
Abstract
Influenza A/H1N1 is widely considered to be a very evolutionary virus causing major public health problems. Since the pandemic of 2009, there has been a rapid rise in human Influenza virus characterization. However, little data is available in Tunisia regarding its genetic evolution. In light of this fact, our paper aim is to genetically characterize the Neuraminidase, known as the target of antiviral inhibitors, in Tunisian isolates circulating in Monastir region during 2017–2018. In total of 31 positive Influenza A/H1N1 detected by multiplex real-time PCR, RT-PCR of neuraminidase was performed. Among the 31 positive samples, 7 samples representing fatal and most severe cases were conducted for sequencing and genetic analysis. The results thus obtained showed genetic evolution of the A/H1N1 neuraminidase between 2009 and 2010 and 2018–2019 outbreaks. All Tunisian isolates were genetically related to the recommended vaccine strain with a specific evolution. Moreover, the phylogenetic analysis demonstrated that France and especially Italian strains were the major related strains. Interestingly, our results revealed a specific cluster of Tunisian isolates where two intragroup were evolved in correlation with the severity and the fatalities cases. From the outcome of our investigation, this study confirms the genetic evolution of the Influenza A virus circulating in Tunisia and gives a preliminary analysis for a better comprehension of new emerging Tunisian strain’s virulence and thus, a more appropriate monitoring of Influenza virus A/H1N1 during each round of outbreaks.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Seasonal Influenza vaccines are the key public health tool against the Influenza virus. However, its efficacy remains limited due to the antigen mismatch between vaccine strains and circulating viruses. The immune status of the host can lower the effectiveness which varies by age group and by the vaccination status (Osterholm et al. 2012; Lewnard and Cobey 2018). Seasonal Influenza epidemics cause up to 650,000 respiratory deaths and 3 to 5 million cases of severe illness per year worldwide and present a real economical problem (Petrova and Russell 2018). Influenza viruses are segmented single-stranded, negative-sense RNA viruses that belong to the Orthomyxoviridae family. Based on their antigenic specificity of the nucleoprotein and matrix protein, Influenza viruses are divided into Influenza A (IAV), Influenza B, and Influenza C (Bouvier and Palese 2008). Several publications have appeared in recent years documenting that the IAV virus exhibits the most circulating type causing disease in humans, and different animal and avian species (Horimoto and Kawaoka 2005). The Influenza genome contains 8 RNA segments (PB2, PB1, PA, HA, NP, NA, M, and NS). The 6th segment encodes for the Neuraminidase (NA), one of the major surface glycoproteins of the Influenza virus (Bouvier and Palese 2008). There are 11 different neuraminidase subtypes found in circulating IAV (N1 through N11, respectively). Only N1 and N2 which are expressed in H1N1 and H3N2 subtypes cause endemic in humans and give rise to seasonal outbreaks (McAuley et al. 2019). In contrast to IAV which is classified into subtypes, Influenza B is classified into two lineages: B/Yamagata and B/Victoria (Hatta and Kawaoka 2003). The IAV can be further classified into specific clades and subclades. For example, IAV (H1N1) belongs to the clade 6B1 and the subtype 6B1.A (CDC 2019). It has now been demonstrated that NA plays several roles during the infection process. It acts in the early stage of infection by enhancing virus entry contributing to an efficient virus-cell association (Guo et al. 2018). In addition, this glycoprotein acts in the final stage of infection by removing sialic acid from cellular glycoconjugates in a specific site. Thus, newly synthesized virions are released from the infected cell to spread the infection (Palese et al. 1974). During the virus infection, and to escape from the immune system, the natural selection favorite the continuous modifications of the antigenic structure of NA by generating mutations. Since NA is an important antiviral target, a single substitution on a specific site can lead to a resistance capacity against NA inhibitors such as zanamivir and oseltamivir (Hurt et al. 2012; Pinilla et al. 2012; Krammer et al. 2018).
In the last few years, there has been a growing interest in the genetic variability of Influenza NA to understand the causes of the immune escape and its impact on the fitness. However, in Tunisia, studies on genetic characterization of Influenza NA are still lacking. In this paper, we focus on a specific region in Tunisia, Monastir, localized in the coastal region of Mediterranean Sea in North Africa and characterized by the presence of sebkha islands, one of the major site for migratory birds with more than 72,368 birds per year (Hamdi and Charfi 2011; Hamza et al. 2015). Based on this case, we suppose that the Tunisian Influenza strains might have a rapid evolution. In line with this, IAV(H1N1) outbreak during the 2017–2018 season was quite spectacular causing severe acute respiratory infections (SARI) leading even to death. The national observatory of new and emerging diseases has reported 48 death cases caused by Influenza where A/H1N1 presents 80 %(ONMNE 2018). In this context, the present work aims to study the virus phylogeny and to explore the evolutionary processes of NA that shape the genetic diversity of Tunisian IAV(H1N1) strains.
Materials and methods
Ethics approval
Samples were collected after the receiving of written informed consent from the subjects or their legal representatives. The study protocol was approved by the research ethics committee of the faculty of medicine, the university of Monastir under the number [IORG0009738 N° 18/OMB0990-0279].
Samples and clinic data collection
During the 2017–2018 Influenza outbreak, nasopharyngeal swab samples were collected from patients suffering from severe acute respiratory illness in the Sahel region of Tunisia (Monastir) in collaboration with the laboratory of medical microbiology of Fatouma Bourguiba university hospital Monastir, Tunisia. In this work, we focus on 31 samples that were detected positive for A/H1N1 by multiplex real-time PCR (RespiFinder® 2Smart PathoFinder) as per the manufacturer’s instructions, during respiratory virus routine detection. Each sample was received with its clinic information then stored directly at -80°C for RT-PCR based detection of NA and genetic analysis. All infected patients were not vaccinated against the Influenza virus.
Viral RNA extraction and RT-PCR of NA gene
RNA extraction from clinical samples was performed using TRIzol™ Reagent (Invitrogen™) according to the manufacturer’s instructions.
The cDNA synthesis of the whole Influenza genome was performed using the universal primer UNIT12 (AGCRAAAGCAGG) (Hoffmann et al. 2001), the viral RNA was mixed with 20 pMol of Unit 12 primer and 12,5 pM of dNTP for a pre-heating step at 65°C during 5 min. The mixture was incubated immediately on ice. 10 U/ µl of Mmlv enzyme (Biogene) and 10 µM of DTT (dithiothreitol) was added to the previous mixture and the total volume was incubated for 60 min at 42°C for the retro-transcription.
PCR reaction for the neuraminidase gene amplification was performed by TaKaRa LA Taq® DNA-Polymerase using specific primers (NA F:5’AGCAGGAGATTAAAATGAATCCAA3’) and (NA-R:5’CCTTCCTATCCAAACACCATT3’) for NA of seasonal IAV (H1N1) amplification (1097pb) (WHO 2017a).
The thermal cycle was programmed for amplification as follows: Incubation at 95°C for 1 min, and then 30 cycles of denaturation at 94°C for 30’, 58°C for 30’ for hybridization, 72°C for 2 min followed by a final elongation step at 72°C for 5 min using a thermocycler 2770 (Applied Biosystems).
An agarose gel (1 %) in 1X Tris-borate-EDTA and ethidium bromide (250 ng/µl) was prepared. Then, 5 µL of NA PCR amplification products were added to 1 µL of 6X loading and loaded. 3 µL of 1 kb DNA ladder (GeneON) were also loaded. Finally, the gel was run at 100 Volts for 30 then NA bands were visualized and photographed under UV light using « Quantity One » software of Gel Doc 2000 (Bio-Rad).
PCR products purification and sequencing
Since we aim to sequence the NA segment and analyze it genetically, the key step is to have a good quality of DNA from the RT-PCR for the NA. Thus, we kept only samples where nasopharyngeal swabs were collected during the first 5 days as the recommendation of the centers for disease control and prevention (CDC 2020). Also, samples from fatal cases were conducted for sequencing to study the virulence of their strains (Methodology is shown in Fig. 1).

Overview of the methodology used to characterize Neuraminidase gene of Influenza A (H1N1) in Monastir region (Tunisia)
The 7 selected PCR amplification products were purified from PCR reagents following the clean-up protocol steps from FavorPrep GEL/ PCR purification Kit as per the manufacturer’s instructions. These 7 samples were subjected for sequencing (commercially) using the big dye terminator method in 3500 ABI Prism Genetic Analyzer (Applied Biosystems) with the same NA primers recommended by the WHO for NA detection and sequencing studies.
Nucleotide sequence submission
The nucleotide sequences data obtained from this study have been submitted to NCBI GenBank. The accession number of the sequences for the NA gene obtained are MT239356 to MT239362.
Phylogenic analysis
Tunisian sequences were aligned using Cluster X software (Larkin et al. 2007) with other European, and Mediterranean strains sequences obtained randomly from online Influenza database from the 2009–2010 season until the 2018–2019 season available on [https://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi?go=genomeset]. WHO recommended vaccine strain A/Michigan/45/2015 (A/H1N1) pdm09-like for the 2017–2018 season was also included in the alignment. The phylogenetic tree was generated in MEGA version 7.0 (Kumar et al. 2016) using the maximum likelihood method with the general time-reversible (GTR) + Gevolutionary model. 1000 bootstrap replicates were used to construct the phylogenetic tree and high bootstrap values above 70 % were visualized. The final tree was visualized and annotated with FigTree v.1.4.2 (http://tree.bio.ed.ac.uk/software/figtree).
Results
Clinical characteristics
Demographic data
The study included data generated from 31 samples in the region of Monastir, Tunisia. The population was composed of 31,3 % males and 38,7 % females. The patient’s ages ranged from 3 months to 80 years with a mean age of 38,16 ± 31,46. The age group with the highest number of SARI was the 0–2 years with (36,5 %), followed by the ⩾65 years with 29 %, lastly the 36–64 years and 20–35 with a rate of 19,4 %, 12,9 % respectively. Among the 31 positive cases, 19 patients were hospitalized in ICU (61,3 %) and 12 patients (38 %). 3 of the 31 patients are passed away (9,68 %) which the ages are ranged from 3 months to 2 years (Table 1). Taking into consideration the fatality cases, the most severe cases, and the period between the first sign of infection (days ≤ 5 days), only 7 positive samples were sequenced which represents (19,35 %).
Clinical outcomes in the totality of infected patients
The clinical outcomes of IAV (H1N1) are shown in Table 2. The majority of patients suffered from fever (96,77 %) and cough (24 %). In total, 22,58 % of patients developed respiratory distress and dyspnea 32,25 %. In total 10/31 (32,25 %) of patients received antiviral treatment and 9,67 % of patients received mechanical ventilation. In addition to the respiratory outcomes, the infection was associated with comorbidity (22,58 %) like diabetes, hypertension, cardiovascular diseases. From the 31 patients, 22,58 % have immunodeficiency and 6,45 % had a pregnancy.’’.
Clinical data of the selected sequenced samples
The genetically analyzed infected cases were predominantly males (6 male and 1 female) aged between 3 months to 70 years. All samples were taken from patients suffering from SARI coming from the Monastir region. Sampling was performed in the 2017/2018 season (One sample during the first trimester and 6 during the second trimester). All the analyzed 7 patients presented Influenza A symptoms (caugh, fever, fatigue), Besides, the 3 fatal cases (SB. G51; SB. G47; SB. G67) were suffering from additional respiratory complications (distress). Real-time RT-PCR revealed IAV (H1N1) as a responsible agent of these acute respiratory infections for all the samples with one case of co-infections with adenovirus. Only 2 patients SB. G70 and SB.G47 were treated with oseltamivir. The overall clinical characteristics of the studied samples are summarized in Table 3.
Sequencing
Sequences obtained for all amplifications were verified, aligned, and completed manually using reference sequences from the GenBank. The accession numbers of the sequences for the NA gene obtained were registered in the Genbank with accession numbers from MT239356 to MT239362.
Phylogenetic analysis
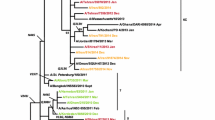
As can be seen from Fig. 2, phylogenetic analysis for the NA gene of IAV(H1N1) Tunisian isolates highlighted two major clades: Clade A and Clade B. On one hand, the NA sequences analysis revealed that all Tunisian strains of the 2017–2018 season had evolved away from IAV(H1N1) pandemic strains circulating during the 2009–2010 and 2010–2011 outbreak (clade A). In contrast, they belonged to clade B with the vaccine strain A/Michigan/45/2015 (A/H1N1) pdm09-like and European IAV strains circulating during the 2017–2018 season in several Mediterranean countries: Spain, Italy, and France. On the other hand, the phylogenetic tree showed clearly that IAV(H1N1) Tunisian isolates were related to the vaccine strain A/Michigan/45/2015 (A/H1N1) pdm09-like virus recommended by the WHO for the northern hemisphere during 2017–2018. However, our isolates showed a genetic variation when related to this vaccine strain.

Phylogenetic tree constructed for the NA gene (1097 nucleotides) of A/H1N1 influenza strains collected in Monastir region (Tunisia) during 2017 to 2018. The tree was constructed using the maximum likelihood method with bootstrap analysis of 1000 replicates. The A/Michigan/45/2015 (A/H1N1) pdm09-like strain was used as the root for the tree and bootstrap values greater than 70 % are shown. The vaccine strain is colored in Blue and Tunisian isolates are colored in Red
Besides, it has been found that Tunisian isolates clustered with IAV strains circulating during the 2018–2019 outbreak such as the Italian strain IAV (A/Italy/7841/2019 H1N1, A/France/180130-1/2018 H1N1, and A/France/180130-2/2018 H1N1) forming together a distinct clade: clade B1 belonging to the clade B (Fig. 2) where they share the same ancestor.
Focusing on Tunisian isolates strains themselves, our results revealed two different intragroup clades B.1.1 and clade B.1.2 in the same population hospitalized in Fatouma Bourguiba Monastir for SARI infections during Influenza 2017–2018 outbreak. Interestingly, the phylogenetic tree demonstrated a proper cluster where mortality cases were assembled, However, other severe cases were assembled in another cluster. These results offer a powerful correlation between phenotypes and genotypes. As far as we know, this is the first time that the NA of Tunisian IAV(H1N1) isolates causing fatalities in the Sahel region were characterized.
Discussion
In this study, we focused on severe acute respiratory illness caused by IAV(H1N1) to investigate the reason for fatal and severe infections. Usually, IAV(H1N1) was described to be associated with fever and coughs symptoms. When infecting IAV(H1N1) can cause inflammation of the upper and lower respiratory tract (Taubenberger and Morens 2008; Jilani et al. 2020). The infection period persists for three to seven days for healthy persons. However, it can be longer (9 to 10 days) for patients with high risk such as children, pregnant women, and aged subjects (Reed et al. 2014). Thus, the inflammation will be associated with further complications such as respiratory distress and dyspnea requiring in most cases intensive care and antiviral treatments. The highest infected age group in all patients was children ≤ 2 years with 3 fatal cases which explains the high risk of infection for this age category. This description supports a previous Tunisian study showing the severity of IAV(H1N1) infection outcomes in hospitalized children with a median age of 12 months (Tinsa et al. 2018).The most likely explanation about infection susceptibility of young children is their fragile immune system which has never been exposed to IAV previously (Pinilla et al. 2012). So, their organisms can develop more severe symptoms and shed a larger number of viruses for a longer time. Besides, the second-highest age group develo** SARI infection is the ≥ 65 years confirming the great prevalence of this category of age to be infected and hospitalized for IAV(H1N1) (Taubenberger and Morens 2008).
In this paper, our attention was directed to only to 31 SARI caused by IAV(H1N1) in the Monastir region. These patients were enrolled in the university hospital of Fatouma Bourguiba in several departments (polyvalent ICU, pediatrics department, emergency department, infection diseases department, and maternity ICU). However, non-Influenza respiratory viruses can be also a serious emerging threat of respiratory infection during the same season. In Tunisia (Sousse Area), it has been demonstrated that the rhinoviruses, respiratory syncytial virus A/B, adenovirus, human coronavirus (NL63, HKU1, OC43, and 229) are the most frequently detected viruses in children. In addition, IAV (H3N2) and can circulate in the same period and cause similar infections (Brini et al. 2017).
The time of sampling is critical for IAV diagnostics. After 5 days of symptoms appearing, the virus titer decreases in the upper respiratory tract which makes the qualitative PCR detection of NA from nasopharyngeal swabs complicated. Thus, only a few samples representing (19,35 %) from the totality were sequenced and analyzed genetically (3 fatal cases, 4 most severe cases).
From the studied samples, we reported fatal cases. We suppose that, once the virus binds to columnar epithelial cells in the respiratory tract, the virus had a high ability of replication leading to a high virus titer. Thus, the Influenza virus may cause severe diseases including apoptosis of the host and adjacent cells origin of respiratory complications described in Table 3 (Otte et al. 2016). From the two treated subjects with NA inhibitor (oseltamivir), one died (SB.47). For that reason, we focused on NA amplification and sequencing for more genetic exploration. Interestingly, the amplification of the NA gene of seasonal IAV(H1N1) has been poorly realized in Tunisia and most interest has been focused on the Hemaglutunine gene for detection and characterization (El Moussi et al. 2013; Ben M’hadheb et al. 2015; Soli et al. 2019).
Although all the patients included in this study were detected positive for IAV (H1N1) by real-time PCR, the qualitative RT-PCR for the neuraminidase showed some weak bands. This can be explained by the time from illness onset to specimen collection, and the type of the sample. To maximize detection of Influenza viruses, respiratory specimens should be collected as close to illness onset as possible (ideally < 3–4 days). However, among the 31 samples only (19,35 %) were sampled ideally. Molecular assays may detect Influenza viral RNA in respiratory tract specimens for longer periods after illness but re-using the same sample for qualitative RT-PCR might impact the quality of bands, and therefore, sequencing.
The phylogenetic study demonstrated a divergence between the pandemic IAV(H1N1) strains isolated in 2009 and 2010 and the Tunisian isolates during 2017–2018 forming a defined clade. The main clades were organized regarding the time which underlined a continuing evolution of the virus since its appearance in 2009. Directly after the infection into humans, the process of natural selection acts on the pandemic strain by adaptation to the new host. Since then, IAV(H1N1) pdm2009 was driven by immunological escape resulting in an antigenic diversity within the virus population. During the infection, and to escape from the immune system, amino acids might be affected and assembled in the head of NA. Although the precise reason for this accumulation remains unknown, it participates in the migration of IAV(H1N1) 2009 viruses’ strain to a seasonal Influenza virus. It’s the antigenic drift phenomenon (Su et al. 2015; Petrova and Russell 2018).
Again, our data shows clearly that all the Tunisian isolates of IAV(H1N1) included in this study, share the same ancestor with the vaccine strain. Every year, the WHO design an Influenza vaccine for the Northern and Southern Hemisphere. During the 2017–2018 season, the vaccine composition included A/Michigan/45/2015 (A/H1N1) pdm09-like virus (WHO 2017). In the present study, we focus on the IAV(H1N1) subtype because of its predominance and severity during this season in North Africa and specifically in Tunisia (WHO 2018a). As mentioned above, our results concur well with the genetic characterization of 32 European IAV(H1N1) strains during 2017–2018 which showed a grou** to the sub-clade 6.B1 presented by the vaccine A/Michigan/45/2015(WHO 2018b). Besides, it has been reported, globally, that IAV(H1N1) viruses are in genetic flux, with substantial heterogeneity declaring a fitness advantage (ECDC 2018). In line with this, the genetic diversity in Europe did not seem to affect the vaccine effectiveness against IAV(H1N1) pdm09 in most studies (Rondy et al. 2018). Interestingly, the analysis for the NA characterization of Tunisian IAV(H1N1) isolates has revealed genetic variations compared to the vaccine strain although they are genetically related. Thus, a probable NA evolution has occurred.
Clade B1 has gathered, mostly, Mediterranean IAV(H1N1) strains circulating in the same period 2017–2018. Tunisian isolates were closely related especially to French and Italian strains. This genetic relation seems to be explained by the virus transmission between individuals in Europe and individuals in Tunisia due to the geographic position. Since Tunisia is located in a central position between North Africa and Europe, frequent movement of individuals, especially between France and Italy, might promote virus transmission. Tourism activity, close economic relationships can argue this hypothesis(France diplomacy 2017). During 2018 the national Tunisian office of tourism reported that 30,8 % of tourists are coming from Europe (ONTT 2018).
Our attention was focused not only on the comparison of the Tunisian isolates with the Mediterranian strains but also on the phylogeny between the circulating viruses in the same Tunisian population. Particular attention is paid to the shape of the tree describing two internal clusters. The remarkable result to emerge from this data is that internal clusters have been distinguished regarding the severe infection effect: fatal cases together, and other severe cases together. Thus, IAV(H1N1) circulating in the Monastir region of Tunisia have different virulence and severity between patients. Our study provides additional support for a possible correlation between the phenotype and genotype that has never been described in this region. Several factors underlying age, patient’s immune status, coinfection, and comorbidity may affect the severity of IAV (H1N1) (Goka et al. 2014). However, it has been reported that mutations in NA and Hemagglutinin (HA) proteins contribute to the increase of pathogenicity, virulence, and immune effectiveness (Pinilla et al. 2012; Russier et al. 2020). The D222G, D293 H, G172R mutations in HA domains were correlated with severe disease outcomes and fatality (Baldanti et al. 2011; Elderfield et al. 2014; Otte et al. 2015; Russier et al. 2020; Nandhini and Sistla 2020). Moreover, the emerging variants in the same population during the same season might reinforce the theory of quasispecies (Domingo et al. 2012). During each replication cycle, the high genetic variability of IAV(H1N1) can enhance its virulence. This dynamic of heterogeneity is not surprising for one of the major RNA viruses characterized by a high rate of mutation and recombination events (Pompei et al. 2012).Despite our finding still a preliminary investigation with a limited number of isolates, it fits with the only previous Tunisian phylogenetic investigation about IAV(H1N1) circulating between 2009 and 2013 showing two major NA populations sharing the same ancestor and potential recombination events of this virus (Su et al. 2015).
The main limitation of this study is the lack of NA sequences in the Influenza database in some Mediterranean regions during 2014–2015 and 2015–2016. Also, a deeper investigation of possible mutations and aminoacid alterations would be very interesting if the sequence covers fully the neuraminidase segment. Moreover, the few numbers of Influenza virus Tunisian isolates seem to present a further limitation of this study.
This work is the first step of IAV(H1N1) genetic evolution understanding in the region of Monastir, Tunisia. The NA characterization will certainly help to solve virus resistance phenomena during antiviral treatment, could potentially help on the monitoring of Influenza virus circulation in Tunisia, and might estimate vaccine efficiency against new emerging strains.
Conclusions
From the research that has been carried out, it is possible to conclude that Tunisian IAV(H1N1) isolated in the Monastir region are genetically related to France strains and especially to Italian strains. A possible genetic heterogeneity and recombination events between the Tunisian circulating strains 2017–2018 might be the origin of such virulent strains. Our results are promising and should be continued with whole-genome sequencing to study deeply the process of evolution in terms of mutations on the genetic and proteomic levels.
Abbreviations
- IAV:
-
Influenza A virus
- NA:
-
Neuraminidase
- SARI:
-
Severe acute respiratory infections
- NCBI:
-
National Center for Biotechnology Information
- WHO:
-
World Health Organization
- PCR:
-
Polymerase chain reaction
- HA:
-
Hemagglutinin
References
Baldanti F, Campanini G, Piralla A et al (2011) Severe outcome of influenza A/H1N1/09v infection associated with 222G/N polymorphisms in the haemagglutinin: a multicentre study. Clin Microbiol Infect 17:1166–1169. https://doi.org/10.1111/j.1469-0691.2010.03403.x
Ben M, Harrabi M, Souii M et al (2015) Multiplex RT-PCR and indirect immunofluorescence assays for detection and subty** of human influenza virus in Tunisia. Curr Microbiol 70:324–329. https://doi.org/10.1007/s00284-014-0719-0
Bouvier NM, Palese P (2008) The biology of influenza viruses. Vaccine 26 Suppl 4:D49–D53. https://doi.org/10.1016/j.vaccine.2008.07.039
Brini I, Guerrero A, Hannachi N et al (2017) Epidemiology and clinical profile of pathogens responsible for the hospitalization of children in Sousse area, Tunisia. PLoS One 12:e0188325. https://doi.org/10.1371/journal.pone.0188325
CDC (2019) Types of influenza viruses. In: Centers for disease control and prevention. https://www.cdc.gov/flu/about/viruses/types.htm. Accessed 21 Jan 2021
CDC (2020) Influenza signs and symptoms and the role of laboratory diagnostics | CDC. https://www.cdc.gov/flu/professionals/diagnosis/labrolesprocedures.htm. Accessed 21 Jan 2021
Domingo E, Sheldon J, Perales C (2012) Viral quasispecies evolution. Microbiol Mol Biol Rev 76:159–216. https://doi.org/10.1128/MMBR.05023-11
ECDC (2018) Joint WHO/ECDC influenza virus characterisation report, summary of TESSy virus characterisation data 2017/18. In: European Centre for Disease Prevention and Control. https://www.ecdc.europa.eu/en/publications-data/influenza-virus-characterisation-report-summary-17-18. Accessed 21 Jan 2021
El Moussi A, Ben Hadj Kacem MA, Pozo F et al (2013) Genetic diversity of HA1 domain of heammaglutinin gene of influenza A(H1N1)pdm09 in Tunisia. Virol J 10:150. https://doi.org/10.1186/1743-422X-10-150
Elderfield RA, Watson SJ, Godlee A et al (2014) Accumulation of human-adapting mutations during circulation of A(H1N1)pdm09 influenza virus in humans in the United Kingdom. J Virol 88:13269–13283. https://doi.org/10.1128/JVI.01636-14
France diplomacy (2017) Tunisia. In: France Diplomacy - Ministry for Europe and Foreign Affairs. https://www.diplomatie.gouv.fr/en/country-files/tunisia/. Accessed 21 Jan 2021
Goka EA, Vallely PJ, Mutton KJ, Klapper PE (2014) Mutations associated with severity of the pandemic influenza A(H1N1)pdm09 in humans: a systematic review and meta-analysis of epidemiological evidence. Arch Virol 159:3167–3183. https://doi.org/10.1007/s00705-014-2179-z
Guo H, Rabouw H, Slomp A et al (2018) Kinetic analysis of the influenza A virus HA/NA balance reveals contribution of NA to virus-receptor binding and NA-dependent rolling on receptor-containing surfaces. PLoS Pathog 14:e1007233. https://doi.org/10.1371/journal.ppat.1007233
Hamdi N, Charfi F (2011) Estimation du nombre total des oiseaux aquatiques hivernant en Tunisie: Période 2001/2002 À 2006/2007. Rev Ecol 66:231
Hamza F, Hammouda A, Selmi S (2015) Species richness patterns of waterbirds wintering in the gulf of Gabès in relation to habitat and anthropogenic features. Estuar Coast Shelf Sci 165:254–260. https://doi.org/10.1016/j.ecss.2015.05.025
Hatta M, Kawaoka Y (2003) The NB protein of influenza B virus is not necessary for virus replication in vitro. J Virol 77:6050–6054. https://doi.org/10.1128/JVI.77.10.6050-6054.2003
Hoffmann E, Stech J, Guan Y et al (2001) Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 146:2275–2289. https://doi.org/10.1007/s007050170002
Horimoto T, Kawaoka Y (2005) Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol 3:591–600. https://doi.org/10.1038/nrmicro1208
Hurt AC, Chotpitayasunondh T, Cox NJ et al (2012) Antiviral resistance during the 2009 influenza A H1N1 pandemic: public health, laboratory, and clinical perspectives. Lancet Infect Dis 12:240–248. https://doi.org/10.1016/S1473-3099(11)70318-8
Jilani TN, Jamil RT, Siddiqui AH (2020) H1N1 influenza. StatPearls. StatPearls Publishing, Treasure Island
Krammer F, Smith GJD, Fouchier RAM et al (2018) Influenza. Nat Rev Dis Primers 4:1–21. https://doi.org/10.1038/s41572-018-0002-y
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Larkin MA, Blackshields G, Brown NP et al (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. https://doi.org/10.1093/bioinformatics/btm404
Lewnard JA, Cobey S (2018) Immune history and influenza vaccine effectiveness. Vaccines 6:28. https://doi.org/10.3390/vaccines6020028
McAuley JL, Gilbertson BP, Trifkovic S et al (2019) Influenza virus neuraminidase structure and functions. Front Microbiol 10. https://doi.org/10.3389/fmicb.2019.00039
Nandhini P, Sistla S (2020) Genetic sequencing of influenza A (H1N1) pdm09 isolates from South India, collected between 2011 and 2015 to detect mutations affecting virulence and resistance to oseltamivir. In: Indian journal of medical microbiology. https://pubmed.ncbi.nlm.nih.gov/33154243/. Accessed 27 Jan 2021
ONMNE (2018) Bulletin mensuel de surveillance de la grippe saisonnière, janvier 2018. https://www.onmne.tn/?page_id=5580. Accessed 21 Jan 2021
ONTT (2018) National office for Tourisme Tunisia. touristics statistics. https://www.ontt.tn/ar/al-ihsaeyet-al-siyahia. Accessed 21 Jan 2021
Osterholm MT, Kelley NS, Sommer A, Belongia EA (2012) Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis 12:36–44. https://doi.org/10.1016/S1473-3099(11)70295-X
Otte A, Sauter M, Daxer MA et al (2015) Adaptive mutations that occurred during circulation in humans of H1N1 influenza virus in the 2009 pandemic enhance virulence in mice. J Virol 89:7329–7337. https://doi.org/10.1128/JVI.00665-15
Otte A, Marriott AC, Dreier C et al (2016) Evolution of 2009 H1N1 influenza viruses during the pandemic correlates with increased viral pathogenicity and transmissibility in the ferret model. Sci Rep 6:1–10. https://doi.org/10.1038/srep28583
Palese P, Tobita K, Ueda M, Compans RW (1974) Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61:397–410. https://doi.org/10.1016/0042-6822(74)90276-1
Petrova VN, Russell CA (2018) The evolution of seasonal influenza viruses. Nat Rev Microbiol 16:47–60. https://doi.org/10.1038/nrmicro.2017.118
Pinilla LT, Holder BP, Abed Y et al (2012) The H275Y neuraminidase mutation of the pandemic A/H1N1 influenza virus lengthens the eclipse phase and reduces viral output of infected cells, potentially compromising fitness in ferrets. J Virol 86:10651–10660. https://doi.org/10.1128/JVI.07244-11
Pompei S, Loreto V, Tria F (2012) Phylogenetic properties of RNA viruses. PLoS One 7:e44849. https://doi.org/10.1371/journal.pone.0044849
Reed C, Chaves SS, Perez A et al (2014) Complications among adults hospitalized with influenza: a comparison of seasonal influenza and the 2009 H1N1 pandemic. Clin Infect Dis 59:166–174. https://doi.org/10.1093/cid/ciu285
Rondy M, Kissling E, Emborg H-D et al (2018) Interim 2017/18 influenza seasonal vaccine effectiveness: combined results from five European studies. Eurosurveillance 23:18–00086. https://doi.org/10.2807/1560-7917.ES.2018.23.9.18-00086
Russier M, Yang G, Briard B et al (2020) Hemagglutinin stability regulates H1N1 influenza virus replication and pathogenicity in mice by modulating type I interferon responses in dendritic cells. J Virol 94. https://doi.org/10.1128/JVI.01423-19
Soli R, Kaabi B, Barhoumi M et al (2019) Bayesian phylogenetic analysis of the influenza-A virus genomes isolated in Tunisia, and determination of potential recombination events. Mol Phylogenet Evol 134:253–268. https://doi.org/10.1016/j.ympev.2019.01.019
Su YCF, Bahl J, Joseph U et al (2015) Phylodynamics of H1N1/2009 influenza reveals the transition from host adaptation to immune-driven selection. Nat Commun 6:7952. https://doi.org/10.1038/ncomms8952
Taubenberger JK, Morens DM (2008) The pathology of influenza virus infections. Annu Rev Pathol Mech Dis 3:499–522. https://doi.org/10.1146/annurev.pathmechdis.3.121806.154316
Tinsa F, Barakizou H, Belhadj I et al (2018) Influenza A/H1N1 pdm 09 in children’s Hospital Bechir Hamza of Tunis. Tunis Med 96:348–352
WHO (2017) WHO | Recommended composition of influenza virus vaccines for use in the 2017–2018 northern hemisphere influenza season. http://www.who.int/influenza/vaccines/virus/recommendations/2017_18_north/en/. Accessed 12 Apr 2020
WHO (2018a) WHO | Weekly epidemiological record, 24 August 2018, vol 93, 34 (pp 429–444). http://www.who.int/wer/2018/wer9334/en/. Accessed 21 Jan 2021
WHO (2018b) Seasonal influenza in the WHO European Region, 2017–2018 early season (2018). https://www.euro.who.int/en/health-topics/communicable-diseases/influenza/publications/2018/seasonal-influenza-in-the-who-european-region,-2017-2018-early-season. Accessed 21 Jan 2021
Acknowledgements
The authors would like to thank the university hospital of Fatouma Bourguiba Monastir: Pediatrics departement, emergency department, polyvalent ICU department, microbiology department. The author’s thank Dr. Issaad Kahwthar Ezzine; Dr. Mohamed Salem Zellama and Mohamed Ali Btach for technical assistance.
Author information
Authors and Affiliations
Contributions
Conceptualization: [Sabrine Ben Hamed, Jawhar Gharbi]; Methodology: [Sabrine Ben Hamed, Myriam Harrabi]. Formal analysis and investigation: [Sabrine Ben Hamed, Aida Elargoub, Haythem Srihi, Oussema souiai]. Writing original draft preparation: [Sabrine Ben Hamed] review and editing: [Jawhar Gharbi, Mohammed A Almalki ]; Supervision: [Jawhar Gharbi, Maha Mastouri, Manel Ben M’hadheb].
Corresponding author
Ethics declarations
Ethical standards
Human Nasopharyngeal swabs samples used in this study were obtained by special physicians for the routine laboratory testing of severe acute respiratory illness using real-time RT-PCR. The study protocol was approved with a formal authorization by the Research Ethics Committee, Faculty of Medicine of Monastir, under the number [IORG0009738 N° 18/OMB0990-0279].
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(XLSX 13.1Springer-Verlag GmbH Germany, part of Springer Nature KB)
Rights and permissions
About this article

Cite this article
Ben Hamed, S., Elargoubi, A., Harrabi, M. et al. Phylogenetic analysis of the neuraminidase segment gene of Influenza A/H1N1 strains isolated from Monastir Region (Tunisia) during the 2017–2018 outbreak. Biologia 76, 1797–1806 (2021). https://doi.org/10.1007/s11756-021-00723-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11756-021-00723-y