
Abstract
Purpose of Review
Cardiac consequences occur in both acute COVID-19 and post-acute sequelae of COVID-19 (PASC). Here, we highlight the current understanding about COVID-19 cardiac effects, based upon clinical, imaging, autopsy, and molecular studies.
Recent Findings
COVID-19 cardiac effects are heterogeneous. Multiple, concurrent cardiac histopathologic findings have been detected on autopsies of COVID-19 non-survivors. Microthrombi and cardiomyocyte necrosis are commonly detected. Macrophages often infiltrate the heart at high density but without fulfilling histologic criteria for myocarditis. The high prevalences of microthrombi and inflammatory infiltrates in fatal COVID-19 raise the concern that recovered COVID-19 patients may have similar but subclinical cardiac pathology. Molecular studies suggest that SARS-CoV-2 infection of cardiac pericytes, dysregulated immunothrombosis, and pro-inflammatory and anti-fibrinolytic responses underlie COVID-19 cardiac pathology. The extent and nature by which mild COVID-19 affects the heart is unknown. Imaging and epidemiologic studies of recovered COVID-19 patients suggest that even mild illness confers increased risks of cardiac inflammation, cardiovascular disorders, and cardiovascular death. The mechanistic details of COVID-19 cardiac pathophysiology remain under active investigation.
Summary
The ongoing evolution of SARS-CoV-2 variants and vast numbers of recovered COVID-19 patients portend a burgeoning global cardiovascular disease burden. Our ability to prevent and treat cardiovascular disease in the future will likely depend on comprehensive understanding of COVID-19 cardiac pathophysiologic phenotypes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
COVID-19 was introduced to the world as a respiratory viral infection ranging from a pneumonia to acute respiratory distress syndrome. As the pandemic spread, its specter proved to extend beyond the respiratory system. Critically ill COVID patients commonly had multi-organ failure. Autopsies revealed histopathology in not only the lungs but also multiple extrapulmonary organs, including the heart, brain, kidney, and liver. The primary and secondary mechanisms by which SARS-CoV-2 infection causes these extrapulmonary manifestations of COVID-19 remain under active investigation. This review aims to highlight current knowledge about the effects of COVID-19 on the heart, as derived from clinical, imaging, autopsy, and molecular studies. Our understanding of COVID-19 and its effects on the heart continues to grow and evolve at a record pace. Still, the insights summarized here can foretell the potential long-term cardiovascular effects of COVID-19 in recovered patients. These data may guide novel approaches to the prevention and treatment of cardiac complications in acute and post-acute COVID-19.
Clinical Presentation of Cardiac Complications in Acute and Post-Acute COVID-19
Cardiac Effects of COVID-19 Are Heterogeneous
It became clear, early in the pandemic, that COVID-19 does not spare the heart [1]. Many acute COVID-19 patients also present with cardiovascular syndromes. These cardiac clinical presentations are heterogenous and include arrhythmias, acute on chronic congestive heart failure, right heart failure, stress cardiomyopathy, cardiogenic shock, myocarditis, and multisystem inflammatory syndrome in adults (MIS-A) [2, 3••, 4,5,6,7,8,9,10]. Occasionally, acute COVID-19 patients develop new-onset acute heart failure or new-onset atrial fibrillation. Despite high suspicion of its causal role, acute COVID-19 associated myocarditis is extraordinarily rare, occurring in 2.4 per 1,000 acute COVID-19 hospitalizations, according to a recent international, retrospective, observational study [11••]. In the rare instances that it does occur, acute COVID-19 related myocarditis commonly presents as fulminant heart failure, requiring inotropic support or temporary mechanical circulatory support [11••]. Phenotypic heterogeneity has also been noted even among patients with acute fulminant COVID-19 myocarditis. Patients with concurrent multisystem inflammatory syndrome in adults (MIS-A) suffered severe systemic inflammation and presented later in the post-COVID-19 period than did those without MIS-A. The MIS-A-positive patients developed myocardial dysfunction more gradually, rarely had detectable SARS-CoV-2 on RT-qPCR, and frequently had positive COVID-19 serology. Strikingly, in-hospital survival rates were better for those patients with acute COVID-19 myocarditis and concurrent MIS-A than those without concurrent MIS-A [3••].
In general, pre-existing cardiac conditions (i.e., atrial fibrillation, coronary artery disease, and congestive heart failure) and risk factors for heart disease (i.e., obesity, diabetes, hypertension, and tobacco abuse) have been associated with an increased mortality risk in COVID-19 patients [12, 13]. Hypothesized mechanisms of the increased mortality risk in this population include limited physiologic reserve, a relatively immunocompromised state, and increased propensity for an inflammatory response [14]. The mechanisms that underlie the clinical heterogeneity of COVID-19 remain under investigation, but studies to date suggest that differential host immune responses yield the wide range of disease acuity and severity.
With most of the US population now protected against severe SARS-CoV-2 via infection- or vaccination-induced immunity, the severity and mortality rate of acute COVID-19 illness have decreased significantly [15, 16]. Since cardiac injury is associated with worse COVID-19 severity, it is unclear whether, how, and for how long mild COVID-19 might affect the heart. Even if its incidence declines, the prevalence of COVID-19 cardiac effects will remain high considering the cumulative millions of people who have been infected with SARS-CoV-2.
Abnormalities in Systemic Inflammatory Biomarkers, ECG, and Cardiac Imaging Are Commonly Detected in Moderate to Severe COVID-19 Illness
Given the heterogeneity of COVID-19 cardiac involvement, diagnostic evaluation may include cardiac biomarkers (i.e., troponin and natriuretic peptides), electrocardiography, and cardiac imaging (e.g., echocardiography and cardiac magnetic resonance imaging). Acute cardiac injury, defined as a serum cardiac troponin (cTn) level above the 99th percentile of the upper reference limit, presented in as many as a third of hospitalized patients during the initial surge of the pandemic. Acute cardiac injury in COVID-19 was associated with increased mortality risk, regardless of any underlying chronic cardiovascular disorders [17]. Moreover, the risk of death in COVID-19 increased with the magnitude of the troponin elevation [18••]. Lower elevations of cTn were associated with approximately twice the mortality risk of those without cardiac injury, while the highest elevations of cTn were associated with a three-fold greater mortality risk. Similarly, elevations in B-type natriuretic peptide and N-terminal pro-B-type natriuretic peptide, to at least half their normal thresholds, have also been associated with increased mortality in COVID-19 [19].
Despite its simplicity as a diagnostic test, an ECG can predict cardiac and respiratory outcomes as well as overall survival [20]. ECG findings of atrial fibrillation/flutter, right ventricular strain, and ST segment abnormalities were associated with a two- to threefold increased risk of death or mechanical ventilation [21]. Despite their association with severe COVID-19 illness, ventricular arrhythmias had an overall prevalence of less than 10% in cases of acute COVID-19 [22].
Cardiac involvement of COVID-19 can also be revealed by imaging, most commonly transthoracic echocardiography (TTE). Nearly half of acute COVID-19 patients without a pre-existing echocardiographic abnormality had an abnormal TTE, most commonly with findings of left and/or right ventricular dysfunction [23, 24]. Abnormal TTE findings reportedly changed the clinical management in a third of hospitalized COVID-19 patients. When coupled with serum troponin level, TTE abnormalities help identify the highest risk patients. COVID-19 patients with elevated cTn and abnormal TTEs were reported to have an in-hospital mortality of 31.7% [25]. Early in the pandemic, concerns about intra-procedural transmission of SARS-CoV-2 led to a marked decline of invasive diagnostic tests. For suspected acute myocarditis, cardiac magnetic resonance (CMR) imaging studies were preferred over endomyocardial biopsies for detecting any myocardial edema and myocardial inflammation/ early fibrosis [26].
Cardiac Complications Are Common in Post-Acute Sequelae of COVID-19 (PASC)
Among individuals who recover from COVID-19, one in 5 may suffer from post-acute sequelae of COVID-19 (PASC) or “long COVID” [27]. Cardiac manifestations of PASC are relatively common. A retrospective study of US veterans found that those who recovered from COVID-19 within 30 days of illness onset had increased burden and risk of cardiovascular disease compared to matched controls [28]. The greatest burden among recovered COVID-19 patients was cardiac arrhythmia, which occurred in 20 per 1,000 patients with 1.7 times the risk of matched controls. The arrhythmias included atrial fibrillation/flutter, ventricular arrhythmias, and persistent sinus tachycardia. Heart failure was the next most common, occurring in 12 per 1,000 recovered patients with 1.7 times the risk of controls. Though less prevalent than either arrhythmias or heart failure at 4 per 1,000 people, venous thromboembolism occurred with a risk 2–3 times that of matched controls. Pericarditis and myocardial infarction each occurred in 1–3 per 1,000 recovered COVID-19 patients with 1.6–1.8 times the risk. Myocarditis was rare (0.3 per 1,000 people) but occurred with the highest risk of cardiovascular disease (5.4 times control) in this cohort of veterans. Overall, there were 23 excess cardiac events per 1,000 patients, with higher risk among those with more severe COVID-19 illness. A CDC study of nearly 2,000,000 patients found similar burden and risk of cardiac disease, supporting a true association between COVID-19 recovery and long-term cardiovascular complications [27]. Proposed mechanisms for the elevated cardiac risk following COVID-19 include a persistent pro-inflammatory state, endothelial dysfunction, hypercoagulopathy, and residual damage from the initial myocardial stress. Longitudinal studies of PASC patients, including analyses of their biospecimens and cardiac imaging, will be vital for testing these theories.
Patients who were previously infected with SARS-CoV-2 may develop cardiac complications with or without cardiovascular symptoms. The American College of Cardiology has devised an expert consensus decision pathway for the clinical management of post-COVID-19 patients suspected to have cardiovascular involvement [29••]. The suggested framework for evaluating and managing these patients takes a multimodal approach that is centered around cardiac imaging.
Impact of COVID-19 on Cardiac Structure and Function
During the initial pandemic surge, cardiac imaging was heavily limited by their relatively elective nature, the overwhelming patient care burden on beleaguered health care systems, and the scarcity of personal protective equipment (PPE). These initial constraints eased over time; clinical management of COVID-19 advanced and PPE and hospital resources became available. These dynamic circumstances impacted the early data that could be obtained about the heart in acute COVID-19 illness due to the original SARS-CoV-2 strain. Nonetheless, cardiac imaging of acutely ill patients and recovered patients has established some of our fundamental understanding about the cardiac effects of COVID-19.
Severity of Ventricular Dysfunction Increases with COVID-19 Severity and Co-Morbidities
Echocardiography studies of acutely ill COVID-19 patients commonly found biventricular dysfunction, typically of greater severity in patients with pre-existing cardiac disease. In a study of approximately 100 patients hospitalized with acute COVID-19, nearly 70% were found to have some echocardiographic abnormality [24]. This cohort was largely comprised of older men (mean age 66 years), most of whom had hypertension, diabetes, obesity, and/or coronary artery disease. Patients with more severe COVID-19 had right ventricular (RV) dilatation and dysfunction. Importantly, baseline TTE findings of lower left ventricular ejection fraction (LVEF) and elevated LV filling pressure (reflected by E/e’) were associated with clinical deterioration and higher death rates, consistent with other reports [30]. Abnormal TTE findings were also found in the majority (55%) of 1272 patients in an international COVID-19 study [23]. This study population was also comprised chiefly of older men (70% male, median age 62 years) with cardiovascular co-morbidities. These patients generally had more severe COVID-19; 60% were critically ill. In this COVID-19 cohort, 39% had LV abnormalities and a third had RV abnormalities. The abnormalities included new myocardial infarction (3%), myocarditis (3%), stress cardiomyopathy (2%), mild (19%) to severe (6%) RV impairment, RV dilatation (15%), elevated pulmonary artery pressures (8%), and RV pressure and/or volume overload (4%). About 1% had cardiac tamponade or endocarditis. Altogether, 15% of patients had severe dysfunction of either the LV or RV. Notably, patients without pre-existing cardiac disease were more likely to have normal echocardiograms, suggesting that COVID-19 may have worse effects on hearts with underlying structural or functional abnormalities.
Widespread, Non-Specific Myocardial Inflammation Is More Common than Myocarditis on CMR
For suspected myocarditis, pericarditis, or myopericarditis, CMR has the advantage over TTE for assessing cardiac structure, function, and tissue characteristics. In one case series, eight out of 10 acute COVID-19 patients with suspected myocarditis were confirmed as such, based upon CMR detection of diffuse myocardial edema and fulfillment of the modified Lake Louise criteria [31]. In a study of 100 symptomatic COVID-19 patients, 33% of whom were hospitalized, CMR at 2 months after illness onset showed cardiac structural and functional abnormalities in most patients [26]. Compared with healthy controls, 78% of the COVID-19 patients were found on CMR to have myocardial edema and lower LVEF; 32% had late gadolinium enhancement (LGE, sign of early fibrosis), and 22% had pericardial enhancement. In some of these patients, endomyocardial biopsy was performed and revealed active and ongoing lymphocytic infiltration. Hence, recently recovered COVID-19 patients with LGE on CMR may have subacute or resolved myocarditis.
Even without fulfilling the modified Lake Louise criteria for myocarditis, abnormal CMR findings can still have clinical and functional significance. Cardiopulmonary exercise testing (CPET) was coupled with CMR for 58 hospitalized COVID-19 patients who had recovered but continued to experience cardiac symptoms beyond 3 months after initial infection [32]. In this prospective, blinded study of PASC patients, lower maximal oxygen consumption (VO2max) on CPET was associated with initial CMR abnormalities suggestive of myocardial edema and inflammation at 2 months. At 6 months following acute COVID-19, all previously detected CMR abnormalities had resolved despite ongoing symptoms in some patients. This discordance remains to be explained but suggests the possibility that CMR is not sensitive enough to detect residual cardiac pathology.
Myocardial Inflammation and Its Resultant Cardiac Dysfunction in COVID-19 Are Transient
Although seldom used for diagnosing myocarditis, 18F-fluoro-deoxy-glucose (FDG) positron emission tomography (PET) is more sensitive than CMR in detecting myocardial inflammation and more specific in detecting chronic myocarditis [33]. In a study of 47 recovered COVID-19 patients, CMR and FDG-PET were concurrently performed at about 2 months following diagnosis and, for those with focal FDG uptake, again at another 2 months thereafter [34]. Only a small proportion (17%) of recovered patients had focal FDG uptake on their initial PET, indicating myocardial inflammation or, more specifically, metabolically active immune cells. CMR findings of myocardial inflammation were stronger for the FDG-PET positive patients; however, a couple FDG-PET positive patients had no CMR abnormalities. Regardless of CMR findings, all FDG-PET positive patients demonstrated worse ventricular systolic function and increased blood levels of systemic inflammatory biomarkers. On subsequent follow-up, cardiac imaging abnormalities, ventricular function, and inflammatory biomarker levels of all FDG-PET positive patients improved or normalized. The resolution of these abnormal findings suggests that inflammatory cardiac dysfunction in recovered COVID-19 patients is transient. Whether and how such transient ventricular dysfunction might still impact upon long-term outcomes, including vulnerability to future cardiac injury and risk of chronic cardiac disease or cardiovascular death, have yet to be determined.
Cardiac Microvascular Dysfunction, Rather than Coronary Artery Occlusion, May Underlie Acute Cardiac Injury in COVID-19
Acute cardiac injury is not only highly prevalent but also prognostic among hospitalized COVID-19 patients. Elevated blood levels of cTn are associated with greater COVID-19 illness severity and worse clinical outcomes, including increased risk of death. For some COVID-19 patients, acute ischemic ECG changes accompanied their elevated cTn levels, thereby suggesting acute myocardial ischemia or infarction (AMI) due to COVID-19. However, studies using invasive coronary angiography or computed tomography coronary angiography (CTCA) have revealed more nuanced vascular pathology. In a case series of 28 Italian COVID-19 patients with suspected AMI, focal culprit coronary obstructions were identified on invasive coronary angiography in most but not all the patients. Forty percent of the study cohort did not have any obstructive coronary artery disease, thereby suggesting MINOCA (myocardial infarction with non-obstructive coronary arteries) as the etiology of their acute cardiac injury [35]. In the general adult population, MINOCA accounts for 6% of all AMIs [36]. The higher prevalence of MINOCA amongst acute COVID-19 patients suggests that coronary microvascular dysfunction is associated with, possibly even causal to, COVID-19 cardiac injury. Likewise, in a study of 52 patients hospitalized for moderate to severe COVID-19, the majority (65%) of patients had normal coronary arteries on CTCA at 3 months following illness onset [37]. Furthermore, 20% of the cohort had mild or moderate, non-obstructive coronary disease. It remains to be confirmed whether underlying subclinical coronary artery and/or microvascular disease predisposes some COVID-19 patients to acute cardiac injury or, conversely, whether SARS-CoV-2 infection induces coronary microvascular dysfunction, thereby injuring the heart.
Characterization of COVID-19 Cardiac Histopathology
The high mortality rate of COVID-19, particularly in the first several months of the pandemic, was a devastating and unfathomable tragedy. Its aftershocks will be felt for years to come. At the same time, our understanding of COVID-19 histopathology grew exponentially because of the availability of autopsy tissue samples. Histologic and molecular analyses of post-mortem tissue samples provided some of the earliest information about the novel coronavirus and its pathology.
Acute Lymphocytic Myocarditis Is Rare in COVID-19
Initial suspicions ran high that SARS-CoV-2 myocarditis was responsible for the cardiac presentations of some acute COVID-19 patients. Two autopsy series early in the pandemic supported this theory. Each reported myocarditis in more than half their cases [38, 39••]. However, by the end of 2020, only 20 of 277 autopsied COVID-19 hearts (7.2%) reported across 22 publications had any histologic evidence of myocarditis [40]. Subsequent single-center studies of post-mortem COVID-19 hearts corroborate the estimated low prevalence of myocarditis in fatal COVID-19. Focal myocarditis was detected in only 2 of 50 (4%) post-mortem hearts in one study and 4 of 69 (6%) in another [41••, 42].
Macrophage Infiltration of Myocardial Interstitium Is Increased in Acute COVID-19 Myocarditis
Whereas acute lymphocytic myocarditis is exceedingly rare in fatal COVID-19, macrophage infiltrates are increased in the interstitium of post-mortem COVID-19 hearts. Macrophage infiltrates were seldom associated with cardiomyocyte injury and thus qualified only as inflammatory infiltrates, not myocarditis as defined by the histology-based Dallas criteria [43]. Yet, the density of interstitial myocardial CD68 + macrophages was significantly higher in post-mortem COVID-19 hearts with myocarditis than in those without myocarditis [44•, 45•]. In the rare cases of COVID-19 lymphocytic myocarditis, the densities of myocardial CD3 + T-cells and CD4 + helper T-cells, but not CD8 + cytotoxic T-cells, were also increased compared to COVID-19 hearts without myocarditis [44•, 45•].
Hence, COVID-19 myocarditis demonstrates the heterogeneity of myocarditides with regards to inflammatory cell type infiltrates. Cell signaling underlying these distinct inflammatory responses in COVID-19 is still being studied. It is widely believed that the infiltrating macrophages in the COVID-19 heart likely derive from circulating monocytes recruited to the myocardium, as is the case in the lungs of COVID-19 non-survivors. In severe COVID-19, SARS-CoV-2 infects the tissue resident macrophages of the lung, alveolar macrophages, and induces pyroptosis. The inflammatory cell death of infected alveolar macrophages further attracts inflammatory monocytes and monocyte-derived macrophages [46, 47•, 48].
Most Commonly Detected Acute Histopathology in Post-Mortem COVID-19 Hearts Is Microthrombi
Other acute histopathologic findings have also been reported in post-mortem COVID-19 hearts, including microvascular endothelial cell damage, cardiomyocyte degeneration, focal cardiac necrosis, focal inflammatory infiltrates, and microthrombi [41••, 44•, 49••, 50, 51••, 52]. Determining the prevalences of these histopathologic findings is difficult given limitations of autopsy studies. Different studies analyzed different pathologies, often focusing on a specific cardiac finding (e.g., cardiac necrosis, endothelial injury, or inflammatory infiltration). Some studies used routine hematoxylin and eosin (H&E) staining alone; others included immunohistochemical staining. Neither uniform methods nor standardized criteria were used across all studies. As such, even systematic reviews are limited by selection and reporting bias. Furthermore, COVID-19 autopsy studies span the multiple waves of the pandemic, and different strains of SARS-CoV-2 may differ in their effect upon the heart. During the initial wave of the pandemic, a single-center study used both H&E and immunohistochemical staining to assess all 6 abovementioned acute cardiac histopathologic findings in post-mortem COVID-19 hearts. Microthrombi were detected in 48 (70%) of the 69 COVID-19 non-survivors, rendering it the most commonly detected acute cardiac histopathology in this fatal COVID-19 cohort. Two thirds of the cohort exhibited 2 or more of the 6 cardiac histopathologic features [41••]. In another COVID-19 study, 14 (35%) of 40 post-mortem hearts had evidence of cardiomyocyte necrosis. Of the 14 hearts with necrosis, 9 also had microthrombi, suggesting that microthrombi are a major cause of cardiac necrosis in COVID-19 [51••]. However, the prevalence and extent of cardiac necrosis, in this and other autopsy studies, appear modest relative to the prevalence and magnitude of cardiac troponin level elevations in COVID-19 non-survivors. The discrepancy suggests that non-necrotic cardiomyocyte cell death (i.e., pyroptosis or necroptosis) may instead underlie the acute cardiac injury associated with fatal COVID-19. The role of these alternate forms of cardiomyocyte cell death remain to be proven.
Elevated Systemic Inflammatory Biomarkers, Extensive Neutrophil Infiltration, and Clot Composition and Structure Highlight Dysregulated Immunothrombosis in Critically Ill COVID-19 Patients
Inflammation plays an important role in both epicardial coronary artery thrombosis and intra-myocardial microthrombosis. Yet, how inflammation initiates clot formation differs significantly between the two processes. In the case of coronary thrombosis, chronic low-level and acute, local coronary inflammation, respectively, initiate and propagate atherosclerosis and cause plaque rupture and erosion. Blood comes into contact with exposed thrombogenic contents of the atherosclerotic plaque—tissue factor within the necrotic plaque core or collagen in areas of plaque endothelial denudation. In contrast, microthrombosis is a thromboinflammatory or immunothrombotic event—a reciprocally, synergistic, physiological response of the innate immune system and the coagulation cascade to defend against circulating pathogens (Fig. 1A). Coronary thrombosis is always pathologic. Microthrombosis is physiologic, unless immunothrombosis becomes dysregulated, as occurs in critical COVID-19.

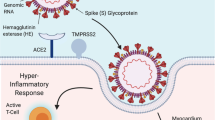
a SARS-CoV-2 infection triggers reciprocal activation of the complement and coagulation cascades. Activation of these pathways induces platelet activation and the release of neutrophil extracellular traps (a processes known as NET-osis). Consequently, a network of NETs and compact fibrin traps activated platelets, erythrocytes, and terminal complement component (membrane attack complex or MAC), thereby forming microthrombi. Green arrows point to cell types that are recruited by specified complement subunit. Solid orange arrow point to cell type activated by specified complement subunit. Dashed arrows highlight the effect of specified activated cell type on the extrinsic (purple) and intrinsic (orange) pathways of the coagulation cascade. b SARS-CoV-2 infection of cardiac and immune cells lead to excessive and dysregulated proinflammatory responses and cell death. Studies thus far suggest that SARS-CoV-2 infects cardiac pericytes and possibly cardiomyocytes through ACE2 receptors and, in the case of cardiomyocytes, type-II transmembrane serine proteases (TMPRSS2) co-receptor. Viral infection of these two cardiac cell types has been shown by in vitro studies to be productive. In vivo evidence of direct infection by SARS-CoV-2 is limited for cardiac pericytes and cardiomyocytes. Infected pericytes and cardiomyocytes are thought to undergo pyroptosis, a highly inflammatory programmed cell death. Endothelial cells are not infected by SARS-CoV-2 but become impaired when infected pericytes die. The resulting release of proinflammatory cytokines and chemoattractants then perpetuate the cycle of innate immune response dysregulation and pyroptosis. SARS-CoV-2 infects, and thereby activates, monocytes/macrophages and neutrophils. Circulating monocyte-derived macrophages and neutrophils are then recruited to the heart. Activated neutrophils release NETs into the circulation. NETs entangle erythrocytes, activated platelets, MAC, and compact fibrin, thereby promoting immunothrombosis/microthrombosis. Color of arrows and labels correspond to the respective cell type of the same color. Created with BioRender.com
The role of systemic inflammation in the pathophysiology of critical COVID-19 was clear early in the pandemic. Critically ill COVID-19 patients were reported to have markedly elevated biomarkers of systemic inflammation (i.e., lactate dehydrogenase, C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and interleukin-6) [53••, 54, 55]. The role of dysregulated immunothrombosis became more evident upon microscopic examinations of microthrombi and clinico-histological analyses of COVID-19 non-survivors [39••, 41••, 51••, 56]. Many COVID-19 non-survivors had extensive inflammation in multiple organs with a disproportionate presence of neutrophil infiltrate, and neutrophil extracellular traps (NETs) were detected in both macro- and microthrombi [39••]. Compared to non-COVID-19 coronary thromboemboli, COVID-19 cardiac microthrombi had more fibrin and abundant terminal complement complex C5b-9 [51••]. Not only was clot composition different but the architecture of COVID-19 fibrin clots was also distinct from that of non-COVID-19 patients and influenza-ARDS patients. Clots formed from plasma of severely ill COVID-19 patients demonstrated increased fibrin density, making them more resistant to fibrinolysis [56].
Mechanistic details of cardiac microthrombosis in COVID-19—the signaling that trigger and then promulgate immunothrombosis—have yet to be fully defined. However, molecular and cellular analyses of cardiac tissue samples and other biospecimens from COVID-19 non-survivors provide some insights.
Understanding Pathophysiology of COVID-19 Cardiac Effects via High-Resolution Molecular and Cellular Analyses
Cardiac injury in COVID-19 is heterogeneous in its causes and extent. Hence, its pathophysiology also varies with regards to the contribution of direct cardiac infection, microthrombi-associated local damage, and/or systemic inflammation. Hereafter, we describe the profile of COVID-19 associated cardiac damage as characterized by state-of-the-art molecular and imaging-based techniques. We also highlight the key insights into the important cellular players, molecular mechanisms, and potential therapeutic targets of COVID-19 cardiac injury.
Infection Potential of Cardiac Cells Is Estimated by Expression of ACE2 and Viral Entry Co-Factors
Now three years into this pandemic, the true extent of direct cardiac infection by SARS-CoV-2 and its subsequent impact on cardiac function remain controversial. Single cell/nucleus RNA sequencing (sc-/snRNA-seq) approaches have been used to identify the cell type populations most vulnerable to SARS-CoV-2 infection. By characterizing the cellular composition of the heart, these techniques have profiled the expression distribution of the primary SARS-CoV-2 receptor (ACE2) and target cell viral entry cofactors (e.g., TMPRSS2 and cathepsins). These expression data help determine the relative “infection potential” for each cell type. The expression of ACE2 is most abundant in the microvasculature-lining pericytes and lower, but still appreciable, in vascular smooth muscle cells, fibroblasts, and cardiomyocytes. Notably, in the failing heart, the relative expression of ACE2 changes—increasing in cardiomyocytes but decreasing in all other cell types [57•]. Whether this altered ACE2 expression is associated with worse clinical outcomes for those with pre-existing cardiovascular disease is unclear. Prior to the pandemic, these snRNA-seq techniques largely focused on studying healthy tissues. Even when employed on COVID-19 cases, snRNA-seq analyses are insensitive to viral RNA due to the cytoplasmic (extra-nuclear) residence of SARS-CoV-2 [58].
Evidence of SARS-CoV-2 Infection of the Heart
Estimates of viral presence in the heart vary widely from cohort to cohort when viral RNA is measured by real-time PCR amplification from tissue samples. This variability may in part be due to the temporal dynamics of viral clearance and/or tissue sampling error. In one study that examined tissue samples of both the left and right ventricles, SARS-CoV-2 was detected in 60% of COVID-19 non-survivors [41••]. In others, however, virus was detected in heart tissue in the minority of cases [49••, 59–63]. The low rates of viral detection in autopsy cardiac tissue mirror those reported in endomyocardial biopsies [64•, 65]. Even though viral particles have been observed across series of organs, the low viral presence in cardiac tissue is consistent with the limited viremia observed in COVID-19 patients [66,67,68]. Microscopic analysis of SARS-CoV-2-positive cardiac tissue show that internalized virions reside locally, around microthrombi or in large regions of immune cell infiltrates [69, 70], rather than diffusely throughout the myocardium. This focused distribution pattern of SARS-CoV-2 in cardiac tissue supports the possibility and likelihood that tissue sampling error contributed to reported low rates of cardiac tissue viral load.
Identifying Cardiac Cellular Tropism of SARS-CoV-2
Cardiomyocytes
While the detection of SARS-CoV-2 in heart tissue has varied across studies of COVID-19 patients, basic cardiovascular scientists have interrogated the infection potential of different cardiac cell types across various experimental platforms. In vitro cardiomyocyte infection via endosomal internalization of SARS-CoV-2 has been shown in stem cell models of human cardiomyocytes [69, 71]. Cardiomyocyte infection leads to viral replication and release, cell death, impaired contractile and electrophysiological function, and increased expression of pro-inflammatory pathways [69, 72,73,74,75,76]. Despite such easy in vitro infection of isolated cardiomyocytes, cardiomyocytes in organoid models, and cardiomyocytes within cardiac tissue slice preparations, in vivo evidence for cardiomyocyte infection in COVID-19 patients is limited. Transmission electron microscopy and in situ labeling of viral RNA have sporadically detected viral particles in cardiomyocytes of a small fraction of post-mortem cardiac samples [72, 77]. Given the elevated cTn levels commonly observed in severely ill COVID-19 patients, the paucity of evidence for direct viral infection of cardiomyocytes suggests the predominance of indirect mechanisms of cardiomyocyte damage.
Vascular Endothelial Cells and Pericytes
A notable pathology of acute COVID-19 is endothelial dysfunction of the coronary microvasculature, but not the larger epicardial coronary vessels. Microvascular endothelial dysfunction observed in COVID-19 hearts includes pro-inflammatory endothelial cell activation, loss of junctional integrity, and cell death [78]. Early studies suggested that direct infection of the vascular endothelium initiated the observed microvascular damage [50, 78]. However, the most recent evidence indicate that endothelial cells in fact have low SARS-CoV-2 “infection potential” since they lack ACE2 expression [74, 79•, 80]. Instead, the detection of viral RNA in the microvasculature may be explained by the direct infection of pericytes—multifunctional, vascular mural cells with multiple processes that wrap around endothelial cells lining the microvasculature. In in vitro studies, mere exposure to the SARS-CoV-2 spike protein is sufficient to alter pericyte function, thereby reducing endothelial cell support, stimulating pro-inflammatory cascades, and promoting endothelial cell death [81]. As detection of SARS-CoV-2 in the heart is significantly associated with endothelial damage [41••], infection of pericytes rather than endothelial cells may in fact serve as the primary mechanism (Fig. 1B). In fact, a recent study demonstrated ex vivo SARS-CoV-2 infection of human primary cardiac pericytes via an endosomal pathway [82••]. The same study reported two cases of COVID-19 myocarditis, wherein SARS-CoV-2 RNA was detected, via in situ hybridization, in PDGFR-β (platelet derived growth factor receptor beta, a receptor tyrosine kinase expressed by mural cells)+ cells located in the perivascular space of the post-mortem hearts. Despite its limitations, this evidence strongly suggests that direct SARS-CoV-2 infection of cardiac pericytes occurs in acute, fatal COVID-19 myocarditis.
Molecular and Cellular Signaling of Cardiac Microthrombosis
A predominant aspect of microvascular pathology in COVID-19 is the presence of microthrombi, a feature of COVID-19 that has been found in the heart, lungs, and other organs [41••, 49••, 51••, 83••, 84]. Hyperactivated platelets, including those which contain viral RNA, express and secrete factors (e.g. S100A8/S100A9) which stimulate microvascular endothelial activation and weaken endothelial junctions, thereby initiating immunothrombosis [85,86,87]. This prothrombotic interaction between endothelial cells and platelets does not exist in isolation; snRNA-seq analysis comparing microthrombi-positive and microthrombi-negative ventricular tissue samples of COVID-19 non-survivors demonstrated specific alterations in fibroblast autocrine signaling and transcriptional response as well [41••]. While canonical fibroblast activation was not widespread, genes encoding prothrombotic, anti-fibrinolytic, and pro-inflammatory responses (e.g., SERPINE1 and THBS2) were specifically upregulated in cardiac fibroblasts of hearts with observable microthrombi.
Importantly, the temporal and causative relationship of these events is challenging to disentangle. To do so, adequate clinical stratification, robust sample sizes, uniform and standardized analytic frameworks, and generous amounts of primary tissue samples will be necessary. Animal models must innately have SARS-CoV-2 infectivity or be genetically modified to be so. More complex human cellular models will need to be developed to reflect the multicellular events that injure the heart. New models and new tools must have the flexibility to assess and incorporate both past strains of SARS-CoV-2 and future variants.
Management of Patients with COVID-19 Cardiac Complications
At present, there is limited literature on the use of guideline directed medical therapy (GDMT), anti-inflammatory agents, or anti-viral drugs specifically for treating COVID-19 cardiac complications. Consensus decision pathway guidelines for management of COVID-19 cardiovascular complications essentially align with major cardiovascular society guidelines, regardless of patient history of COVID-19 [29••]. Whether during acute illness or post-acute sequelae of COVID-19, the cardiac presentation determines clinical management. As such, COVID-19 patients presenting with acute myocardial ischemia and infarction should be managed with anticoagulation, anti-platelet therapy, β-blocker, statin, and immediate coronary angiography and coronary reperfusion or revascularization, as indicated by early risk stratification [88, 89]. Critically ill COVID-19 patients with fulminant myocarditis should receive intensive care as aggressive as that for non-COVID-19 fulminant myocarditis patients [90]. In fact, case series have reported that early intervention with veno-arterial extracorporeal membrane oxygenation (VA ECMO) improved the survival of COVID-19 patients with fulminant myocarditis [91,92,93]. Similarly, the four major pillars of heart failure GDMT—-β-blockers, mineralocorticoid receptor antagonists (MRA), angiotensin receptor/neprilysin inhibitor (ARNI), and sodium-glucose co-transporter-2 inhibitors (SGLT2i)—should be initiated in patients with new onset heart failure, whether associated with acute COVID-19 illness, PASC, or recovery from acute COVID-19 [94, 95].
Despite increasing insights into the pathophysiological mechanisms of COVID-19 microvascular dysfunction and microthrombosis, effective therapeutic interventions remain elusive. In a retrospective, multi-center study of 178 critically ill COVID-19 patients in Europe, treatment with dexamethasone was associated with reduced peak biomarker levels of inflammation and cardiac injury, as well as decreased rates of pulmonary embolism [96]. Such findings suggest that an anti-inflammatory approach may be necessary as an adjunctive therapy against COVID-19 cardiac complications.
Given the relationship between cardiac injury severity and acute COVID-19 illness severity, the most effective management of COVID-19 cardiac complications may in fact be vaccination against SARS-CoV-2 which lowers COVID-19 hospitalization rates [17, 18••, 97]. Only time will tell whether cardiac effects are less likely or less significant with “breakthrough” COVID-19. In the meanwhile, clinical and translational scientists continue to investigate the epidemiology and pathophysiological mechanisms of COVID-19 cardiac effects.
Conclusion
This review only touches upon a small portion of all that researchers have learned about COVID-19 and its cardiac effects. Even as our scientific understanding rapidly grows, much remains unknown regarding clinical and immune phenotypes, optimal imaging modalities and diagnostic tests, risk stratification, and long-term cardiac consequences. Hundreds of millions of people around the world have recovered from acute COVID-19 and, on average, almost a million new cases of COVID-19 worldwide continue to be reported daily [98, 99]. Our current understanding of the cardiac effects of COVID-19 portend a potentially overwhelming, global cardiovascular disease burden in the not-so-distant future. Multi-disciplinary collaborations between clinicians and scientists that employ wide-ranging approaches will be essential for staving off and overcoming this challenge. The global response to COVID-19 has shown that collectively and collaboratively, we can accelerate scientific and medical advances at an historic rate.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, et al. COVID-19 and cardiovascular disease. Circulation. 2020;141(20):1648–55. https://doi.org/10.1161/circulationaha.120.046941.
Alvarez-Garcia J, Jaladanki S, Rivas-Lasarte M, Cagliostro M, Gupta A, Joshi A, et al. New heart failure diagnoses among patients hospitalized for COVID-19. J Am Coll Cardiol. 2021;77(17):2260–2. https://doi.org/10.1016/j.jacc.2021.03.006.
•• Barhoum P, Pineton de Chambrun M, Dorgham K, Kerneis M, Burrel S, Quentric P, et al. Phenotypic heterogeneity of fulminant COVID-19--related myocarditis in adults. J Am Coll Cardiol. 2022;80(4):299–312. https://doi.org/10.1016/j.jacc.2022.04.056. This single-center retrospective study analyzed clinical, biological, and immunological characteristics of 38 patients who were admitted to the intensive care unit for suspected fulminant myocarditis and laboratory-confirmed SARS-CoV-2 infection. Findings revealed two distinct phenotypes of COVID-19 related myocarditis differentiated by the presence of absence of multisystem inflammatory syndrome in adults (MIS-A). Patients without concurrent MIS-A had worse morbidity and mortality than those with MIS-A. The hyperinflammatory cytokine profiles of each also differed, suggesting that distinct immunologic pathophysiology underlies distinct phenotypes of COVID-19 cardiac sequelae.
Bangalore S, Sharma A, Slotwiner A, Yatskar L, Harari R, Shah B, et al. ST-segment elevation in patients with COVID-19 — a case series. N Engl J Med. 2020;382(25):2478–80. https://doi.org/10.1056/NEJMc2009020.
Giustino G, Croft LB, Oates CP, Rahman K, Lerakis S, Reddy VY, et al. Takotsubo cardiomyopathy in COVID-19. J Am Coll Cardiol. 2020;76(5):628–9. https://doi.org/10.1016/j.jacc.2020.05.068.
Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020;41(32):3038–44. https://doi.org/10.1093/eurheartj/ehaa623.
Morris SB, Schwartz NG, Patel P, Abbo L, Beauchamps L, Balan S, et al. Case series of multisystem inflammatory syndrome in adults associated with SARS-CoV-2 infection - United Kingdom and United States, March-August 2020. MMWR Morb Mortal Wkly Rep. 2020;69(40):1450–6. https://doi.org/10.15585/mmwr.mm6940e1.
Rosenblatt AG, Ayers CR, Rao A, Howell SJ, Hendren NS, Zadikany RH, et al. New-onset atrial fibrillation in patients hospitalized with COVID-19: results from the American Heart Association COVID-19 Cardiovascular Registry. Circ Arrhythm Electrophysiol. 2022;15(5):e010666. https://doi.org/10.1161/CIRCEP.121.010666.
Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med. 2020;173(4):268–77. https://doi.org/10.7326/m20-2003.
Zheng YY, Ma YT, Zhang JY, **e X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17(5):259–60. https://doi.org/10.1038/s41569-020-0360-5.
•• Ammirati E, Lupi L, Palazzini M, Hendren NS, Grodin JL, Cannistraci CV, et al. Prevalence, characteristics, and outcomes of COVID-19-associated acute myocarditis. Circulation. 2022;145(15):1123–39. https://doi.org/10.1161/CIRCULATIONAHA.121.056817. This international, multicenter, retrospective observational study of patients hospitalized for COVID-19 with definite or probable myocarditis found that the prevalence of myocarditis was rare among COVID-19 patients. Though rare, acute COVID-19 myocarditis commonly presented as fulminant heart failure . Those with concurrent COVID-19 pneumonia were more likely to rapidly develop hemodynamic compromise and die compared to those without lung injury.
Figliozzi S, Masci PG, Ahmadi N, Tondi L, Koutli E, Aimo A, et al. Predictors of adverse prognosis in COVID-19: a systematic review and meta-analysis. Eur J Clin Invest. 2020;50(10):e13362. https://doi.org/10.1111/eci.13362.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62. https://doi.org/10.1016/S0140-6736(20)30566-3.
Zhang J, Lu S, Wang X, Jia X, Li J, Lei H, et al. Do underlying cardiovascular diseases have any impact on hospitalised patients with COVID-19? Heart. 2020;106(15):1148–53. https://doi.org/10.1136/heartjnl-2020-316909.
Adjei S, Hong K, Molinari NM, Bull-Otterson L, Ajani UA, Gundlapalli AV, et al. Mortality risk among patients hospitalized primarily for COVID-19 during the omicron and delta variant pandemic periods - United States, April 2020-June 2022. MMWR Morb Mortal Wkly Rep. 2022;71(37):1182–9. https://doi.org/10.15585/mmwr.mm7137a4.
Plumb ID, Feldstein LR, Barkley E, Posner AB, Bregman HS, Hagen MB, et al. Effectiveness of COVID-19 mRNA vaccination in preventing COVID-19-associated hospitalization among adults with previous SARS-CoV-2 infection - United States, June 2021-February 2022. MMWR Morb Mortal Wkly Rep. 2022;71(15):549–55. https://doi.org/10.15585/mmwr.mm7115e2.
Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5(7):811–8. https://doi.org/10.1001/jamacardio.2020.1017.
•• Lala A, Johnson KW, Januzzi JL, Russak AJ, Paranjpe I, Richter F, et al. Prevalence and impact of myocardial injury in patients hospitalized with COVID-19 infection. J Am Coll Cardiol. 2020;76(5):533–46. https://doi.org/10.1016/j.jacc.2020.06.007. This study was the first to characterize hospitalized COVID-19 patients by various clinical traits, including cardiac troponin levels, and to identify associations between elevated troponin levels and clinical outcomes. Acute cardiac injury was common, but troponin levels were generally low at presentation to the hospital. Still, even small elevations of cardiac troponin levels conferred increased risk of death, and the greater the elevation, the higher the mortality risk.
Qin JJ, Cheng X, Zhou F, Lei F, Akolkar G, Cai J, et al. Redefining cardiac biomarkers in predicting mortality of inpatients with COVID-19. Hypertension. 2020;76(4):1104–12. https://doi.org/10.1161/HYPERTENSIONAHA.120.15528.
Poterucha TJ, Elias P, Jain SS, Sayer G, Redfors B, Burkhoff D, et al. Admission cardiac diagnostic testing with electrocardiography and troponin measurement prognosticates increased 30-day mortality in COVID-19. J Am Heart Assoc. 2021;10(1):e018476. https://doi.org/10.1161/JAHA.120.018476.
Elias P, Poterucha TJ, Jain SS, Sayer G, Raikhelkar J, Fried J, et al. The prognostic value of electrocardiogram at presentation to emergency department in patients with COVID-19. Mayo Clin Proc. 2020;95(10):2099–109. https://doi.org/10.1016/j.mayocp.2020.07.028.
Bhatla A, Mayer MM, Adusumalli S, Hyman MC, Oh E, Tierney A, et al. COVID-19 and cardiac arrhythmias. Heart Rhythm. 2020;17(9):1439–44. https://doi.org/10.1016/j.hrthm.2020.06.016.
Dweck MR, Bularga A, Hahn RT, Bing R, Lee KK, Chapman AR, et al. Global evaluation of echocardiography in patients with COVID-19. Eur Heart J Cardiovasc Imaging. 2020;21(9):949–58. https://doi.org/10.1093/ehjci/jeaa178.
Szekely Y, Lichter Y, Taieb P, Banai A, Hochstadt A, Merdler I, et al. Spectrum of cardiac manifestations in COVID-19: a systematic echocardiographic study. Circulation. 2020;142(4):342–53. https://doi.org/10.1161/circulationaha.120.047971.
Giustino G, Croft LB, Stefanini GG, Bragato R, Silbiger JJ, Vicenzi M, et al. Characterization of myocardial injury in patients with COVID-19. J Am Coll Cardiol. 2020;76(18):2043–55. https://doi.org/10.1016/j.jacc.2020.08.069.
Puntmann VO, Carerj ML, Wieters I, Fahim M, Arendt C, Hoffmann J, et al. Outcomes of cardiovascular magnetic resonance imaging in patients recently recovered from coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5(11):1265–73. https://doi.org/10.1001/jamacardio.2020.3557.
Bull-Otterson L, Baca S, Saydah S, Boehmer TK, Adjei S, Gray S, et al. Post–COVID conditions among adult COVID-19 survivors aged 18–64 and ≥65 years — United States, March 2020–November 2021. MMWR Morb Mortal Wkly Rep. 2022;71.
**e Y, Xu E, Bowe B, Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med. 2022;28(3):583–90. https://doi.org/10.1038/s41591-022-01689-3.
•• Gluckman TJ, Bhave NM, Allen LA, Chung EH, Spatz ES, Ammirati E, et al. 2022 ACC expert consensus decision pathway on cardiovascular sequelae of COVID-19 in adults: myocarditis and other myocardial involvement, post-acute sequelae of SARS-CoV-2 infection, and return to play. J Am Coll Cardiol. 2022;79(17):1717–56. https://doi.org/10.1016/j.jacc.2022.02.003. This expert concensus document provides guidance on the clinical assessment and management of patients with history of COVID-19 illness. It describes various clinical decisions about COVID-19 cardiovascular sequelae that internistss and cardiologists will now frequently encounter.
Faridi KF, Hennessey KC, Shah N, Soufer A, Wang Y, Sugeng L, et al. Left ventricular systolic function and inpatient mortality in patients hospitalized with coronavirus disease 2019 (COVID-19). J Am Soc Echocardiogr. 2020;33(11):1414–5. https://doi.org/10.1016/j.echo.2020.08.016.
Esposito A, Palmisano A, Natale L, Ligabue G, Peretto G, Lovato L, et al. Cardiac magnetic resonance characterization of myocarditis-like acute cardiac syndrome in COVID-19. JACC Cardiovasc Imaging. 2020;13(11):2462–5. https://doi.org/10.1016/j.jcmg.2020.06.003.
Cassar MP, Tunnicliffe EM, Petousi N, Lewandowski AJ, **e C, Mahmod M, et al. Symptom persistence despite improvement in cardiopulmonary health - insights from longitudinal CMR, CPET and lung function testing post-COVID-19. EClinical Medicine. 2021;41:101159. https://doi.org/10.1016/j.eclinm.2021.101159.
Chen W, Jeudy J. Assessment of myocarditis: cardiac MR, PET/CT, or PET/MR? Curr Cardiol Rep. 2019;21(8):76. https://doi.org/10.1007/s11886-019-1158-0.
Hanneman K, Houbois C, Schoffel A, Gustafson D, Iwanochko RM, Wintersperger BJ, et al. Combined cardiac fluorodeoxyglucose-positron emission tomography/magnetic resonance imaging assessment of myocardial injury in patients who recently recovered from COVID-19. JAMA Cardiol. 2022;7(3):298–308. https://doi.org/10.1001/jamacardio.2021.5505.
Stefanini GG, Montorfano M, Trabattoni D, Andreini D, Ferrante G, Ancona M, et al. ST-elevation myocardial infarction in patients with COVID-19: clinical and angiographic outcomes. Circulation. 2020;141(25):2113–6. https://doi.org/10.1161/CIRCULATIONAHA.120.047525.
Pasupathy S, Air T, Dreyer RP, Tavella R, Beltrame JF. Systematic review of patients presenting with suspected myocardial infarction and nonobstructive coronary arteries. Circulation. 2015;131(10):861–70. https://doi.org/10.1161/CIRCULATIONAHA.114.011201.
Singh T, Kite TA, Joshi SS, Spath NB, Kershaw L, Baker A, et al. MRI and CT coronary angiography in survivors of COVID-19. Heart. 2022;108(1):46–53. https://doi.org/10.1136/heartjnl-2021-319926.
Falasca L, Nardacci R, Colombo D, Lalle E, Di Caro A, Nicastri E, et al. Post-mortem findings in Italian patients with COVID-19 - a descriptive full autopsy study of cases with and without co-morbidities. J Infect Dis. 2020. https://doi.org/10.1093/infdis/jiaa578.10.1093/infdis/jiaa578.
•• Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe. 2020;1(7):e290–9. https://doi.org/10.1016/S2666-5247(20)30144-0. This single-center prospective autopsy study detected extensive inflammation and SARS-CoV-2 in multiple organs of 11 (52%) of 21 fatal COVID-19 cases. SARS-CoV-2 positive cells were most abundant in the lungs, but their numbers declined over disease time course. Thrombi, neutrophilic infiltrates, and neutrophilic plugs--- aggregates of neutrophil extracellular traps (NETs) with neutrophils or platelets--- were frequently found and persisted late into disease time course, suggesting that a maladaptive immune reponse underlies fatal COVID-19.
Halushka MK, Vander Heide RS. Myocarditis is rare in COVID-19 autopsies: cardiovascular findings across 277 postmortem examinations. Cardiovasc Pathol. 2021;50:107300. https://doi.org/10.1016/j.carpath.2020.107300.
•• Brener MI, Hulke ML, Fukuma N, Golob S, Zilinyi RS, Zhou Z, et al. Clinico-histopathologic and single-nuclei RNA-sequencing insights into cardiac injury and microthrombi in critical COVID-19. JCI Insight. 2022;7(2). https://doi.org/10.1172/jci.insight.154633. This single-center prospective COVID-19 autopsy study was a comprehensive clinico-histologic analysis of 69 hearts of COVID-19 non-survivors. It incorporated high resolution molecular analysis of a subset of COVID-19 hearts differentiated by the presence or absence of cardiac microthrombi. Findings indicated that a profound systemic inflammatory response was independently associated with increased odds of COVID-19 cardiac microthrombi. Moreover, cardiac fibroblast autocrine signaling involving prothrombotic / anti-fibrinolytic pathways and innate immunity activation were identified as being salient to the pathophysiology of COVID-19 cardiac microthrombi.
Sang CJ, 3rd, Burkett A, Heindl B, Litovsky SH, Prabhu SD, Benson PV, et al. Cardiac pathology in COVID-19: a single center autopsy experience. Cardiovasc Pathol. 2021;54:107370. https://doi.org/10.1016/j.carpath.2021.107370.
Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJ, Jr., et al. Myocarditis. A histopathologic definition and classification. Am J Cardiovasc Pathol. 1987;1(1):3–14.
• Basso C, Leone O, Rizzo S, De Gaspari M, van der Wal AC, Aubry MC, et al. Pathological features of COVID-19-associated myocardial injury: a multicentre cardiovascular pathology study. Eur Heart J. 2020;41(39):3827–35. https://doi.org/10.1093/eurheartj/ehaa664. This retrospective autopsy of 21 COVID-19 patients characterized the cardiac immune response to SARS-CoV-2 infection. While lymphocytic myocarditis was uncommon, macrophage infiltration at high cell density and without acute myocardial injury was frequently detected. The small number of myocarditis was disproportionately low compared to the high prevalence of elevated cardiac troponin levels in this cohort.
• Bearse M, Hung YP, Krauson AJ, Bonanno L, Boyraz B, Harris CK, et al. Factors associated with myocardial SARS-CoV-2 infection, myocarditis, and cardiac inflammation in patients with COVID-19. Mod Pathol. 2021;34(7):1345–57. https://doi.org/10.1038/s41379-021-00790-1. This retrospective autopsy study detected SARS-CoV-2 virus in 30 (73%) of 41 COVID-19 non-survivors. SARS-CoV-2 presence in cardiac tissue was associated with more cardiac inflammation and electrocardiographic changes. Moreover, densities of CD68+ macrophage and CD3+ lymphocyte cardiac infiltrates increased with longer COVID-19 symptom duration.
Chen ST, Park MD, Del Valle DM, Buckup M, Tabachnikova A, Thompson RC, et al. A shift in lung macrophage composition is associated with COVID-19 severity and recovery. Sci Transl Med. 2022;14(662):eabn5168. https://doi.org/10.1126/scitranslmed.abn5168.
• Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature. 2022;606(7914):585–93. https://doi.org/10.1038/s41586-022-04802-1. Using a humanizezd mouse model of COVID-19, this study found that SARS-CoV-2 infects and replicates in lung resident human macrophages, Infected macrophages then activate inflammasomes, release proinflammatory cytokines, and undergo an inflammatory cell death. This process serves to limit the productive viral cycle but contributes to the hyperinflammation that characterizes severe/critical COVID-19.
Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Querrey M, Guzman ER, et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature. 2021;590(7847):635–41. https://doi.org/10.1038/s41586-020-03148-w.
•• Bois MC, Boire NA, Layman AJ, Aubry MC, Alexander MP, Roden AC, et al. COVID-19-associated nonocclusive fibrin microthrombi in the heart. Circulation. 2021;143(3):230–43. https://doi.org/10.1161/CIRCULATIONAHA.120.050754. This study examined 15 post-mortem hearts of COVID-19 non-survivors and compared them to those of fatal influenza-A cases and non-virally mediated deaths. Fibrin cardiac microthrombi without any acute ischemic injury was frequently deteced in COVID-19 hearts, while myocarditis was uncommon and limited in extent when present.
Fox SE, Li G, Akmatbekov A, Harbert JL, Lameira FS, Brown JQ, et al. Unexpected features of cardiac pathology in COVID-19 infection. Circulation. 2020;142(11):1123–5. https://doi.org/10.1161/CIRCULATIONAHA.120.049465.
•• Pellegrini D, Kawakami R, Guagliumi G, Sakamoto A, Kawai K, Gianatti A, et al. Microthrombi as a major cause of cardiac injury in COVID-19: a pathologic study. Circulation. 2021;143(10):1031–42. https://doi.org/10.1161/CIRCULATIONAHA.120.051828. This COVID-19 autopsy study detected cardiac necrosis in 14 (35%) of 40 hearts and microthrombi in 9 (62.3%) of those 14 hearts with necrosis. COVID-19 cardiac microthrombi were found to have more fibrin and terminal complement C5b-9 than coronary thrombus aspirates from non-COVID-19 and COVID-19 decedents of acute ST elevation myocardial infarction.
Schaller T, Hirschbuhl K, Burkhardt K, Braun G, Trepel M, Markl B, et al. Postmortem examination of patients with COVID-19. JAMA. 2020;323(24):2518–20. https://doi.org/10.1001/jama.2020.8907.
•• Cummings MJ, Baldwin MR, Abrams D, Jacobson SD, Meyer BJ, Balough EM, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet. 2020;395(10239):1763–70. https://doi.org/10.1016/s0140-6736(20)31189-2. This prospective observational cohort study was the first and largest in the US to characterise the epidemiology, clinical course, and outcomes of critically ill COVID-19 patients. It identified independent associations between biomarkers of inflammation (IL-6) and thrombosis (D-dimer) and in-hospital death. It also identified a high incidence of critical COVID-19 among racial and ethnic minorities.
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620–9. https://doi.org/10.1172/JCI137244.
Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368:m1091. https://doi.org/10.1136/bmj.m1091.
Wygrecka M, Birnhuber A, Seeliger B, Michalick L, Pak O, Schultz AS, et al. Altered fibrin clot structure and dysregulated fibrinolysis contribute to thrombosis risk in severe COVID-19. Blood Adv. 2022;6(3):1074–87. https://doi.org/10.1182/bloodadvances.2021004816.
• Tucker NR, Chaffin M, Bedi KC Jr, Papangeli I, Akkad AD, Arduini A, et al. Myocyte-specific upregulation of ACE2 in cardiovascular disease: implications for SARS-CoV-2-mediated myocarditis. Circulation. 2020;142(7):708–10. https://doi.org/10.1161/CIRCULATIONAHA.120.047911. This study assessed myocardial expression of SARS-CoV-2 receptor ACE2 transcript using bulk and single nucleus RNA-sequencing of left ventricular myocardial tissue from individuals with dilated cardiomyopathy and hypertrophic cardiomyopathy. Myocardial tissue ACE2 expression was similar across samples of dilated cardiomyopathy, hypertrophic cardiomyopathy, and non-failing controls. However, ACE2 expression at the cellular level was highest in pericytes and appreciable in vascular smooth muscle cells, fibroblasts, and cardiomyocytes. ACE2 expression decreased in fibroblasts, pericytes, and vascular smooth muscle and increased in cardiomyocytes of dilated and hypertrophic cardiomyopathy samples.
V’Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19(3):155–70. https://doi.org/10.1038/s41579-020-00468-6.
Ferrer-Gomez A, Pian-Arias H, Carretero-Barrio I, Navarro-Cantero A, Pestana D, de Pablo R, et al. Late cardiac pathology in severe COVID-19. A postmortem series of 30 patients. Front Cardiovasc Med. 2021;8:748396. https://doi.org/10.3389/fcvm.2021.748396.
Brauninger H, Stoffers B, Fitzek ADE, Meissner K, Aleshcheva G, Schweizer M, et al. Cardiac SARS-CoV-2 infection is associated with pro-inflammatory transcriptomic alterations within the heart. Cardiovasc Res. 2022;118(2):542–55. https://doi.org/10.1093/cvr/cvab322.
Haslbauer JD, Tzankov A, Mertz KD, Schwab N, Nienhold R, Twerenbold R, et al. Characterisation of cardiac pathology in 23 autopsies of lethal COVID-19. J Pathol Clin Res. 2021;7(4):326–37. https://doi.org/10.1002/cjp2.212.
Kawakami R, Sakamoto A, Kawai K, Gianatti A, Pellegrini D, Nasr A, et al. Pathological evidence for SARS-CoV-2 as a cause of myocarditis: JACC review topic of the week. J Am Coll Cardiol. 2021;77(3):314–25. https://doi.org/10.1016/j.jacc.2020.11.031.
Lindner D, Fitzek A, Brauninger H, Aleshcheva G, Edler C, Meissner K, et al. Association of cardiac infection with SARS-CoV-2 in confirmed COVID-19 autopsy cases. JAMA Cardiol. 2020;5(11):1281–5. https://doi.org/10.1001/jamacardio.2020.3551.
• Escher F, Pietsch H, Aleshcheva G, Bock T, Baumeier C, Elsaesser A, et al. Detection of viral SARS-CoV-2 genomes and histopathological changes in endomyocardial biopsies. ESC Heart Fail. 2020;7(5):2440–7. https://doi.org/10.1002/ehf2.12805. This study was the first to detect SARS-CoV-2 genome in endomyocardial biopsies of patients with suspected myocarditis or unexplained heart failure. Five of the 104 cases had SARS-CoV-2 positive endomyocardial biopsies. One showed active myocarditis, another borderline myocarditis, and the remaining 3 had inflammatory cardiomyopathy. The SARS-CoV-2 positive endomyocardial biopsies also showed extensive myocardial inflammation that also extended into vascular wall, leading to small arterial obliteration.
Wenzel P, Kopp S, Gobel S, Jansen T, Geyer M, Hahn F, et al. Evidence of SARS-CoV-2 mRNA in endomyocardial biopsies of patients with clinically suspected myocarditis tested negative for COVID-19 in nasopharyngeal swab. Cardiovasc Res. 2020;116(10):1661–3. https://doi.org/10.1093/cvr/cvaa160.
Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary manifestations of COVID-19. Nat Med. 2020;26(7):1017–32. https://doi.org/10.1038/s41591-020-0968-3.
Peiris S, Mesa H, Aysola A, Manivel J, Toledo J, Borges-Sa M, et al. Pathological findings in organs and tissues of patients with COVID-19: a systematic review. PLoS One. 2021;16(4):e0250708. https://doi.org/10.1371/journal.pone.0250708.
Andersson MI, Arancibia-Carcamo CV, Auckland K, Baillie JK, Barnes E, Beneke T, et al. SARS-CoV-2 RNA detected in blood products from patients with COVID-19 is not associated with infectious virus. Wellcome Open Res. 2020;5:181. https://doi.org/10.12688/wellcomeopenres.16002.2.
Bailey AL, Dmytrenko O, Greenberg L, Bredemeyer AL, Ma P, Liu J, et al. SARS-CoV-2 infects human engineered heart tissues and models COVID-19 myocarditis. JACC Basic Transl Sci. 2021;6(4):331–45. https://doi.org/10.1016/j.jacbts.2021.01.002.
Tavazzi G, Pellegrini C, Maurelli M, Belliato M, Sciutti F, Bottazzi A, et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur J Heart Fail. 2020;22(5):911–5. https://doi.org/10.1002/ejhf.1828.
Navaratnarajah CK, Pease DR, Halfmann PJ, Taye B, Barkhymer A, Howell KG, et al. Highly efficient SARS-CoV-2 infection of human cardiomyocytes: spike protein-mediated cell fusion and its inhibition. J Virol. 2021;95(24):e0136821. https://doi.org/10.1128/JVI.01368-21.
Bojkova D, Wagner JUG, Shumliakivska M, Aslan GS, Saleem U, Hansen A, et al. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. Cardiovasc Res. 2020;116(14):2207–15. https://doi.org/10.1093/cvr/cvaa267.
Li Y, Renner DM, Comar CE, Whelan JN, Reyes HM, Cardenas-Diaz FL, et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc Natl Acad Sci U S A. 2021;118(16). https://doi.org/10.1073/pnas.2022643118.
Perez-Bermejo JA, Kang S, Rockwood SJ, Simoneau CR, Joy DA, Silva AC, et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci Transl Med. 2021;13(590). https://doi.org/10.1126/scitranslmed.abf7872.
Sharma A, Garcia G, Jr., Wang Y, Plummer JT, Morizono K, Arumugaswami V, et al. Human iPSC-derived cardiomyocytes are susceptible to SARS-CoV-2 infection. Cell Rep Med. 2020;1(4):100052. https://doi.org/10.1016/j.xcrm.2020.100052.
Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, et al. A human pluripotent stem cell-based platform to study SARS-CoV-2 tropism and model virus infection in human cells and organoids. Cell Stem Cell. 2020;27(1):125–36 e7. https://doi.org/10.1016/j.stem.2020.06.015.
Bulfamante GP, Perrucci GL, Falleni M, Sommariva E, Tosi D, Martinelli C, et al. Evidence of SARS-CoV-2 transcriptional activity in cardiomyocytes of COVID-19 patients without clinical signs of cardiac involvement. Biomedicines. 2020;8(12). https://doi.org/10.3390/biomedicines8120626.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–8. https://doi.org/10.1016/s0140-6736(20)30937-5.
• McCracken IR, Saginc G, He L, Huseynov A, Daniels A, Fletcher S, et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation. 2021;143(8):865–8. https://doi.org/10.1161/CIRCULATIONAHA.120.052824. Using transcriptomic and epigenomic data, this study found that the transcript encoding SARS-Co-V2 receptor ACE2 is not expressed in human endothelial cells. Endothelial cells do express other coreceptors of SARS-CoV-2, and in experiments in which endothelial cells were exposed to very high virus concentration, low levels of SARS-CoV-2 replication could be observed in endothelial cells. This is most likely due to non-ACE2- dependent viral entry. Regardless, the overall data indicates that direct infection of endothelial cells by SARS-CoV-2 is highly unlikely.
Schimmel L, Chew KY, Stocks CJ, Yordanov TE, Essebier P, Kulasinghe A, et al. Endothelial cells are not productively infected by SARS-CoV-2. Clin Transl Immunology. 2021;10(10):e1350. https://doi.org/10.1002/cti2.1350.
Avolio E, Carrabba M, Milligan R, Kavanagh Williamson M, Beltrami AP, Gupta K, et al. The SARS-CoV-2 spike protein disrupts human cardiac pericytes function through CD147 receptor-mediated signalling: a potential non-infective mechanism of COVID-19 microvascular disease. Clin Sci (Lond). 2021;135(24):2667–89. https://doi.org/10.1042/CS20210735.
•• Brumback BD, Dmytrenko O, Robinson AN, Bailey AL, Ma P,Liu J, Hicks SC, Ng S, et al. (2022) Human Cardiac Pericytes Are Susceptible to SARS-CoV-2 Infection. JACC: Basic to Translational Science. https://doi.org/10.1016/j.jacbts.2022.09.001. Using organotypic slices and primary cell culture, this translational study demonstrated for the first time that human cardiac pericytes can be infected by SARS-CoV-2 and that viral entry occurs through an endosomal pathway. Experimental findings also revealed downstream cellular effects of viral infection of pericytes, including eliciting inflammation, vasoactive responses, and cell death. Lastly, the gene encoding SARS-CoV-2 protein S was detected by in situ hybridization in 2 post-mortem hearts of COVID-19 myocarditis patients. The viral gene was detected in perivascular regions of the myocardium and co-localized with known markers of pericyte cells.
•• Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl J Med. 2020;383(2):120–8. https://doi.org/10.1056/NEJMoa2015432. This study found that post-mortem lungs of COVID-19 non-survivors exhibited severe endothelial injury associated with intracellular detection of SARS-CoV-2 and disrupted vascular endothelium. Additionally, vascular thrombosis with microangiopathy and alveolar capillary occlusion was detected throughout the post-mortem COVID-19 lungs. COVID-19 lungs also showed evidence of intussusceptive angiogenesis. These features were unique to COVID-19 lungs and not present in the lungs of patients who died from H1N1 influenza-acute respiratory distress syndrome or of uninfected controls.
Parra-Medina R, Herrera S, Mejia J. Systematic review of microthrombi in COVID-19 autopsies. Acta Haematol. 2021;144(5):476–83. https://doi.org/10.1159/000515104.
Barrett TJ, Cornwell M, Myndzar K, Rolling CC, **a Y, Drenkova K, et al. Platelets amplify endotheliopathy in COVID-19. Sci Adv. 2021;7(37):eabh2434. https://doi.org/10.1126/sciadv.abh2434.
Ji W, Chen L, Yang W, Li K, Zhao J, Yan C, et al. Transcriptional landscape of circulating platelets from patients with COVID-19 reveals key subnetworks and regulators underlying SARS-CoV-2 infection: implications for immunothrombosis. Cell Biosci. 2022;12(1):15. https://doi.org/10.1186/s13578-022-00750-5.
Koupenova M, Corkrey HA, Vitseva O, Tanriverdi K, Somasundaran M, Liu P, et al. SARS-CoV-2 initiates programmed cell death in platelets. Circ Res. 2021;129(6):631–46. https://doi.org/10.1161/CIRCRESAHA.121.319117.
Chieffo A, Stefanini GG, Price S, Barbato E, Tarantini G, Karam N, et al. EAPCI position statement on invasive management of acute coronary syndromes during the COVID-19 pandemic. Eur Heart J. 2020;41(19):1839–51. https://doi.org/10.1093/eurheartj/ehaa381.
Mahmud E, Dauerman HL, Welt FGP, Messenger JC, Rao SV, Grines C, et al. Management of acute myocardial infarction during the COVID-19 pandemic: a position statement from the Society for Cardiovascular Angiography and Interventions (SCAI), the American College of Cardiology (ACC), and the American College of Emergency Physicians (ACEP). J Am Coll Cardiol. 2020;76(11):1375–84. https://doi.org/10.1016/j.jacc.2020.04.039.
Kociol RD, Cooper LT, Fang JC, Moslehi JJ, Pang PS, Sabe MA, et al. Recognition and initial management of fulminant myocarditis: a scientific statement from the American Heart Association. Circulation. 2020;141(6):e69–92. https://doi.org/10.1161/CIR.0000000000000745.
Bhardwaj A, Kirincich J, Rampersad P, Soltesz E, Krishnan S. Fulminant myocarditis in COVID-19 and favorable outcomes with VA-ECMO. Resuscitation. 2022;175:75–6. https://doi.org/10.1016/j.resuscitation.2022.04.021.
Buitrago DH, Munoz J, Finkelstein ER, Mulinari L. A case of fulminant myocarditis due to COVID-19 in an adolescent patient successfully treated with venous arterial ECMO as a bridge to recovery. J Card Surg. 2022;37(5):1439–43. https://doi.org/10.1111/jocs.16313.
Rajpal S, Kahwash R, Tong MS, Paschke K, Satoskar AA, Foreman B, et al. Fulminant myocarditis following SARS-CoV-2 infection: JACC patient care pathways. JACC Case Rep. 2022;4(10):567–75. https://doi.org/10.1016/j.jaccas.2022.03.013.
Zhang Y, Coats AJS, Zheng Z, Adamo M, Ambrosio G, Anker SD, et al. Management of heart failure patients with COVID-19: a joint position paper of the Chinese Heart Failure Association & National Heart Failure Committee and the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2020;22(6):941–56. https://doi.org/10.1002/ejhf.1915.
Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022;79(17):e263–421. https://doi.org/10.1016/j.jacc.2021.12.012.
Jirak P, van Almsick V, Dimitroulis D, Mirna M, Seelmaier C, Shomanova Z, et al. Dexamethasone improves cardiovascular outcomes in critically ill COVID-19, a real world scenario multicenter analysis. Front Med (Lausanne). 2022;9:808221. https://doi.org/10.3389/fmed.2022.808221.
Heidecker B, Dagan N, Balicer R, Eriksson U, Rosano G, Coats A, et al. Myocarditis following COVID-19 vaccine: incidence, presentation, diagnosis, pathophysiology, therapy, and outcomes put into perspective. Eur J Heart Fail. 2022. https://doi.org/10.1002/ejhf.2669.10.1002/ejhf.2669.
Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20(5):533–4. https://doi.org/10.1016/S1473-3099(20)30120-1.
COVID-19 Global Map. https://coronavirus.jhu.edu/map.html. (2020 Jan 22). Accessed 2022 Aug 24.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Heart Failure
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sewanan, L.R., Clerkin, K.J., Tucker, N.R. et al. How Does COVID-19 Affect the Heart?. Curr Cardiol Rep 25, 171–184 (2023). https://doi.org/10.1007/s11886-023-01841-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-023-01841-6