaling pathways and can enhance dendritic cell (DC) activation and Th1 polarization [129]. The translation efficiency and half-life of mRNA are influenced by the length of the poly(A) tail. Protein expression was elevated and prolonged in our mRNA vaccine with an A120 tail [130]. The 5′ UTR of the human β-globin gene, which can improve mRNA translation efficiency, and the 3′ UTR of the rabbit β-globin gene, which can modify antibody titer, seroconversion, and cytokine profiles, were also added to the vaccine construct [130, 131]. The 7-methyl(3-O-methyl)GpppG Cap (also known as ARCA [132, 133]) was used to cap the 5′ end. It is founded that the 5′ end cap and the 3′ end poly(A) tail synergistically regulate mRNA translational efficiency [134]. The MITD sequence was added as it has been shown to enhance CD4 + T cell antigen presentation efficiency [135]. The signal peptide of the tissue plasminogen activator (tPA) of Homo sapiens was also added into the vaccine to improve the immunogenicity of that [136].
The selection of an appropriate epitope-specific linker is a crucial step in designing a multi-epitope vaccine, so that the domains can work independently, avoiding interference between them. Linkers used in this study were chosen based on their length and rigidity-flexibility properties [137, 138]. The EAAAK linker was incorporated between epitopes and adjuvant to improve the bioactivity of fused protein and to reach a high level of expression and an increase in the stability of the vaccine construct [138]. Previous studies indicated that GPGPG and AAY linkers added between predicted the HTL and CTL epitopes, respectively, produced synergistic immunogenicity, consequently allowing the rational design construction of a potent multi-epitope vaccine [139, 140]. Our designed mRNA vaccine construct has a total length of 1077 nucleotides.
The computational analysis of the translated peptide from our mRNA construct predicted that the vaccine is almost neutral, stable, highly antigen, non-allergen, hydrophilic, and thermostable with a longer half-life in the human reticulocyte, making it as a potential candidate for mRNA vaccine (Table 4).
Analyses of various post-translational modification sites within the target protein indicated that there were only 19 sites of phosphorylation modification. Some studies indicate that the phosphorylation of some proteins causes them to be degraded by the ATP-dependent ubiquitin/proteasome pathway [141]. Since this construct contains several sites for aforesaid modification, it may improve its degradation, and subsequent entry into the MHC-I pathway (Supplementary Table S3).
In the context of the 3D model of the vaccine, according to ERRAT, Verify 3D, Ramachandran plot, and ProSA-web evaluations, the quality of the vaccine 3D model prepared by the trRosetta web server, has a high quality and there was no need for refinement (Fig. 4 and Supplementary Table S4).
The structure of the vaccine peptide was docked against TLR 4, MHC-I and MHC-II receptors, next the refinement of the best complex was performed. The lowest docking energy score was used to select the top-ranked vaccine-TLR4 complex structure for the MD simulation analysis. The MD simulation study of the vaccine-TLR4 and complex confirmed its stable nature at atomistic conditions (Tables 5 and 6).
The codon improvement of the designed mRNA sequence for better expression in the human host was performed using a codon optimization tool. Several properties of the optimized sequence demonstrated that the mRNA would be expressed efficiently by the host cells. The predicted CAI value of 0.92 suggested that the codons of the vaccine can efficiently express in the human cells (Fig. 6A). Moreover, the predicted CFD value suggested that the mRNA sequence contained no unusual tandem codons that can reduce the translation efficiency in the host cell (Fig. 6B). The free energies and secondary structures results of the optimized sequence indicated that the mRNA construct will be stable as well (Fig. 6D and A).
In silico immune simulation was performed to evaluate the immunogenic potential of our target antigen. The vaccination regime of the only two FDA-approved mRNA-based vaccines developed by Pfizer/BioNTech (BNT162b2) and Moderna (mRNA-1273) has widely been established at day 0 and day 21 or 28, respectively and followed by at least 6 months resting period between the second and the third dose [142,143,144,145,146]. Therefore, we conducted another study according to the Pfizer/BioNTech (BNT162b2) and Moderna (mRNA-1273) vaccination regime, where the designed vaccine was administered at 1, 63, 873 time-steps (the first dose was given at time = 0, and each time-step equals to 8 h of real life). The other parameters were as same as the main conducted study. The results indicated that the main vaccination scheme, including 1, 84, and 168 time-steps had overall superior immunological responses. The results indicated a general increase in immune responses following repeated exposure to the antigen (Fig. 7). B-cell and T-cell populations both responded remarkably with corresponding memory development. The levels of IL-2 and IFN-γ were also increased, which are cardinal for immune system activation in the case of viral infection.
Conclusion
Our immunoinformatic-based approach for designing a multi-epitope mRNA vaccine against BKV demonstrated the proposed novel vaccine model as an efficient candidate. However, it should be tested and determined by meticulous in vitro and in vivo studies and clinical trials for further validation of its efficacy.
Supplementary information
Data Availability
All data and material of this study are publicly available.
References
Gardner S, et al. New human papovavirus (BK) isolated from urine after renal transplantation. The Lancet. 1971;297(7712):1253–7.
Polz, D., A. Stec, and M. Polz-Dacewicz, BK-virus (BKV)–structure, epidemiology and pathogenesis. Journal of Pre-Clinical and Clinical Research, 2013. 7(2).
Mäntyjärvi R, et al. A human papovavirus (BK), biological properties and seroepidemiology. Ann Clin Res. 1973;5(5):283–7.
Shah KV, Daniel RW, Warszawski RM. High prevalence of antibodies to BK virus, an SV40-related papovavirus, in residents of Maryland. J Infect Dis. 1973;128(6):784–7.
Portolani M, et al. Prevalence in Italy of antibodies to a new human papovavirus (BK virus). J Med Microbiol. 1974;7(4):543–6.
Gardner SD. Prevalence in England of antibody to human polyomavirus (BK). Br Med J. 1973;1(5845):77–8.
Das D, Shah RB, Imperiale MJ. Detection and expression of human BK virus sequences in neoplastic prostate tissues. Oncogene. 2004;23(42):7031–46.
Knowles WA, et al. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol. 2003;71(1):115–23.
McCaffrey J, Bhute VJ, Shenoy M. BK virus infection and outcome following kidney transplantation in childhood. Sci Rep. 2021;11(1):1–8.
Knowles, W.A., Discovery and epidemiology of the human polyomaviruses BK virus (BKV) and JC virus (JCV). Polyomaviruses and human diseases, 2006: p. 19–45.
Corallini, A., et al., Evidence for BK virus as a human tumor virus. Human polyomaviruses: molecular and clinical perspectives, 2001: p. 431–60.
Ambalathingal GR, et al. BK polyomavirus: clinical aspects, immune regulation, and emerging therapies. Clin Microbiol Rev. 2017;30(2):503–28.
Portolani M, et al. Restricted replication of BK virus in human lymphocytes. Microbiologica. 1985;8(1):59–66.
Bofill-Mas S, Pina S, Girones R. Documenting the epidemiologic patterns of polyomaviruses in human populations by studying their presence in urban sewage. Appl Environ Microbiol. 2000;66(1):238–45.
Chatterjee M, Weyandt TB, Frisque RJ. Identification of archetype and rearranged forms of BK virus in leukocytes from healthy individuals. J Med Virol. 2000;60(3):353–62.
Boldorini R, et al. BK virus sequences in specimens from aborted fetuses. J Med Virol. 2010;82(12):2127–32.
Taguchi F, et al. Transplacental transmission of BK virus in human. Jpn J Microbiol. 1975;19(5):395–8.
Taguchi F, Kajioka J, Shimada N. Presence of interferon and antibodies to BK virus in amniotic fluid of normal pregnant women. Acta Virol. 1985;29(4):299–304.
Saade A, Styczynski J, Cesaro S. BK virus infection in allogeneic hematopoietic cell transplantation: an update on pathogenesis, immune responses, diagnosis and treatments. J Infect. 2020;81(3):372–82.
Goudsmit J, et al. The role of BK virus in acute respiratory tract disease and the presence of BKV DNA in tonsils. J Med Virol. 1982;10(2):91–9.
Sawinski D, Goral S. BK virus infection: an update on diagnosis and treatment. Nephrol Dial Transplant. 2015;30(2):209–17.
Dörries K. Molecular biology and pathogenesis of human polyomavirus infections. Dev Biol Stand. 1998;94:71–9.
Shah, K. and P. Howley, Papillomaviruses. Virology. 1995, Raven Press Ltd., New York.
Reploeg MD, Storch GA, Clifford DB. BK virus: a clinical review. Clin Infect Dis. 2001;33(2):191–202.
Drachenberg CB, Papadimitriou JC, Ramos E. Histologic versus molecular diagnosis of BK polyomavirus–associated nephropathy: a shifting paradigm? Clin J Am Soc Nephrol. 2006;1(3):374–9.
Vago L, et al. JCV-DNA and BKV-DNA in the CNS tissue and CSF of AIDS patients and normal subjects Study of 41 cases and review of the literature. J Acquir Immune Defic Syndr. 1996;12(2):139–46.
Daveson KL, et al. BK virus-associated progressive multifocal leukoencephalopathy. Med J Aust. 2013;198(4):216–8.
Cabrejo L, et al. Progressive BK virus associated multifocal leukoencephalopathy in an immunocompromised patient treated with corticosteroids. Revue Neurologique. 2005;161(3):326–30.
Randhawa PS, et al. Human polyoma virus-associated interstitial nephritis in the allograft kidney1. Transplantation. 1999;67(1):103–9.
Purighalla R, et al. BK virus infection in a kidney allograft diagnosed by needle biopsy. Am J Kidney Dis. 1995;26(4):671–3.
Santeusanio AD, Lukens BE, Eun J. Antiviral treatment of BK virus viremia after kidney transplantation. Am J Health Syst Pharm. 2017;74(24):2037–45.
Wu S-W, Chang H-R, Lian J-D. The effect of low-dose cidofovir on the long-term outcome of polyomavirus-associated nephropathy in renal transplant recipients. Nephrol Dial Transplant. 2009;24(3):1034–8.
Brennan DC, et al. Incidence of BK with tacrolimus versus cyclosporine and impact of preemptive immunosuppression reduction. Am J Transplant. 2005;5(3):582–94.
Pezzotti P, et al. The impact of immunization programs on 10 vaccine preventable diseases in Italy: 1900–2015. Vaccine. 2018;36(11):1435–43.
Tomar, N. and R.K. De, Immunoinformatics: a brief review. Immunoinformatics, 2014: p. 23–55.
Khalili S, et al. Computational vaccinology and epitope vaccine design by immunoinformatics. Acta Microbiol Immunol Hung. 2014;61(3):285–307.
María R, et al. The impact of bioinformatics on vaccine design and development. Vaccines. 2017;2:3–6.
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8(1):1–7.
Doytchinova IA, Flower DR. Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine. 2007;25(5):856–66.
Magnan CN, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936–43.
Reynisson B, et al. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–54.
Stern LJ, Wiley DC. Antigenic peptide binding by class I and class II histocompatibility proteins. Structure. 1994;2(4):245–51.
Reche PA, Glutting J-P, Reinherz EL. Prediction of MHC class I binding peptides using profile motifs. Hum Immunol. 2002;63(9):701–9.
Calis JJ, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol. 2013;9(10):e1003266.
Gupta S, et al. In silico approach for predicting toxicity of peptides and proteins. Plos One. 2013;8(9):e73957.
Dimitrov I, et al. AllerTOP v. 2—a server for in silico prediction of allergens. J Mol Model. 2014;20(6):1–6.
Dimitrov, I., D.R. Flower, and I. Doytchinova. AllerTOP-a server for in silico prediction of allergens. in BMC bioinformatics. 2013. BioMed Central.
Dimitrov I, et al. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30(6):846–51.
Nielsen M, Lund O. NN-align An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10(1):1–10.
Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8(1):1–12.
Sidney J, et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 2008;4(1):1–14.
Sturniolo T, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17(6):555–61.
Andreatta M, et al. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics. 2015;67(11):641–50.
Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8(1):1–15.
Dhanda, S.K., et al., Prediction of IL4 inducing peptides. Clinical and Developmental Immunology, 2013. 2013.
Nagpal G, et al. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci Rep. 2017;7(1):1–10.
Jespersen MC, et al. BepiPred-20: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45(W1):W24-w29.
Bui HH, et al. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics. 2007;8:361.
Burley SK, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019;47(D1):D464–74.
Berman HM, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–42.
Lamiable A, et al. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016;44(W1):W449–54.
Shen Y, et al. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J Chem Theory Comput. 2014;10(10):4745–58.
Thévenet P, et al. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012;40(W1):W288-93.
Kozakov D, et al. The ClusPro web server for protein–protein docking. Nat Protoc. 2017;12(2):255–78.
Bui HH, et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics. 2006;7:153.
Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44(2):283–92.
Schlake T, et al. Develo** mRNA-vaccine technologies. RNA Biol. 2012;9(11):1319–30.
Liu Q. Comparative analysis of base biases around the stop codons in six eukaryotes. Biosystems. 2005;81(3):281–9.
Lima KM, et al. Vaccine adjuvant: it makes the difference. Vaccine. 2004;22(19):2374–9.
Consortium U. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–15.
Gasteiger, E., et al., Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook, 2005: p. 571–607.
Maurer-Stroh S, Eisenhaber B, Eisenhaber F. N-terminal N-myristoylation of proteins: refinement of the sequence motif and its taxon-specific differences. J Mol Biol. 2002;317(4):523–40.
Eisenhaber B, Bork P, Eisenhaber F. Prediction of potential GPI-modification sites in proprotein sequences. J Mol Biol. 1999;292(3):741–58.
Du Z, et al. The trRosetta server for fast and accurate protein structure prediction. Nat Protoc. 2021;16(12):5634–51.
Su, H., et al., Improved protein structure prediction using a new multi‐scale network and homologous templates. Advanced Science, 2021: p. 2102592.
Yang J, et al. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci. 2020;117(3):1496–503.
Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–9.
Bowie JU, Lüthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253(5016):164–70.
Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356(6364):83–5.
Laskowski RA, et al. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283–91.
Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(2):W407–10.
Sippl MJ. Recognition of errors in three-dimensional structures of proteins: Proteins Struct Funct Bioinforma. 1993;17(4):355–62.
Eisenberg D, Lüthy R, Bowie JU. [20] VERIFY3D: assessment of protein models with three-dimensional profiles. In: Methods in enzymology. Elsevier; 1997. p. 396–404.
Xue LC, et al. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics. 2016;32(23):3676–8.
Andrusier N, Nussinov R, Wolfson HJ. FireDock fast interaction refinement in molecular docking. Proteins Struct Funct Bioinforma. 2007;69(1):139–59.
Mashiach E, et al. FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008;36(2):W229–32.
Laskowski RA, et al. PDBsum: structural summaries of PDB entries. Protein Sci. 2018;27(1):129–34.
López-Blanco JR, et al. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 2014;42(W1):W271–6.
Gruber AR, et al. The vienna RNA websuite. Nucleic Acids Res. 2008;36(2):W70–4.
Rapin N, et al. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. Plos One. 2010;5(4):e9862.
Castiglione F, et al. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput Math Methods Med. 2012;2012:842329.
Walker, J.M., The proteomics protocols handbook. 2005: Springer.
Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem. 1980;88(6):1895–8.
Kesherwani V, Tarang S. An immunoinformatic approach to universal therapeutic vaccine design against BK virus. Vaccine. 2019;37(26):3457–63.
Plotkin SA. Vaccines: the fourth century. Clin Vaccine Immunol. 2009;16(12):1709–19.
Tandrup Schmidt S, et al. Liposome-based adjuvants for subunit vaccines: formulation strategies for subunit antigens and immunostimulators. Pharmaceutics. 2016;8(1):7.
Li W, et al. Peptide vaccine: progress and challenges. Vaccines. 2014;2(3):515–36.
Ahammad I, Lira SS. Designing a novel mRNA vaccine against SARS-CoV-2: An immunoinformatics approach. Int J Biol Macromol. 2020;162:820–37.
Redding L, Weiner DB. DNA vaccines in veterinary use. Expert Rev Vaccines. 2009;8(9):1251–76.
Harrison R, Bianco A. DNA immunization with Onchocerca volvulus genes, Ov-tmy-1 and OvB20: serological and parasitological outcomes following intramuscular or GeneGun delivery in a mouse model of onchocerciasis. Parasite Immunol. 2000;22(5):249–57.
Pardi N, et al. mRNA vaccines — a new era in vaccinology. Nat Rev Drug Discov. 2018;17(4):261–79.
Karikó K, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16(11):1833–40.
Kauffman KJ, Webber MJ, Anderson DG. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J Control Release. 2016;240:227–34.
Guan S, Rosenecker J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. 2017;24(3):133–43.
Thess A, et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol Ther. 2015;23(9):1456–64.
Kariko K, et al. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011;39(21):e142–e142.
Sahin U, Karikó K, Türeci Ö. mRNA-based therapeutics—develo** a new class of drugs. Nat Rev Drug Discov. 2014;13(10):759–80.
Fotin-Mleczek M, et al. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J Immunother. 2011;34(1):1–15.
Tsui NB, Ng EK, Lo YD. Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clin Chem. 2002;48(10):1647–53.
Chen N, et al. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life. 2017;69(5):297–304.
Petsch B, et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol. 2012;30(12):1210–6.
Geall AJ, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci. 2012;109(36):14604–9.
Pardi N, et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. 2017;543(7644):248–51.
Anand P, Stahel VP. Review the safety of Covid-19 mRNA vaccines: a review. Patient Saf Surg. 2021;15(1):1–9.
Negahdaripour M, et al. Administration of COVID-19 vaccines in immunocompromised patients. Int Immunopharmacol. 2021;99:108021.
Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;357(19):1903–15.
Kareko BW, et al. Persistence of neutralizing antibody responses among yellow fever virus 17D vaccinees living in a nonendemic setting. J Infect Dis. 2020;221(12):2018–25.
Combadiere B, et al. Distinct time effects of vaccination on long-term proliferative and IFN-γ–producing T cell memory to smallpox in humans. J Exp Med. 2004;199(11):1585–93.
Rosendahl Huber S, et al. T cell responses to viral infections–opportunities for peptide vaccination. Front Immunol. 2014;5:171.
Dittmer U, et al. Role of interleukin-4 (IL-4), IL-12, and gamma interferon in primary and vaccine-primed immune responses to Friend retrovirus infection. J Virol. 2001;75(2):654–60.
Chesler DA, Reiss CS. The role of IFN-γ in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002;13(6):441–54.
Rojas, J.M., et al., IL-10: a multifunctional cytokine in viral infections. Journal of immunology research, 2017. 2017.
Luckheeram, R.V., et al., CD4+ T cells: differentiation and functions. Clinical and developmental immunology, 2012. 2012.
Dörner T, Radbruch A. Antibodies and B cell memory in viral immunity. Immunity. 2007;27(3):384–92.
Do RK, et al. Attenuation of apoptosis underlies B lymphocyte stimulator enhancement of humoral immune response. J Exp Med. 2000;192(7):953–64.
Janeway Jr, C.A., et al., B-cell activation by armed helper T cells, in Immunobiology: the immune system in health and disease. 5th edition. 2001, Garland Science.
Ahmad TA, Eweida AE, Sheweita SA. B-cell epitope map** for the design of vaccines and effective diagnostics. Trials Vaccinology. 2016;5:71–83.
EL‐Manzalawy, Y., D. Dobbs, and V. Honavar, Predicting linear B‐cell epitopes using string kernels. Journal of Molecular Recognition: An Interdisciplinary Journal, 2008. 21(4): p. 243–255.
Lee SJ, et al. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. Plos One. 2014;9(8):e104351.
Holtkamp S, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108(13):4009–17.
Zinckgraf JW, Silbart LK. Modulating gene expression using DNA vaccines with different 3′-UTRs influences antibody titer, seroconversion and cytokine profiles. Vaccine. 2003;21(15):1640–9.
Stepinski J, et al. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl (3′-O-methyl) GpppG and 7-methyl (3′-deoxy) GpppG. Rna. 2001;7(10):1486–95.
Pasquinelli A, Dahlberg JE, Lund E. Reverse 5’caps in RNAs made in vitro by phage RNA polymerases. RNA. 1995;1(9):957–67.
Gallie DR. The cap and poly (A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991;5(11):2108–16.
Kreiter S, et al. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol. 2008;180(1):309–18.
Kou Y, et al. Tissue plasminogen activator (tPA) signal sequence enhances immunogenicity of MVA-based vaccine against tuberculosis. Immunol Lett. 2017;190:51–7.
Chen X, Zaro JL, Shen W-C. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357–69.
Arai R, et al. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001;14(8):529–32.
Meza B, et al. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: an in silico approach. Infect Genet Evol. 2017;49:309–17.
Khatoon N, Pandey RK, Prajapati VK. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep. 2017;7(1):1–12.
Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30(1):405–39.
Park JW, et al. mRNA vaccines for COVID-19: what, why and how. Int J Biol Sci. 2021;17(6):1446.
Jackson, L.A., et al., An mRNA vaccine against SARS-CoV-2—preliminary report. New England journal of medicine, 2020.
Polack, F.P., et al., Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. New England journal of medicine, 2020.
Falsey AR, et al. SARS-CoV-2 neutralization with BNT162b2 vaccine dose 3. N Engl J Med. 2021;385(17):1627–9.
Hogan MJ, Pardi N. mRNA vaccines in the COVID-19 pandemic and beyond. Annu Rev Med. 2022;73:17–39.
Funding
This study was supported by a Grant from the Shiraz University of Medical Sciences, Shiraz, Iran.
Author information
Authors and Affiliations
Contributions
N. N. and M. N. designed the study. Y. M. and S. E. performed bioinformatic analyses. Y. M. and M. Z. wrote the first draft of the manuscript. All authors commented on and edited previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable. No procedures requiring ethics approval were performed in this study.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.

Supplemantary Fig S1
(PNG 238 KB)

Supplemantary Fig S2
(PNG 163 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
Cite this article
Mohammadi, Y., Nezafat, N., Negahdaripour, M. et al. In silico design and evaluation of a novel mRNA vaccine against BK virus: a reverse vaccinology approach. Immunol Res 71, 422–441 (2023). https://doi.org/10.1007/s12026-022-09351-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-022-09351-3