
Abstract
Efforts to fully understand pharmacological differences between G protein-coupled receptor (GPCR) species homologues are generally not pursued in detail during the drug development process. To date, many GPCRs that have been successfully targeted are relatively well-conserved across species in amino acid sequence and display minimal variability of biological effects. However, the A3 adenosine receptor (AR), an exciting drug target for a multitude of diseases associated with tissue injury, ischemia, and inflammation, displays as little as 70% sequence identity among mammalian species (e.g., rodent vs. primate) commonly used in drug development. Consequently, the pharmacological properties of synthetic A3AR ligands vary widely, not only in binding affinity, selectivity, and signaling efficacy, but to the extent that some function as agonists in some species and antagonists in others. Numerous heterocyclic antagonists that have nM affinity at the human A3AR are inactive or weakly active at the rat and mouse A3ARs. Positive allosteric modulators, including the imidazo [4,5-c]quinolin-4-amine derivative LUF6000, are only active at human and some larger animal species that have been evaluated (rabbit and dog), but not rodents. A3AR agonists evoke systemic degranulation of rodent, but not human mast cells. The rat A3AR undergoes desensitization faster than the human A3AR, but the human homologue can be completely re-sensitized and recycled back to the cell surface. Thus, comprehensive pharmacological evaluation and awareness of potential A3AR species differences are critical in studies to further understand the basic biological functions of this unique AR subtype. Recombinant A3ARs from eight different species have been pharmacologically characterized thus far. In this review, we describe in detail current knowledge of species differences in genetic identity, G protein-coupling, receptor regulation, and both orthosteric and allosteric A3AR pharmacology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
G protein-coupled receptors (GPCRs) play essential physiological roles as signal transducers for regulation of organ function and sensory perception and are proven targets for therapeutic drugs [1]. In general, the amino acid sequence of a given GPCR is fairly well-conserved among mammalian species homologs, typically ~ 90%, particularly for most GPCRs that have been successfully targeted with therapeutic drugs. For example, the amino acid sequence identity for all five Class A muscarinic acetylcholine receptors (M1–M5) is 89–98% identical among mammalian species that are commonly used in drug development research (dog, rabbit, rat, mouse), and Class A human and rat β1 adrenergic receptors are 88% identical. Nevertheless, awareness of significant species differences among various druggable GPCRs has emerged, including the Class A histamine receptor family [2] and the Class C metabotropic glutamate receptor family [3]. As an example, subtle differences in the functionally enhancing activity of the M4 positive allosteric modulator (PAM) LY2033298 between human and mouse receptors has been described [4]. More striking species differences have been noted for D1 dopamine receptor allosteric modulators [5, 6]. An evolutionary approach has been used to analyze conserved GPCR sequence-function relationships between distant species homologues [7].
Species differences among adenosine receptors
There are four subtypes of Class A adenosine receptors (ARs), termed A1, A2A, A2B, and A3. The A1 and A3ARs couple to Gi/o inhibitory proteins, whereas A2AARs couple to Gs proteins. Uniquely, the A2BAR dually couples to Gs and Gi in many cell types [8, 9] and couples to Gq proteins as well [10, 11]. For A1, A2A, and A2BARs, the sequence identity among human, dog, rat, and mouse is greater than 90%, so accordingly, there is relatively little pharmacological variability among these receptors, and only relatively small differences in binding affinity of agonists and antagonists have been described. The A1AR is a good example, where the binding affinity of N6-adenine-substituted agonists such as R-PIA (Fig. 1) and C-8-substituted xanthine antagonist derivatives such as DPCPX (Fig. 2) remains potent across species, having only approximately 20-fold differences among human, dog, bovine, and rat. These species differences have been attributed to two amino acids in transmembrane helical domain (TM)7 using site-directed mutagenesis [12]. A well-studied A1AR PAM, PD81,723, retains activity across all species that have been tested [13,14,15]. Other characteristics are also common to all A1AR species, including activation and desensitization, which both occur slowly over several hours for this receptor [16, 17].

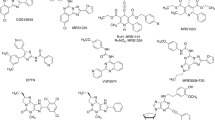
Chemical structures of representative A3AR agonists and allosteric modulators, including those that are commercially available (see text)

Chemical structures of representative A3AR antagonists, including those that are commercially available (see text). The four 4'-truncated nucleosides (bottom row, right) can display residual low efficacy agonism, depending on the model
However, for the A3AR, the amino acid sequence identity between human and rat homologues is only 72% (Table 1), and stark species differences in pharmacology have been observed for agonists, antagonists, and allosteric modulators [16,17,18,19]. Pharmacological differences between human and rodent (rat and mouse) A3ARs are especially prominent (Table 2). For example, the binding affinities of N6-methyladenosine at human and rat A3ARs are 9.0 nM and 6.4 µM, respectively [16], a difference of ~ 700-fold. The binding affinity of the potent human A3AR antagonist MRS1220 is 0.6 nM, i.e., 50,000-fold higher affinity than at the rat A3AR, which is 30 µM [20]. Caffeine, theophylline, and other alkylxanthines are weak antagonists of all the human ARs but are inactive at rat or mouse A3ARs at concentrations up to 100 µM [21,22,23].
The dramatic species-dependent variation in ligand potency between human and rodent A3ARs is most likely because of the large genetic difference among species, as the amino acid identity between human and mouse A3ARs is 73%. A higher degree of identity is found between human and canine A3ARs (88%), between human and sheep A3ARs (86%), and between rat and mouse A3ARs (89%). The genetic differences may be reflected in functional variation among different A3AR species homologues. Generally, A3AR function is similar among more closely related mammalian species such as larger mammals, and among smaller mammals such as rodents, but differences are more pronounced between the two. This point should be earnestly considered in develo** drugs that target the A3AR. The application of various genetically modified small animal models, including global [28] and conditional mouse A3AR gene knockout (KO) models [29], is necessary to implicate the A3AR in specific human diseases and to validate effectiveness and specificity of potential clinical leads. The difference in A3AR antagonist pharmacology between human and mouse led to the introduction of an “A3AR functionally humanized” mouse model in which a human/mouse chimera that maintains faithful coupling to phosphoinositol 3-kinase (PI3K) Υ-signaling in mast cells replaced the mouse A3AR [30]. This is a particularly useful model to test the utility of antagonists for inflammatory and allergic diseases.
In this review, we first compare the protein sequence differences among the eight A3AR species homologues that have been cloned and studied pharmacologically. We then assess the pharmacological profile of agonists, antagonists, and allosteric modulators at the A3AR of various species where data are available. We also explore differences in receptor activation, desensitization, re-sensitization, internalization and downregulation, and the mechanistic differences behind these A3AR-mediated events using human and rat A3ARs as models. Finally, we discuss examples where lack of attention to fundamental A3AR species differences may have led to data misinterpretation in A3AR studies in experimental animals.
Discovery and genetic differences of A3AR from various species
The original observation of an A3AR pharmacological action was from Ali et al. [31], who reported that the relatively nonselective AR agonists NECA and R-PIA induced release of inositol phosphates (IP) and increased cytosolic Ca2+ levels in RBL-2H3 cells, a rat basophilic leukemia cell line. This activity was later ascribed to the cloned rat A3AR [32]. Both NECA and R-PIA enhanced antigen (DNP-BSA)-induced IP and Ca2+ release in RBL-2H3 cells in a pertussis toxin-sensitive manner, whereas the effect of DNP-BSA was pertussis toxin-insensitive [31]. The prototypical AR antagonist theophylline did not substantially inhibit the stimulatory effects of NECA and R-PIA at concentrations up to 100 µM. 8-Phenyltheophylline, which binds to native rat A1 and A2AARs with affinities of 86 and 848 nM [33], respectively, was also ineffective (10 µM). Thus, the effects of NECA and R-PIA were sensitive to pertussis toxin but insensitive to alkyl xanthine antagonists. The authors proposed that NECA and R-PIA act via a novel AR subtype (termed “A3 adenosine receptor”) [31]. Since then, RBL-2H3 cells have been widely used as a model for studying the A3AR, especially its role in mediating mast cell degranulation [34,35,36]. Even prior to the report by Ali et al. [31], Marquardt et al. [37] reported that adenosine (100 µM) enhanced cation ionophore (A23187)-induced histamine release in primary mast cells collected from rat thoracic and abdominal cavities. Adenosine-induced potentiation was partially inhibited by a theophylline concentration of 1 µM and completely blocked at 100 µM. The relatively xanthine-resistant effect of adenosine was suggestive of involvement of an atypical adenosine receptor.
Ribeiro and Sebastiao [38] studied the role of adenosine in the frog neuromuscular junction and proposed that an “A3 adenosine receptor is a voltage-dependent calcium channel, which changes its conformation after binding adenosine.” However, the term “A3 adenosine receptor” described in this work does not seem to be related to the A3AR identified in RBL-2H3 cells [31] or the cloned rat A3AR, which will be described below. The term “A3 adenosine receptor” has also been used in other instances in past years, but none are related to the one reported by Ali et al. [31] and Zhou et al. [32].
At the beginning of the molecular cloning era of GPCRs in the late 1980s and early 1990s, the rat A3AR cDNA was first isolated from a rat testis cDNA library by Meyerhof et al. [39], but adenosine derivatives were not included among a panel of a dozen bioactive compounds that were initially tested as potential ligands, and thus the receptor was not deorphanized until later. Zhou et al. [32] re-cloned the rat A3AR (318 amino acids) in 1992 from a rat brain cDNA library and then first discovered that it responded to adenosine and comprised a new AR subtype. Using in situ hybridization, the authors found highest expression of transcript in the testis, implicating for the first time a role of adenosine in reproduction. Zhou et al. [32] expressed the newly discovered cDNA in CHO cells and detected specific binding in isolated cell membranes using the agonist radioligand [125I]I-APNEA. In subsequent competition binding assays with [125I]I-APNEA, the rank order of potency of agonist AR ligands was as follows: R-PIA≈NECA > S-PIA > adenosine. Xanthine antagonists including xanthine amine congener (XAC) and DPCPX displaced [125I]I-APNEA binding with very low affinity. In functional assays with transfected cells, R-PIA and NECA potently inhibited forskolin-stimulated cAMP production in a pertussis toxin-dependent manner, demonstrating Gi protein coupling. Since the sequence and functional characterization of this receptor were unlike any previously cloned AR subtypes at the time, Zhou et al. [32] termed this receptor the A3AR. In later studies, the A3AR initially described in RBL-2H3 cells and the receptor encoded by the cDNA cloned by Meyerhof and colleagues from a rat testis cDNA library were confirmed to be the same A3AR [32]. Both rat RBL-2H3 cells and mouse bone marrow-derived mast cells (BMMCs) express the A3AR at high levels, as measured by both transcript (Northern blotting, RT-PCR) and protein (radioligand binding) expression [24], which will be described below.
As described previously, the A3AR was first studied in the rat where transcripts were initially detected in testis [32, 39] by in situ hybridization. Later studies using the more sensitive technique of RT-PCR identified transcripts in the heart, lung, and a limited number of CNS regions [32] (Table 3). In a follow-up study, Dixon et al. [40] also found that rat A3AR mRNA only highly expressed in testis using in situ hybridization. However, by using RT-PCR, A3AR gene expression was also found to be widespread in the rat. In peripheral tissues, the A3AR was found highly expressed in lung, spleen, uterus, and testis. Moderate levels occurred in liver and bladder and very low levels in the heart, aorta, stomach, jejunum, proximal colon, kidney, and eye [40]. RT-PCR also revealed the presence of low levels of A3AR mRNA in all brain regions [40]. Efforts to detect A3AR protein expression by radioligand binding have not been overly informative, because the available A3AR radioligands lacked sufficient selectivity for this purpose. While Shearman and Weaver [41] detected specific binding using [125I]I-AB-MECA using rat brain membranes, > 95% of binding was displaced by XAC at a concentration (1 µM) that would not be expected to displace A3AR binding. However, in similar studies, Ji et al. [42] detected residual [125I]I-AB-MECA binding in the presence of 1 µM XAC in rabbit, gerbil, and rat brain. The AR expression profile in rat testis has been characterized in functional assays of receptor-induced regulation of cAMP generation, and the presence of A3ARs has been confirmed in germ cells (spermatocytes and spermatids) but not spermatozoa [43]. A role of the A3AR in spermatogenesis or sperm motility in rats has not been determined.
Haeusler et al. [48], using [125I]I-AB-MECA, studied A3AR distribution in rat and human tissues. Displacement of [125I]I-AB-MECA specific binding by the A3AR-selective 4-aryl-3,5-diacylpyridine derivatives MRS1523 and FE@SUPPY, first reported in Li et al. [27], was found in all rat peripheral tissues examined with the highest amounts in the spleen, lung, heart, and testes; lower amounts were found in rat brain and human brain. These observations are consistent with assays of mRNA expression. Kiesewetter et al. [49] showed that positron-emitting [76Br]-labeled nucleoside A3AR antagonist MRS5147 (designed based on the corresponding 3-chlorobenzyl analogue MRS5127) was taken up rapidly in the rat testes. Wadsak et al. [50] attempted to use [18F]5-(2-fluoroethyl) 2,4-diethyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate (FE@SUPPY) to label rat brain A3ARs. In this study, autoradiography was performed on rat brain slices in the presence or absence of Cl-IB-MECA. The authors suggested that [18F]FE@SUPPY has the potential to serve as the first positron emission tomography (PET) tracer for the A3AR, but the results were not definitive.
Linden et al. [51] cloned the sheep A3AR (317 amino acids) in 1993 and found that the sequence is only 72% identical to the rat A3AR. The largest differences between sheep and rat were found in the C-terminal segment and the second and third extracellular loops (EL2 and EL3). These interspecies differences suggested that desensitization, signaling, and ligand binding might differ between these two species homologues. In contrast to the distribution observed in rat, where A3AR mRNA transcript was found primarily in testis, the A3AR transcripts in sheep were found to be most abundant in lung, spleen, and pineal gland. Moderate levels were detected in the brain, kidney, and testis [51]. Agonists were found to inhibit forskolin-stimulated cAMP accumulation in CHO cells stably expressing the sheep A3AR, confirming coupling to Gi proteins. The potency order of agonists in displacing [125I]I-ABA binding was NECA > R-PIA > S-PIA > CPA. However, several xanthine derivatives, which do not bind to the rat A3AR, were potent sheep A3AR antagonists with a potency order of I-ABOPX > XAC > DPCPX > theophylline. Thus, the sheep A3AR showed clear differences from the rat A3AR in ligand binding, amino acid sequence, tissue distribution, and possibly receptor regulation.
Salvatore et al. [52] cloned the human A3AR in 1993 from a striatal cDNA library (318 amino acids). The human A3AR exhibits 72% and 85% overall identity with the rat and sheep A3AR sequences, respectively. In CHO cells expressing the human A3AR, agonists inhibited forskolin-stimulated cAMP accumulation with the order of potencies of NECA > R-PIA > CPA > S-PIA. In human A3AR radioligand binding assays, several xanthine antagonists bound with high affinity with a rank order of potency of I-ABOPX > XAC > DPCPX. The human A3AR transcript is widespread with the most abundant expression in the lung and liver, which was similar to the profile found in sheep. Abundant A3AR expression in lung tissue raised the possibility for the first time that the A3AR may play a prominent role in pulmonary pathophysiology in some species including humans.
Sajjadi and Firestein [53] cloned a human A3AR homolog from a human heart cDNA library. The receptor shared a low degree of sequence homology to the rat A3AR (71%). Northern analysis of various human tissues showed the human A3AR gene to be expressed primarily in the lung, liver, kidney and heart, with very low expression in the brain and skeletal muscle. Comparable variants have not been identified in other species. It is generally viewed that this variant likely plays no physiological role and likely is not functionally expressed. Murrison et al. [54] isolated the human A3AR gene from a HepG2 cell genomic DNA library, which encodes a protein of 319 amino acids identical in sequence to that of the A3AR described by Salvatore et al. [52] but differing by a single nucleotide (G rather than A at position 310) from that described by Sajjadi and Firestein [53]. Atkinson et al. [55] also cloned the human A3AR gene using fluorescence in situ hybridization to map the ADORA3 gene to chromosomal locus 1p13.3. Northern blot studies showed that the A3AR is widely expressed and is most abundant in brain and some endocrine tissues. The authors found no evidence of alternate splicing in the 5′ untranslated region of the ADORA3 gene. Thus, the observed A3AR distribution patterns in humans from different reports were not entirely consistent. The finding of high A3AR expression in human brain deserves further exploration.
Hill et al. [56] cloned the rabbit A3AR (320 amino acids). The amino acid identities with human, sheep and rat A3ARs were 76%, 75%, and 68%, respectively. Rabbit A3AR mRNA was identified in lung, liver, brain, and heart. In CHO cells expressing the rabbit A3AR, R-PIA inhibited forskolin-stimulated cAMP accumulation with an EC50 of 25 nM. The binding affinities (Ki, nM) of selected ligands based on the inhibition of [125I]I-ABA binding were IB-MECA (2), NECA (27), R-PIA (49), CPA (49), XAC (329), and DPCPX (1120). The rabbit A3AR pharmacological profile is closer to the human A3AR in comparison to the rat A3AR.
Auchampach et al. [25] cloned and characterized the canine A3AR (314 amino acids), which was 88%, 86%, 72%, and 77% identical to the human, sheep, rat, and rabbit A3ARs, respectively. Of the six A3AR species homologs cloned before 1997, the human A3AR amino acid sequence was most similar to the canine and least similar to the rat A3AR. Canine A3AR transcripts were found predominantly in the lungs, spleen, liver, and testes. In COS-7 cells transiently expressing the canine A3AR, it is interesting that [125I]I-ABA bound to two affinity states of the receptor with Kd values of 0.7 and 16 nM, respectively. Since [125I]I-ABA bound to a single low-affinity state with a Kd of 17.4 nM when GTPγS was included to effect receptor-G protein-uncoupling, the two affinity states likely reflect binding of [125I]I-ABA to high affinity G protein-coupled receptors and low-affinity G protein-uncoupled receptors. The potency order of agonists for the canine A3AR was IB-MECA > I-ABA > R-PIA. The canine A3AR bound xanthine antagonists with a potency order of I-ABOPX > XAC≈DPCPX > 8-SPT.
Salvatore et al. [24] cloned the mouse A3AR gene encoding a 319-amino acid protein, which exhibits 89% overall identity to the rat, 76% to the sheep, and 73% to the human A3AR. Although A3AR transcripts are abundant in rat testis and a role in reproduction has been suggested [32], the A3AR KO mice bred and matured normally, indicating that the mouse A3AR probably does not play a critical role in reproduction. Also, specific A3AR binding of a newly developed species-general A3AR antagonist radioligand [3H]MRS7799 (DPTN) to membranes from mouse testis or brain was below the detection limit (Gao and Jacobson, unpublished data). However, Burnett et al. [45] reported that both IB-MECA and Cl-IB-MECA accelerate mouse sperm motility. We are not aware of any substantial progress toward the development of A3AR ligands applied to reproduction or contraception. It is important to consider that disparities in results of mouse A3AR expression studies (and perhaps other species) may be explained by the discovery in 2010 that the mouse A3AR gene is embedded within the gene encoding transmembrane and immunoglobulin domain containing 3 (TMIGD3) [57]. Both genes share coding exons that create at least one TMIGD3/A3AR hybrid variant (Adora3i3). Interestingly, TMIGD3 is abundantly expressed in mouse testes, and the Adora3i3 variant, which is encoded by exon 1 of the TMIGD3 gene and exon 2 of the A3AR gene, is possibly controlled by the TMIGD3 promoter. Functionality of the hybrid TMIGD3 splice variant remains highly questionable since it is unlikely to bind adenosine or to transduce signals to G proteins. Expression studies based on probes or PCR primer pairs targeting areas that are shared between TMIGD3 and A3AR splice variants may produce misleading information on A3AR distribution. Notably, TMIGD3 and A3AR splice variants are also found in other species including humans.
Alnouri et al. [18] reported the binding affinity of commonly used agonists and antagonists at the mouse A3AR expressed in CHO cells. The agonist affinities (nM) were as follows: NECA (13.2), R-PIA (9.98), IB-MECA (0.21), and Cl-IB-MECA (0.18). The antagonist affinities (nM) at mouse A3AR were as follows: CGS15943 (2970), MRS1523 (1980), and xanthine antagonists, i.e., caffeine, theophylline, and DPCPX (all > 100,000 nM). Thus, the ligand potency profile at the mouse A3AR is similar to that of the rat A3AR, a conclusion confirmed by Gao et al. [17].
To further investigate mouse A3AR distribution, Yamano et al. [30, 46] generated a transgenic mouse with whole-body expression of jellyfish apoaequorin under control of the A3AR gene promoter. The authors succeeded in detecting adenosine ligand-induced light emission by aequorin in response to increases in cytoplasmic Ca2+ in the pancreas, brain, and testis. Yamano et al. [46] suggested that the pancreas is one of the main tissues that expresses the A3AR.
Durand and Green [47] cloned the chick A3AR cDNA showing 52.1%, 57.0%, and 52.2% homology to the rat, sheep, and human A3ARs, respectively. As a comparison, the chick A3AR was 43.8% homologous to the chick A1AR. In HEK293 cells expressing the chick A3AR, CHA and Cl-IB-MECA inhibited isoproterenol-stimulated cAMP accumulation with EC50s of 16.2 and 25.1 nM, respectively, confirming Gi coupling. In radioligand binding assays using [125I]I-ABA, the following Ki values (nM) were determined: IB-MECA, 2.8; Cl-IB-MECA, 14.9; NECA, 37; CPA, 2.6; R-PIA, 1.1; DPCPX, 53; and MRS1191, 1704. Thus, the binding profile of agonists and antagonists at the chick A3AR differed from other cloned A3ARs. Most notably, the chick A3AR bound both A3 (IB-MECA, Cl-IB-MECA) and A1AR (CPA, R-PIA) agonist ligands with high affinity. We are not aware of any further reports regarding the chick A3AR ligand binding profile.
Other A3AR species homologues have been reported, but with less extensive pharmacological characterization. Brandon et al. [44] cloned and pharmacologically characterized the equine A3AR, although no data related to the tissue distribution of this receptor has been provided. The equine A3AR amino acid sequence is 82.5% and 84.7% identical to human and sheep A3ARs, respectively. In radioligand binding studies with transfected HEK293 cells, the rank order of potency of agonists to displace [125I]I-AB-MECA binding was as follows: IB-MECA (5.2 nM) > NECA (7.9 nM) > CGS21680 (110 nM), which roughly agreed with published findings with all other mammalian nonrodent A3ARs. The rank order of antagonist potency was as follows: MRS1220 (1.2 µM) > ZM241385 > 8-p-sulfophenyltheophylline, which was inconsistent with other nonrodent A3ARs.
Thus, among the eight species that have been characterized, it is of note that the A3AR tissue distribution varies substantially. For example, A3AR expression is high in rat testes but low in mouse and sheep testes. Expression of A3AR transcripts in human liver is abundant, whereas it is low in sheep [32, 51, 52, 56, 58]. In general, A3AR abundance in the lungs is consistent across many species, including rabbit, dog, sheep, and human. However, even within the same species, reports of the A3AR distribution profile vary (especially in the rat and human). It must be kept in mind that many earlier studies utilized techniques (in situ hybridization, Northern blotting) that are less sensitive compared to modern molecular techniques that are now used widely (PCR, qPCR, direct sequencing). Regarding new technology, exon expression by RNA sequencing for the A3AR gene in postmortem human tissues is now available [58]; interestingly, high expression was detected in the spinal cord. Moreover, several public single-cell RNA sequencing databases have become available in recent years. Interestingly, the Tabula Muris database (https://tabula-muris.ds.czbiohub.org/), a compendium of single-cell transcriptome data from the mouse, reports that A3AR mRNA expression is most abundant in microglial cells in the brain and is also found in a number of different immune cell populations including granulocytic cells (basophils, neutrophils) and other myeloid cell populations (monocytes and tissue-resident macrophages). The Human Protein Atlas (www.proteinatlas.org) reports predominant A3AR expression in myeloid cells (granulocytes, monocytes, eosinophils, basophils) and tissue resident macrophages as well as in Kuppfer cells and Hofbauer cells. These findings raise the possibility that prior expression analyses using whole-tissue samples may reflect A3AR presence in immune cell populations within the tissues. With earlier published studies, expression levels in different species were not compared directly using the same methodology under the same experimental conditions. As suggested previously, variability may be related to detection of TMIGD3/A3AR splice variants that are likely non-functional.
To summarize thus far, the initial A3AR molecular cloning studies raised awareness of large differences in A3AR protein sequence, tissue distribution, and pharmacology. These early studies in particular identified stark differences in A3AR properties between humans and rodents, species commonly used during the course of drug development. Thus, it was soon appreciated that observations in rodent models, must be confirmed in additional larger mammalian species, such as dogs, sheep, or rabbits [20, 25, 59,60,61].
Species dependence of A3AR agonists
In most initial studies with recombinant A3ARs from various species, the 5′-substituted adenosine derivative NECA and the N6-substituted adenosine derivative R-PIA were found to be full, relatively potent agonists. The non-selective radioligands [125I]I-ABA [25, 56] and [125I]I-APNEA, used in the initial A3AR cloning [32], are bound with high affinity at rat, mouse, canine, rabbit, and human homologues. The 5′- and N6-substituted radioligand [125I]I-AB-MECA was found to bind with nearly nM affinity at the A3ARs from various species [42, 44, 62, 63]. In subsequent studies after 1997, IB-MECA and/or Cl-IB-MECA, which exhibit moderate A3AR selectivity, were also used to initially characterize cloned A3ARs from a number of different species. It is notable that IB-MECA and Cl-IB-MECA have been demonstrated to act as human A3AR partial agonists in several independent assay systems [64,65,66]. However, they are likely full agonists for rat and mouse A3ARs [58, 67], although this has not been systematically studied or strictly compared.
Adenosine derivatives that act as agonists generally display much less human/rat species variability as compared to non-nucleoside antagonists, but more subtle differences have been noted [16, 68]. The affinity of adenosine itself cannot easily be measured precisely, because it is rapidly degraded by adenosine deaminase found in all cells and tissues. Alternatively, 2-chloroadenosine (CADO), which is resistant to the actions of adenosine deaminase, has been used as a surrogate. The Ki values of CADO are 1890 nM at the rat A3AR and 87 nM at the human A3AR; thus, under the assumption that CADO faithfully replicates the actions of adenosine, these results predict A3AR species variability for its endogenous agonist, which may have evolved as a survival advantage to attenuate A3AR-mediated mast cell degranulation that occurs in rodents. Various N6-substituted agonists have been demonstrated to be > 100-fold more potent at human than at rat A3ARs (Table 4). For example, N6-[(1S,2R)-2-phenyl-1-cyclopropyl]adenosine (Ki = 0.63 nM for hA3AR) is > 1000-fold more potent for human than rat A3ARs. A small N6-alkyl substituent, as with N6-methyladenosine, has been demonstrated to be extremely important for the species difference between human and rat A3ARs (Ki = 9.3 and 6390 nM, for human and rat A3AR, respectively) [16]. Some N6-benzyl substituted agonists, such as 5′-methylamides IB-MECA and Cl-IB-MECA and the corresponding 5′-CH2OH nucleosides, are potent at both human and rat A3ARs. Furthermore, in the (North, N)-methanocarba series containing a sterically constrained bicyclic ribose substitution, an N6-benzyl group was incorporated in adenosine 5′-uronamides to achieve species-independent A3AR receptor-selectivity [68]. Thus, it seems that the N6-benzyl group promotes potency at rodent A3ARs [16,17,18,19,20,21,22,23,24,25, 27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68]. More recently, Alnouri et al. [18] compared some commonly used agonists in radioligand binding studies using membranes from CHO cells expressing four AR subtypes of three species: human, rat, and mouse. The authors confirmed that IB-MECA and Cl-IB-MECA are indeed potent agonists for human, rat or mouse A3ARs, although the A3AR selectivity is variable at different species. Ge et al. [69] and Carlin et al. [23], using HEK293 cells expressing mouse A3AR, also showed that both IB-MECA and Cl-IB-MECA are very highly potent A3AR agonists in the mouse (Ki 87 and 180 pM, respectively). Therefore, it should be kept in mind that it is best to use a concentration of ~ 1 nM or lower to activate the mouse or rat A3AR, since IB-MECA and Cl-IB-MECA are also potent mouse A1AR agonists with Ki values of 5.9 and 35 nM, respectively [69]. At a higher concentration, such as 1 µM, IB-MECA can also act as an A2AAR agonist, which will be described later in this review. The commercially available and moderately selective A3AR agonist HEMADO is potent at human A3AR (1.1 nM). Since the N6-substitution of HEMADO is a methyl group, it is very weak at rat or mouse A3ARs, and the tritiated radioligand was unsuitable for these species [70]. Its potency at mouse A3AR has not been examined, but HEMADO binds to rat A3AR with a Ki of 6.44 ± 1.18 µM (unpublished data). The introduction of H-bonding groups (4-hydroxy-3-methoxy) in a truncated N6-(2-phenylethyl)adenosine analogue leading to MRS7591 was found to dramatically increase the mouse A3AR affinity, as predicted using molecular modeling based on the multiple polar H-bonding side chains in mouse A3AR EL residues. There has been no report of selective A3AR agonists from the non-ribose class compounds [71, 72].
The (N)-methanocarba ring system (consisting of a fused cyclopropyl and cyclopentyl ring) in place of the furanose ring of adenosine is a general method for enhancing both A3AR affinity selectivity, as in a recently available commercially N6-(3-chlorobenzyl) derivative, MRS5698. MRS5698, containing a 5′-methylamide, was shown to be potent and selective at human, mouse and rat A3ARs with Ki values of ~ 3 nM, and > 3000-fold selectivity versus A1, A2A, and A2BARs [17, 23]. Recently reported analogues based on that observation include the highly potent MRS5980 (> 10,000-fold selective for human A3AR compared to A1/A2A, Ki = 0.7 nM) [80]. However, as an N6-methyl analogue, the mouse A3AR affinity of MRS5980 (Ki = 36 nM) is 51-fold lower than at human A3AR [23], but it remains highly selective. The human A3AR affinity is further enhanced by substitution of Cl with F in MRS7334 (Ki = 0.28 nM) [80]. Thionucleoside LJ529, an analogue of Cl-IB-MECA, is a highly potent A3AR agonist [81].
The residues of the human and mouse A3AR sequences that are predicted to be in proximity (distance ≤ 4 Å) to the partial agonist nucleoside ligand MRS7591 [82, Fig. 2] are shown in bold in Fig. 3 for both the human and mouse sequences. Residues of the human A3AR sequence that are shown in red were found to diminish agonist binding or activation [83]. There is a close commonality across adenosine receptor subtypes in the residues that are involved in orthosteric ligand recognition [78, 82]. For example, the following residues that were identified as important for agonist action at the human A3AR [83] are conserved in all 13 species in Fig. 3, including chick: T3.36, F168 (EL2), M5.38, W6.48, L6.51, N6.55, I7.39, S7.42, and H7.43 (using Ballesteros-Weinstein numbering, shown in red in the aligned human sequence). Recently, the binding site for the A3AR-selective PAM LUF6000 was localized to the intracellular side of the receptor [84], although specific residues involved were not identified. LUF6000 is inactive at the mouse A3AR, which allowed use of chimeric mouse-human A3ARs to identify the allosteric binding region on the half of the receptor facing the cytosol. Thus, there is need for additional mutagenesis experiments to better characterize the basis for species differences in both orthosteric ligand recognition and allosteric modulation.

Sequence alignment of thirteen A3AR species homologues (Species identifiers (https://www.ncbi.nlm.nih.gov/): NP_000668.1, Homo sapiens (human); NP_001289704.2, Pan troglodytes (chimpanzee); NP_001075527.1, Oryctolagus cuniculus (rabbit); NP_001003178.1, Canis lupus familiaris (dog); XP_006182940.1, Camelus ferus (camel); NP_001289720.1, Callithrix jacchus (marmoset); XP_006935064.2, Felis catus (cat); XP_012998623.1, Cavia porcellus (guinea pig); XP_035293911.1, Cricetulus griseus (hamster); XP_021514447.1, Meriones unguiculatus (Mongolian gerbil); mouse (source: Fisher et al. [84]); NP_001289680.1, Rattus novegicus (Norwegian rat); NP_989482.2, Gallus gallus (chicken). TMs are indicated in cyan, and extracellular loops in yellow highlight. The definition of helical regions and residues (bold, human and mouse sequences) corresponding to those in contact (distance ≤ 4Å) with partial agonist ligand MRS7591 (Figure 2) are according to Tosh et al. [82]. Ala mutation of red residues in the human A3AR sequence diminished agonist binding or activation [85]: L3.32, T3.36, F168 (EL2), M5.38, W6.48, L6.51, N6.55, L7.35, I7.39, S7.42, H7.43. Note: The green highlighted L in the mouse sequence is the correct residue [82]. On GPCRdb (https://gpcrdb.org/) this residue is incorrectly shown as a valine (V3018.61). Note that residue numbers shown in this alignment may differ from other reports
In summary, two prototypical AR agonists R-PIA and NECA, although nonselective, were used in the initial pharmacological characterization of A3ARs cloned from various species. N6-methyl substituted adenosine derivatives in the ribose series, such as N6-methyladenosine and HEMADO, are potent at human but weak at rat A3ARs. N6-Methyladenosine is a 690-fold weaker in binding to the rat A3AR compared to the human homologue [16]. N6-Benzyl substituted derivatives such as IB-MECA and Cl-IB-MECA are potent across species, although somewhat more potent at rat and mouse A3AR than at human A3AR. The A3AR selectivity of IB-MECA and Cl-IB-MECA is limited; this limitation should be kept in mind when used in studies with mice or rats. For example, the affinity of IB-MECA for the mouse A1AR is 5.9 nM [69]. Accordingly, IB-MECA should be used at a concentration of ≤1 nM in studies with mouse cells/tissues to achieve an A3AR-selective concentration. MRS5698 is a potent and selective agonist across all species tested thus far and is the best agonist that is readily available to study the A3AR in mice or rats. Potent and selective A3AR agonists from the class of non-nucleoside agonists [72] have not been developed, which should be a future effort.
Species dependence of A3AR antagonists
Methylxanthines, such as caffeine and theophylline, are generally weak antagonists for all human and rodent AR subtypes. However, caffeine and theophylline function as weak human A3AR antagonists but are essentially inactive at the rat A3AR [18, 32, 52]. At human, rat, and mouse A1, A2A, and A2BARs, the selective antagonists PSB-36 (A1), preladenant (A2A), and PSB-603 (A2B) displayed high selectivity across species [18]. Thus, there remains a need to discover pan-species, highly potent, and selective non-nucleoside A3AR antagonists.
Pyridine derivative MRS1523 is one of the few A3AR antagonists suitable for rat (Ki = 0.5 µM; mean value of all published data) or mouse (Ki = 0.35 [17], 0.70 [86], 1.98 µM [18]) studies, with limited selectivity versus A1, A2A, and A2BARs [17, 18]. Another known AR antagonist CGS15943 was also demonstrated to be a weak antagonist at rat (1.3 µM) and mouse (3.0 µM), but it is more potent at A1, A2A, and A2BARs [18]. Other commonly used A3AR antagonists, such as MRS1220 (Ki = 0.65 nM for human A3AR [87], a derivative of the triazoloquinazoline antagonist CGS15943, and 1,4-dihydropyridine MRS1191 [88, 89]), exhibit low affinity or are inactive (< 30% inhibition of radioligand binding at 10 µM) at the mouse or rat A3AR. Carlin et al. [187] reported that FM101 proposed for the treatment of glaucoma is an agonist in cAMP pathway, but an antagonist in arrestin translocation. The mechanism behind the agonist-decreased IOP could be a result of desensitization, which is similar in a way to A3AR antagonism, but this remains to be further confirmed [188]. Thus, it seems convincing that A3AR antagonists rather than agonists may reduce IOP, since it has been demonstrated in four species: mouse, rat, monkey and human.
A3AR in pancreatic islets
The A3AR has been found highly expressed in mouse pancreas [46]. Amisten et al. [189] systematically compared the GPCR transcriptome of human and mouse pancreatic islets to determine to what extent mouse islets can be used as surrogates to investigate the roles of specific GPCRs in human islet cell biology. Interestingly, from these analyses, the A3AR was identified as one of several GPCRs with species-specific expression, as it was found to only be expressed in mouse islets and not human islets. In accord with these findings, MRS5698 was found to only inhibit glucose-induced insulin secretion from mouse islets but not from human islets [189].
Examples of data interpretation based on ambiguous use of A3AR agonists and antagonists
As mentioned earlier, the commonly used A3AR agonist IB-MECA is potent at the mouse A3AR (Ki = 0.2 nM) (Table 4), but it is also potent for mouse A1 (Ki = 5.9 nM) and mouse A2AARs (Ki = 300 nM). Thus, if a concentration of 10 nM is used, it can activate both A1 and A3ARs. If a concentration of 1 µM is used in any experiments, mouse A1, A2A, and A3ARs all can be fully activated, and mouse A2BARs can be partially activated [69]. Thus, many controversies from earlier publications may have stemmed from agonist selectivity and affinity at different species. On the other hand, MRS1220 is sometimes used as an A3AR antagonist in mouse or rat, assuming incorrectly that it is an A3AR antagonist across multiple species (Ki = 0.65 nM for human A3AR). For example, MRS1220 was used to antagonize the antinociceptive action of adenosine in rat, Cl-IB-MECA-facilitated epileptiform discharges in rat hippocampal slices, cordycepin (3′-deoxyadenosine)-inhibited B16-BL6 mouse melanoma cell growth, and IB-MECA’s effect on bone destruction in rats [170, 190,191,192]. However, it should be kept in mind that MRS1220 is an antagonist of mouse and rat A1ARs with respective binding Ki values of 81 and 305 nM, respectively, and mouse and rat A2AARs with Ki values of 9.1 and 52 nM, respectively. Its affinity at the mouse or rat A3AR is > 10 µM [17, 193]. In addition to MRS1220, there is also some controversy about the selectivity of another commonly used A3AR antagonist MRS1523. Unlike MRS1220, MRS1523 is a confirmed antagonist of both mouse and rat A3ARs [17, 18], although with variable selectivity versus other AR subtypes (Table 4), which could be due to the use of different expression systems or radioligands (especially agonist vs. antagonist radioligands), etc. in the various studies (Table 4).
It is notable that that not all studies in the past using IB-MECA or Cl-IB-MECA drew a conclusion related to a role of the A3AR, reinforcing the need for applying appropriately selective AR antagonists. For example, Lasley et al. [194] observed that IB-MECA-induced rat coronary dilation was completely blocked by the A2AAR antagonist SCH58261 (50 nM) and thus the authors concluded that coronary dilatory effect of IB-MECA in the rat heart appear to be caused by A2AAR activation. IB-MECA was found to inhibit LPS-induced TNF-α production via A2AARs but not A3ARs in equine monocytes [195]. In mouse macrophages, LPS-induced TNF-α production was potently inhibited by IB-MECA (1 µM) via activation of A2A but not A3ARs [86].
Conclusions
There are large genetic differences of the A3AR from various species, especially between human and mouse or rat, achieving only ~ 70% amino acid similarity. A3AR tissue distribution varies substantially among species. Different mechanisms are involved in human and rat A3AR activation, desensitization and re-sensitization. Some agonist structures, such as N6-methyladenosine, are ~ 700-fold more potent at human compared to rat A3ARs, while some agonists, such as N6-(3-halobenzyl)-substituted adenosines (IB-MECA, Cl-IB-MECA and MRS5698), maintain high affinity at human, mouse, and rat A3ARs. However, most A3AR antagonists are inactive or weakly active at mouse and rat A3ARs; DPTN and MRS1523 are the only two confirmed A3AR antagonists albeit with moderate selectivity against A1, A2A, or A2BARs. Most allosteric modulators, including LUF6000 and LUF6096, are only active at human and large animal species, but not rodents, but amiloride analogs retain allosteric activity across species. Considering that many species differences were identified between human and mouse or rat, some A3AR functions identified using mouse or rat models need to be confirmed in larger animals, such as dogs or sheep. These species differences in the pharmacological properties of synthetic A3AR ligands, e.g., their binding affinity, selectivity, and signaling efficacy, as well as the species-dependent role and regulation of the receptor, must be taken into consideration during drug development.
Data Availability
Not applicable.
Abbreviations
- AR:
-
Adenosine receptor
- CHO:
-
Chinese hamster ovary
- Cl-IB-MECA:
-
2-Chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide
- GPCR:
-
G protein-coupled receptor
- [35S]GTPγS:
-
Guanosine 5′-[γ-[35S]thio]triphosphate
- HEK293:
-
Human embryonic kidney 293
- [125I]I-AB-MECA:
-
N6-(4-amino-3-[125I]iodobenzyl)adenosine-5′-N-methylcarboxamide
- LUF6000:
-
2-Cyclohexyl-N-(3,4-dichlorophenyl)-1H-imidazo [4,5-c]quinolin-4-amine
- LUF6096:
-
N-{2-[(3,4-Dichlorophenyl)amino]quinolin-4-yl}cyclohexanecarboxamide
- NECA:
-
Adenosine-5′-N-ethylcarboxamide
- VUF5455:
-
4-Methoxy-N-[7-methyl-3-(2-pyridyl)-1-isoquinoyl]-benzamide
- PAM:
-
Positive allosteric modulator
References
Congreve M, de Graaf C, Swain NA, Tate CG (2020) Impact of GPCR structures on drug discovery. Cell 181(1):81–91. https://doi.org/10.1016/j.cell.2020.03.003
Strasser A, Wittmann HJ, Buschauer A, Schneider EH, Seifert R (2013) Species-dependent activities of G-protein-coupled receptor ligands: lessons from histamine receptor orthologs. Trends Pharmacol Sci 34(1):13–32. https://doi.org/10.1016/j.tips.2012.10.004
O’Brien DE, Conn PJ (2016) Neurobiological insights from mGlu receptor allosteric modulation. Int J Neuropsychopharmacol. 19(5):133. https://doi.org/10.1093/ijnp/pyv133
Suratman S, Leach K, Sexton P, Felder C, Loiacono Christopoulos A (2011) Impact of species variability and ‘probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br J Pharmacol 162:1659–70. https://doi.org/10.1111/j.1476-5381.2010.01184.x
Wang X, Heinz BA, Qian YW, Carter JH, Gadski RA, Beavers LS, Little SP, Yang CR, Beck JP, Hao J, Schaus JM, Svensson KA, Bruns RF (2018) Intracellular binding site for a positive allosteric modulator of the dopamine D1 receptor. Mol Pharmacol 94(4):1232–1245. https://doi.org/10.1124/mol.118.112649
Svensson KA, Hao J, Bruns RF (2019) Positive allosteric modulators of the dopamine D1 receptor: a new mechanism for the treatment of neuropsychiatric disorders. Adv Pharmacol 86:273–305. https://doi.org/10.1016/bs.apha.2019.06.001
Schöneberg T, Hofreiter M, Schulz A, Römpler H (2007) Learning from the past: evolution of GPCR functions. Trends Pharmacol. Sci 28(3):117–21. https://doi.org/10.1016/j.tips.2007.01.00
Gao ZG, Inoue A, Jacobson KA (2018) On the G protein-coupling selectivity of the native A2B adenosine receptor. Biochem Pharmacol 151:201–213. https://doi.org/10.1016/j.bcp.2017.12.003
Yang X, **n W, Yang XM, Kuno A, Rich TC, Cohen MV, Downey JM (2011) A2B adenosine receptors inhibit superoxide production from mitochondrial complex I in rabbit cardiomyocytes via a mechanism sensitive to pertussis toxin. Br J Pharmacol 163(5):995–1006. https://doi.org/10.1111/j.1476-5381.2011.01288.x
Feoktistov I, Biaggioni I (1995) Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells An enprofylline-sensitive mechanism with implications for asthma. J Clin Invest 96(4):1979–86. https://doi.org/10.1172/JCI118245
Linden J, Thai T, Figler H, ** X, Robeva AS (1999) Characterization of human A2B adenosine receptors: radioligand binding, western blotting, and coupling to Gq in human embryonic kidney 293 cells and HMC-1 mast cells. Mol Pharmacol 56(4):705–713
Tucker AL, Robeva AS, Taylor HE, Holeton D, Bockner M, Lynch KR, Linden J (1994) A1 adenosine receptors Two amino acids are responsible for species differences in ligand recognition. J Biol Chem. 269(45):27900–6
Bruns RF, Fergus JH (1990) Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol Pharmacol 38(6):939–949
Mizumura T, Auchampach JA, Linden J, Bruns RF, Gross GJ (1996) PD 81,723, an allosteric enhancer of the A1 adenosine receptor, lowers the threshold for ischemic preconditioning in dogs. Circ Res 79(3):415–423. https://doi.org/10.1161/01.res.79.3.415
Guo D, Venhorst SN, Massink A, van Veldhoven JP, Vauquelin G (2014) IJzerman AP, Heitman LH Molecular mechanism of allosteric modulation at GPCRs: insight from a binding kinetics study at the human A1 adenosine receptor. Br J Pharmacol. 171(23):5295–312. https://doi.org/10.1111/bph.12836
Gao ZG, Blaustein JB, Gross AS, Melman N, Jacobson KA (2003) N6-Substituted adenosine derivatives: selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem Pharmacol 65(10):1675–1684. https://doi.org/10.1016/S0006-2952(03)00153-9
Gao ZG, Suresh RR, Jacobson KA (2021) Pharmacological characterization of DPTN and other selective A3 adenosine receptor antagonists. Purinergic Signal 17(4):737–746. https://doi.org/10.1007/s11302-021-09823-5
Alnouri MW, Jepards S, Casari A, Schiedel AC, Hinz S, Muller CE (2015) Selectivity is species-dependent: characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal 11(3):389–407. https://doi.org/10.1007/s11302-015-9460-9
Du L, Gao ZG, Paoletta S, Wan TC, Gizewski ET, Barbour S, van Veldhoven JPD, IJzerman AP, Jacobson KA, Auchampach JA. (2018) Species differences and mechanism of action of A3 adenosine receptor allosteric modulators. Purinergic Signal 14(1):59–71. https://doi.org/10.1007/s11302-017-9592-1
Jacobson KA (1998) Adenosine A3 receptors: novel ligands and paradoxical effects. Trends Pharmacol Sci 19(5):184–191. https://doi.org/10.1016/s0165-6147(98)01203-6
van Galen PJ, van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME (1994) IJzerman AP, Stiles GL, Jacobson KA A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol Pharmacol. 45(6):1101–11
Wang Z, Do CW, Avila MY, Peterson-Yantorno K, Stone RA, Gao ZG, Joshi B, Besada P, Jeong LS, Jacobson KA, Civan MM (2010) Nucleoside-derived antagonists to A3 adenosine receptors lower mouse intraocular pressure and act across species. Exp Eye Res 90(1):146–154. https://doi.org/10.1016/j.exer.2009.10.001
Carlin JL, Jain S, Gizewski E, Wan TC, Tosh DK, **ao C, Auchampach JA, Jacobson KA, Gavrilova O, Reitman ML (2017) Hypothermia in mouse is caused by adenosine A1 and A3 receptor agonists and AMP via three distinct mechanisms. Neuropharmacol 114:101–113. https://doi.org/10.1016/j.neuropharm.2016.11.026
Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA (2000) Disruption of the A3 adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem 275(6):4429–4434. https://doi.org/10.1074/jbc.275.6.4429
Auchampach JA, ** X, Wan TC, Caughey GH, Linden J (1997) Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2b receptor. Mol Pharmacol 52(5):846–860. https://doi.org/10.1124/mol.52.5.846
Takano H, Bolli R, Black RG Jr, Kodani E, Tang XL, Yang Z, Bhattacharya S, Auchampach JA (2001) A1 or A3 adenosine receptors induce late preconditioning against infarction in conscious rabbits by different mechanisms. Circ Res 88(5):520–528. https://doi.org/10.1161/01.res.88.5.520
Li AH, Moro S, Melman N, Ji XD, Jacobson KA (1998) Structure-activity relationships and molecular modeling of 3, 5-diacyl-2,4-dialkylpyridine derivatives as selective A3 adenosine receptor antagonists. J Med Chem 41(17):3186–3201. https://doi.org/10.1021/jm980093j
Lopes CR, Lourenço VS, Tomé ÂR, Cunha RA, Canas PM (2021) Use of knockout mice to explore CNS effects of adenosine. Biochem Pharmacol 187:114367
Wan TC, Tampo A, Kwok WM, Auchampach JA (2019) Ability of CP-532,903 to protect mouse hearts from ischemia/reperfusion injury is dependent on expression of A3 adenosine receptors in cardiomyoyctes. Biochem Pharmacol 163:21–31. https://doi.org/10.1016/j.bcp.2019.01.022
Yamano K, Inoue M, Masaki S, Saki M, Ichimura M, Satoh M (2006) Generation of adenosine A3 receptor functionally humanized mice for the evaluation of the human antagonists. Biochem Pharmacol 71(3):294–306. https://doi.org/10.1016/j.bcp.2005.10.028
Ali H, Cunha-Melo JR, Saul WF, Beaven MA (1990) Activation of phospholipase C via adenosine receptors provides synergistic signals for secretion in antigen-stimulated RBL-2H3 cells Evidence for a novel adenosine receptor. J Biol Chem. 265(2):745–53
Zhou QY, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O (1992) Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc Natl Acad Sci USA 89(16):7432–7436. https://doi.org/10.1073/pnas.89.16.7432
Bruns RF, Lu GH, Pugsley TA (1986) Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol Pharmacol 29(4):331–346
Ramkumar V, Stiles GL, Beaven MA, Ali H (1993) The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J Biol Chem 268(23):16887–16990. https://doi.org/10.1016/S0021-9258(19)85277-8
** X, Shepherd RK, Duling BR, Linden J (1997) Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest 100(11):2849–2857. https://doi.org/10.1172/JCI119833
Hannon JP, Tigani B, Schuurman HJ, Fozard JR (2002) Suppression of adenosine A3 receptor-mediated hypotension and mast cell degranulation in the rat by dexamethasone. J Pharmacol Exp Ther 302(2):725–730. https://doi.org/10.1124/jpet.102.035790
Marquardt DL, Parker CW, Sullivan TJ (1978) Potentiation of mast cell mediator release by adenosine. J Immunol 120(3):871–878
Ribeiro JA, Sebastião AM (1986) Adenosine receptors and calcium: basis for proposing a third (A3) adenosine receptor. Prog Neurobiol 26(3):179–209. https://doi.org/10.1016/0301-0082(86)90015-8
Meyerhof W, Müller-Brechlin R, Richter D (1991) Molecular cloning of a novel putative G-protein coupled receptor expressed during rat spermiogenesis. FEBS Lett 284(2):155–160. https://doi.org/10.1016/0014-5793(91)80674-r
Dixon AK (1996) A K Gubitz, D J Sirinathsinghji, P J Richardson, T C Freeman Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol. 118(6):1461–8. https://doi.org/10.1111/j.1476-5381.1996.tb15561.x
Shearman LP, Weaver DR (1997) [125I]4-aminobenzyl-5′-N-methylcarboxamidoadenosine (125I)AB-MECA) labels multiple adenosine receptor subtypes in rat brain. Brain Res 745(1–2):10–20. https://doi.org/10.1016/s0006-8993(96)01120-1
Ji XD, Gallo-Rodriguez C, Jacobson KA (1994) A selective agonist affinity label for A3 adenosine receptors. Biochem Biophys Res Commun 203(1):570–576. https://doi.org/10.1006/bbrc.1994.2220
Rivkees SA (1994) Localization and characterization of adenosine receptor expression in rat testis. Endocrinology 135(6):2307–2313. https://doi.org/10.1210/endo.135.6.7988413
Brandon CI, Vandenplas M, Dookwah H, Murray TF (2006) Cloning and pharmacological characterization of the equine adenosine A3 receptor. J Vet Pharmacol Ther 29(4):255–263. https://doi.org/10.1111/j.1365-2885.2006.00748.x
Burnett LA, Blais EM, Unadkat JD, Hille B, Tilley SL, Babcock DF (2010) Testicular expression of Adora3i2 in Adora3 knockout mice reveals a role of mouse A3Ri2 and human A3Ri3 adenosine receptors in sperm. J Biol Chem 285(44):33662–33670. https://doi.org/10.1074/jbc.M110.156075
Yamano K, Mori K, Nakano R, Kusunoki M, Inoue M, Satoh M (2007) Identification of the functional expression of adenosine A3 receptor in pancreas using transgenic mice expressing jellyfish apoaequorin. Transgenic Res 16(4):429–435. https://doi.org/10.1007/s11248-007-9084-0
Durand IH, Green RD (2001) Cloning of a Chick A3 adenosine receptor: characterization of ligand binding and receptor-effector coupling of chick A1 and A3 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol 363(1):81–86
Haeusler D, Grassinger L, Fuchshuber F, Hörleinsberger WJ, Höftberger R, Leisser I, Girschele F, Shanab K, Spreitzer H, Gerdenitsch W, Hacker M, Wadsak W, Mitterhauser Markus (2015) Hide and Seek: A comparative autoradiographic in vitro investigation of the adenosine A3 receptor. Eur J Nucl Med Mol Imaging 42(6):928–39. https://doi.org/10.1007/s00259-014-2985-2
Kiesewetter DO, Lang L, Ma Y, Bhattacharjee AK, Gao ZG, Joshi BV, Melman A, de Castro S, Jacobson KA (2009) Synthesis and characterization of [76Br]-labeled high-affinity A3 adenosine receptor ligands for positron emission tomography. Nucl Med Biol 36(1):3–10. https://doi.org/10.1016/j.nucmedbio.2008.10.003
Wadsak W, Mien LK, Shanab K, Ettlinger DE, Haeusler D, Sindelar K, Lanzenberger RR, Spreitzer H, Viernstein H, Keppler BK, Dudczak R, Kletter K, Mitterhauser M (2008) Preparation and first evaluation of [18F]FE@SUPPY: a new PET tracer for the adenosine A3 receptor. Nucl Med Biol 35(1):61–66. https://doi.org/10.1016/j.nucmedbio.2007.09.004
Linden J, Taylor HE, Robeva AS, Tucker AL, Stehle H, Rivkees SA, Fink JS, Reppert SM (1993) molecular cloning and functional expression of a sheep A3 adenosine receptor with widespread tissue distribution. Mol Pharmacol 44(3):524–532
Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG (1993) Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci USA 90(21):10365–10369. https://doi.org/10.1073/pnas.90.21.10365
Sajjadi FG, Firestein GS (1993) cDNA Cloning and sequence analysis of the human A3 adenosine receptor. Biochim Biophys Acta 1179(1):105–107. https://doi.org/10.1016/0167-4889(93)90077-3
Murrison EM, Goodson SJ, Edbrooke MR, Harris CA (1996) Cloning and characterisation of the human adenosine A3 receptor gene. FEBS Lett 384(3):243–246. https://doi.org/10.1016/0014-5793(96)00324-9
Atkinson MR, Townsend-Nicholson A, Nicholl JK, Sutherland GR, Schofield PR (1997) Cloning, characterisation and chromosomal assignment of the human adenosine A3 receptor (ADORA3) gene. Neurosci Res 29(1):73–79. https://doi.org/10.1016/s0168-0102(97)00073-4
Hill RJ, Oleynek JJ, Hoth CF, Kiron MA, Weng W, Wester RT, Tracey WR, Knight DR, Buchholz RA, Kennedy SP (1997) Cloning, expression and pharmacological characterization of rabbit adenosine A1 and A3 receptors. J Pharmacol Exp Ther 280(1):122–128
Ranjan A, Iyer SV, Iwakuma T (2017) Suppressive roles of A3AR and TMIGD3 i1 in osteosarcoma malignancy. Cell Cycle 16(10):903–904. https://doi.org/10.1080/15384101.2017.1308153
Jacobson KA, Merighi S, Varani K, Borea PA, Baraldi S, Aghazadeh Tabrizi M, Romagnoli R, Baraldi PG, Ciancetta A, Tosh DK, Gao ZG, Gessi S (2018) A3 adenosine receptors as modulators of inflammation: from medicinal chemistry to therapy. Med Res Rev 38(4):1031–1072. https://doi.org/10.1002/med.21456
Linden J (1994) Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions. Trends Pharmacol Sci 15(8):298–306. https://doi.org/10.1016/0165-6147(94)90011-6
Jacobson KA, Gao ZG (2006) Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5(3):247–264. https://doi.org/10.1038/nrd1983
Müller CE, Jacobson KA (2011) Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Biophys Acta 1808(5):1290–1308. https://doi.org/10.1016/j.bbamem.2010.12.017
Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL (1994) 125I-4-aminobenzyl-5’-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A d adenosine receptor. Mol Pharmacol 45(5):978–982
Kim HO, Hawes C, Towers P, Jacobson KA (1996) Radiolabeling and efficient synthesis of 2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide, a highly selective and potent A3 adenosine receptor agonist. J Labelled Comp Radiopharm 38(6):547–560. https://doi.org/10.1002/(SICI)1099-
Gao ZG, Verzijl D, Zweemer A, Ye K, Göblyös A, IJzerman AP, Jacobson KA. (2011) Functionally biased modulation of A3 adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem Pharmacol 82(6):658–68. https://doi.org/10.1016/j.bcp.2011.06.017
Gao ZG, Ye K, Göblyös A, IJzerman AP, Jacobson KA. (2008) Flexible modulation of agonist efficacy at the human A3 adenosine receptor by the imidazoquinoline allosteric enhancer LUF6000. BMC Pharmacol 8:20. https://doi.org/10.1186/1471-2210-8-20
Fossetta J, Jackson J, Deno G, Fan X, Du XK, Bober L, Soudé-Bermejo A, de Bouteiller O, Caux C, Lunn C, Lundell D, Palmer RK (2003) Pharmacological analysis of calcium responses mediated by the human A3 adenosine receptor in monocyte-derived dendritic cells and recombinant cells. Mol Pharmacol 63(2):342–350. https://doi.org/10.1124/mol.63.2.342
Borea PA, Varani K, Vincenzi F, Baraldi PG, Tabrizi MA, Merighi S, Gessi S (2015) The A3 adenosine receptor: history and perspectives. Pharmacol Rev 67(1):74–102. https://doi.org/10.1124/pr.113.008540
Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA (2008) Design of (N)-methanocarba adenosine 5′-uronamides as species-independent A3 receptor-selective agonists. Bioorg Med Chem Lett 18:2813–2819
Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA (2006) Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5’-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther 319(3):1200–1210. https://doi.org/10.1124/jpet.106.111351
Klotz KN, Falgner N, Kachler S, Lambertucci C, Vittori S, Volpini R, Cristalli G (2007) [3H]HEMADO–a novel tritiated agonist selective for the human adenosine A3 receptor. Eur J Pharmacol 556(1–3):14–18. https://doi.org/10.1016/j.ejphar.2006.10.048
Beukers MW, Chang LC, von Frijtag Drabbe Künzel JK, Mulder-Krieger T, Spanjersberg RF, Brussee J, IJzerman AP. (2004) New, non-adenosine, high-potency agonists for the human adenosine A2B receptor with an improved selectivity profile compared to the reference agonist N-ethylcarboxamidoadenosine. J Med Chem 47(15):3707–9. https://doi.org/10.1021/jm049947s
Guo M, Gao ZG, Tyler R, Stodden T, Li Y, Ramsey J, Zhao WJ, Wang GJ, Wiers CE, Fowler JS, Rice KC, Jacobson KA, Kim SW, Volkow ND (2018) Preclinical evaluation of the first adenosine A1 receptor partial agonist radioligand for positron emission tomography imaging. J Med Chem 61(22):9966–9975. https://doi.org/10.1021/acs.jmedchem.8b01009
Volpini R, Costanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G (2002) N(6)-alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3 receptor and a starting point for searching A2B ligands. J Med Chem 45(15):3271–3279. https://doi.org/10.1021/jm0109762
Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Klotz KN, Leung E, Varani K, Gessi S, Merighi S, Borea PA (1999) Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A3 adenosine receptor antagonists. J Med Chem 42:4473–4478. https://doi.org/10.1021/jm991114s
Ozola V, Thorand M, Diekmann M, Qurishi R, Schumacher B, Jacobson KA, Müller CE (2003) 2-Phenylimidazo[2,1–i]purin-5-ones: structure-activity relationships and characterization of potent and selective inverse agonists at human A3 adenosine receptors. Bioorg Med Chem 11(3):347–356. https://doi.org/10.1016/S0968-0896(02)00456-X
van Muijlwijk-Koezen JE, Timmerman H, van der Goot H, Menge WM, Von Drabbe F, Künzel J, de Groote M, IJzerman AP. Isoquinoline and quinazoline urea analogues as antagonists for the human adenosine A3 receptor. J Med Chem. 2000;43(11):2227–38. https://doi.org/10.1021/jm000002u
Göblyös A, Gao ZG, Brussee J, Connestari R, Santiago SN, Ye K, IJzerman AP, Jacobson KA. (2006) Structure-activity relationships of new 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric enhancers of the A3 adenosine receptor. J Med Chem 49(11):3354–61. https://doi.org/10.1021/jm060086s
Suresh RR, Gao ZG, Salmaso V, Chen E, Campbell RG, Poe RB, Liston TE, Jacobson KA (2022) Selective A3 adenosine receptor antagonist radioligand for human and rodent species. ACS Med Chem Lett 13(4):623–631. https://doi.org/10.1021/acsmedchemlett.1c00685
IJzerman AP, Jacobson KA, Müller CE, Cronstein BN, Cunha RA. (2022) International Union of Basic and Clinical Pharmacology CXII: Adenosine receptors: a further update. Pharmacol Rev 74(2):340–72. https://doi.org/10.1124/pharmrev.12.000445
Tosh DK, Salmaso V, Rao H, Campbell R, Bitant A, Gao ZG, Auchampach JA, Jacobson KA (2020) Direct comparison of (N)-methanocarba and ribose-containing 2-arylalkynyladenosine derivatives as A3 receptor agonists. ACS Med Chem Lett 11:1935–1941. https://doi.org/10.1021/acsmedchemlett.9b00637
Choi IY, Lee JC, Ju C, Hwang S, Cho GS, Lee HW, Choi WJ, Jeong LS, Kim WK. 2011 A3 adenosine receptor agonist reduces brain ischemic injury and inhibits inflammatory cell migration in rats. Am J Pathol.179(4):2042-52. https://doi.org/10.1016/j.ajpath.2011.07.006
Tosh DK, Salmaso V, Rao H, Bitant A, Fisher CL, Lieberman DI, Vorbrüggen H, Reitman ML, Gavrilova O, Gao ZG, Auchampach JA, Jacobson KA (2020) Truncated (N)-methanocarba nucleosides as partial agonists at mouse and human A3 adenosine receptors: Affinity enhancement by N6-(2-phenylethyl) substitution. J Med Chem 63(8):4334–4348. https://doi.org/10.1021/acs.jmedchem.0c00235
Gao ZG, Chen A, Barak D, Kim SK, Müller CE, Jacobson KA (2002) Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J Biol Chem 277:19056–19063
Fisher CL, Fallot LB, Wan TC, Keyes RF, Suresh RR, Rothwell AC, Gao ZG, McCorvy JD, Smith BC, Jacobson KA, Auchampach JA (2022) characterization of dual-acting A3 adenosine receptor positive allosteric modulators that preferentially enhance adenosine-induced Gαi3 and GαoA isoprotein activation. ACS Pharmacol Transl Sci 5(8):625–641. https://doi.org/10.1021/acsptsci.2c00076
Maldonado C, Qiu Y, Tang XL, Cohen MV, Auchampach J, Bolli R (1997) Role of adenosine receptors in late preconditioning against myocardial stunning in conscious rabbits. Am J Physiol 273:H1324–H1332. https://doi.org/10.1152/ajpheart.1997.273.3.H1324
Kreckler LM, Wan TC, Ge ZD, Auchampach JA (2006) Adenosine inhibits tumor necrosis factor-alpha release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptor. J Pharmacol Exp Ther 317(1):172–180. https://doi.org/10.1124/jpet.105.096016
Kim YC, Ji XD, Jacobson KA (1996) Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J Med Chem 39(21):4142–4148. https://doi.org/10.1021/jm960482i
Jiang JL, van Rhee AM, Melman N, Ji XD, Jacobson KA (1996) 6-Phenyl-1,4-dihydropyridine derivatives as potent and selective A3 adenosine receptor antagonists. J Med Chem 39(23):4667–4675. https://doi.org/10.1021/jm960457c
Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, Ji XD (1997) Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology 36:1157–1165. https://doi.org/10.1016/S0028-3908(97)00104-4
Jiang, J.-l, van Rhee, A.M., Chang, L, Patchornik, A, Evans, P, Melman, N, Jacobson, KA. (1997) Structure activity relationships of 4-phenylethynyl-6-phenyl-1,4-dihydropyridines as highly selective A3 adenosine receptor antagonists. J Med Chem 40:2596–608
Miwatashi S, Arikawa Y, Matsumoto T, Uga K, Kanzaki N, Imai YN, Ohkawa S (2008) Synthesis and biological activities of 4-phenyl-5-pyridyl-1,3-thiazole derivatives as selective adenosine A3 antagonists. Chem Pharm Bull (Tokyo) 56(8):1126–1137. https://doi.org/10.1248/cpb.56.1126
Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA (2002) Structural determinants of A3 adenosine receptor activation: nucleoside ligands at the agonist/antagonist boundary. J Med Chem 45(20):4471–4484. https://doi.org/10.1021/jm020211+
Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM (2005) The cross-species A3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Curr Eye Res 30(9):747–754. https://doi.org/10.1080/02713680590953147
Gao ZG, Jacobson KA (2008) Translocation of arrestin induced by human A3 adenosine receptor ligands in an engineered cell line: comparison with G protein-dependent pathways. Pharmacol Res 57(4):303–311. https://doi.org/10.1016/j.phrs.2008.02.008
Gao ZG, Van Muijlwijk-Koezen JE, Chen A, Müller CE, IJzerman AP, Jacobson KA. Allosteric modulation of A3 adenosine receptors by a series of 3-(2-pyridinyl)isoquinoline derivatives. Mol Pharmacol. 2001;60(5):1057-63
Gao ZG, Melman N, Erdmann A, Kim SG, Müller CE, IJzerman AP, Jacobson KA. (2003) Differential allosteric modulation by amiloride analogues of agonist and antagonist binding at A1 and A3 adenosine receptors. Biochem Pharmacol 65(4):525–34. https://doi.org/10.1016/s0006-2952(02)01556-3
Gao ZG, Gross AS, Jacobson KA (2004) Effects of the allosteric modulator SCH-202676 on adenosine and P2Y receptors. Life Sci 74(25):3173–3180. https://doi.org/10.1016/j.lfs.2003.11.014
May LT, Briddon SJ, Hill SJ (2010) Antagonist selective modulation of adenosine A1 and A3 receptor pharmacology by the food dye Brilliant Black BN: evidence for allosteric interactions. Mol Pharmacol 77(4):678–686. https://doi.org/10.1124/mol.109.063065
Heitman LH, Göblyös A, Zweemer AM, Bakker R, Mulder-Krieger T, van Veldhoven JP, de Vries H, Brussee J, IJzerman AP. (2009) A series of 2,4-disubstituted quinolines as a new class of allosteric enhancers of the adenosine A3 receptor. J Med Chem 52(4):926–31. https://doi.org/10.1021/jm8014052
Du L, Gao ZG, Nithipatikom K, IJzerman AP, Veldhoven JP, Jacobson KA, Gross GJ, Auchampach JA. Protection from myocardial ischemia/reperfusion injury by a positive allosteric modulator of the A3 adenosine receptor. J Pharmacol Exp Ther. 2012;340(1):210-7. https://doi.org/10.1124/jpet.111.187559.
Deganutti G, Cuzzolin A, Ciancetta A, Moro S (2015) Understanding allosteric interactions in G protein-coupled receptors using supervised molecular dynamics: a prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000. Bioorg Med Chem 23:4065–4071
Fallot LB, Suresh RR, Fisher CL, Kaufman N, Gao ZG, Auchampach JA, Jacobson KA (2022) Structure activity studies of 1H-imidazo[4,5-c]quinolin-4-amine derivatives as A3 adenosine receptor positive allosteric modulators. J Med Chem. https://doi.org/10.1021/acs.jmedchem.2c01170
Wise A, Sheehan M, Rees S, Lee M, Milligan G (1999) Comparative analysis of the efficacy of A1 adenosine receptor activation of Gi/o alpha G proteins following coexpression of receptor and G protein and expression of A1 adenosine receptor-Gi/o alpha fusion proteins. Biochemistry 38(8):2272–2278. https://doi.org/10.1021/bi982054f
Wall MJ, Hill E, Huckstepp R, Barkan K, Deganutti G, Leuenberger M, Preti B, Winfield I, Carvalho S, Suchankova A, Wei H, Safitri D, Huang X, Imlach W, La Mache C, Dean E, Hume C, Hayward S, Oliver J, Zhao FY, Spanswick D, Reynolds CA, Lochner M, Ladds G, Frenguelli BG (2022) Selective activation of Gαob by an adenosine A1 receptor agonist elicits analgesia without cardiorespiratory depression. Nat Commun 13:4150. https://doi.org/10.1038/s41467-022-31652-2
Yang SJ, An JY, Shim JO, Park CH, Huh IH, Sohn UD (2000) The mechanism of contraction by 2-chloroadenosine in cat detrusor muscle cells. J Urol 163(2):652–658
Murthy KS, Makhlouf GM (1995) Adenosine A1 receptor-mediated activation of phospholipase C-beta 3 in intestinal muscle: dual requirement for alpha and beta gamma subunits of Gi3. Mol Pharmacol 47(6):1172–1179
Tateyama M, Kubo Y (2016) Stabilizing effects of G protein on the active conformation of adenosine A1 receptor differ depending on G protein type. Eur J Pharmacol 788:122–131. https://doi.org/10.1016/j.ejphar.2016.06.025
Palmer TM, Benovic JL, Stiles GL (1995) Agonist-dependent phosphorylation and desensitization of the rat A3 adenosine receptor Evidence for a G-protein-coupled receptor kinase-mediated mechanism. J Biol Chem 270(49):29607–13. https://doi.org/10.1074/jbc.270.49.29607
Baram D, Dekel O, Mekori YA, Sagi-Eisenberg R (2010) Activation of mast cells by trimeric G protein Gi3; Coupling to the A3 adenosine receptor directly and upon t cell contact. J Immunol 184(7):3677–3688. https://doi.org/10.4049/jimmunol.0901333
Yamano K, Inoue M, Masaki S, Saki M, Ichimura M, Satoh M (2005) Human adenosine A3 receptor activation leads to intracellular Ca2+ mobilization but is insufficient to activate the signaling pathway via phosphoinositide 3-kinase gamma in mice. Biochem Pharmacol 70(10):1487–1496. https://doi.org/10.1016/j.bcp.2005.08.003
Klaasse EC, IJzerman AP, de Grip WJ, Beukers MW. (2008) Internalization and desensitization of adenosine receptors. Purinergic Signal 4(1):21–37. https://doi.org/10.1007/s11302-007-9086-7
Olah ME, Stiles GL (2000) The role of receptor structure in determining adenosine receptor activity. Pharmacol Ther 85(2):55–75. https://doi.org/10.1016/s0163-7258(99)00051-0
Trincavelli ML, Tuscano D, Marroni M, Falleni A, Gremigni V, Ceruti S, Abbracchio MP, Jacobson KA, Cattabeni F, Martini C (2002) A3 adenosine receptors in human astrocytoma cells: agonist-mediated desensitization, internalization, and down-regulation. Mol Pharmacol 62(6):1373–1384. https://doi.org/10.1124/mol.62.6.1373
Palmer TM, Benovic JL, Stiles GL (1996) Molecular basis for subtype-specific desensitization of inhibitory adenosine receptors Analysis of a Chimeric A1–A3 Adenosine Receptor. J Biol Chem. 271(25):15272–8. https://doi.org/10.1074/jbc.271.25.15272
Ramkumar V, Olah ME, Jacobson KA, Stiles GL (1991) Distinct pathways of desensitization of A1- and A2-adenosine receptors in DDT1 MF-2 cells. Mol Pharmacol 40(5):639–647
Liang BT, Donovan IA (1990) Differential desensitization of A1 adenosine receptor-mediated inhibition of cardiac myocyte contractility and adenylate cyclase activity relation to the regulation of receptor affinity and density. Circ Res. 67(2):406–14. https://doi.org/10.1161/01.res.67.2.406
Tsutsui S, Vergote D, Shariat N, Warren K, Ferguson SSG, Power C (2008) Glucocorticoids regulate innate immunity in a model of multiple sclerosis: reciprocal interactions between the A1 adenosine receptor and beta-arrestin-1 in monocytoid cells. FASEB J 22(3):786–796. https://doi.org/10.1096/fj.07-9002com
Jajoo S, Mukherjea D, Kumar S, Sheth S, Kaur T, Rybak LP, Ramkumar V (2010) Role of beta-arrestin1/ERK MAP kinase pathway in regulating adenosine A1 receptor desensitization and recovery. Am J Physiol Cell Physiol 298(1):C56-65. https://doi.org/10.1152/ajpcell.00190.2009
Ferguson G, Watterson KR, Palmer TM (2000) Subtype-specific kinetics of inhibitory adenosine receptor internalization are determined by sensitivity to phosphorylation by G protein-coupled receptor kinases. Mol Pharmacol 57(3):546–552
Ferguson G, Watterson KR, Palmer TM (2002) Subtype-specific regulation of receptor internalization and recycling by the carboxyl-terminal domains of the human A1 and rat A3 adenosine receptors: consequences for agonist-stimulated translocation of arrestin3. Biochemistry 41(50):14748–14761. https://doi.org/10.1021/bi0262911
Santini F, Penn RB, Gagnon AW, Benovic JL, Keen JH (2000) Selective recruitment of arrestin-3 to clathrin coated pits upon stimulation of G protein-coupled receptors. J Cell Sci 113(Pt 13):2463–2470
Stoddart LA, Kilpatrick LE, Corriden R, Kellam B, Briddon SJ, Hill SJ (2021) Efficient G protein coupling is not required for agonist-mediated internalization and membrane reorganization of the adenosine A3 receptor. FASEB J 35(4):e21211. https://doi.org/10.1096/fj.202001729RR
Stoddart LA, Kellam B, Briddon SJ, Hill SJ (2014) Effect of a toggle switch mutation in TM6 of the human adenosine A3 receptor on Gi protein-dependent signalling and Gi-independent receptor internalization. Br J Pharmacol 171(16):3827–3844. https://doi.org/10.1111/bph.12739
Stoddart LA, Vernall AJ, Briddon SJ, Kellam B, Hill SJ (2015) Direct visualisation of internalization of the adenosine A3 receptor and localization with arrestin3 using a fluorescent agonist. Neuropharmacology 98:68–77. https://doi.org/10.1016/j.neuropharm.2015.04.013
Storme J, Cannaert A, Van Craenenbroeck K, Stove CP (2018) Molecular dissection of the human A3 adenosine receptor coupling with β-arrestin2. Biochem Pharmacol 148:298–307. https://doi.org/10.1016/j.bcp.2018.01.008
Trincavelli ML, Tuscano D, Cecchetti P, Falleni A, Benzi L, Klotz KN, Gremigni V, Cattabeni F, Lucacchini A, Martini C (2000) Agonist-induced internalization and recycling of the human A3 adenosine receptors: role in receptor desensitization and resensitization. J Neurochem 75(4):1493–1501. https://doi.org/10.1046/j.1471-4159.2000.0751493.x
Holgate ST, Lewis RA, Austen KF (1980) Role of adenylate cyclase in immunologic release of mediators from rat mast cells: agonist and antagonist effects of purine- and ribose-modified adenosine analogs. Proc Natl Acad Sci U S A 77(11):6800–6804. https://doi.org/10.1073/pnas.77.11.6800
Cushley MJ, Tattersfield AE, Holgate ST (1983) Inhaled adenosine and guanosine on airway resistance in normal and asthmatic subjects. Br J Clin Pharmacol 15(2):161–165. https://doi.org/10.1111/j.1365-2125.1983.tb01481.x
Gao ZG, Jacobson KA (2017) Purinergic signaling in mast cell degranulation and asthma. Front Pharmacol 8:947. https://doi.org/10.3389/fphar.2017.00947
Tilley SL, Tsai M, Williams CM, Wang ZS, Erikson CJ, Galli SJ, Koller BH (2003) Identification of A3 receptor- and mast cell-dependent and -independent components of adenosine-mediated airway responsiveness in mice. J Immunol 171(1):331–337. https://doi.org/10.4049/jimmunol.171.1.331
Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG (2006) ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314(5806):1792–1795. https://doi.org/10.1126/science.1132559
Van Schaick EA, Jacobson KA, Kim HO, IJzerman AP, Danhof M. (1996) Hemodynamic effects and histamine release elicited by the selective adenosine A3 receptor agonist 2-Cl-IB-MECA in conscious rats. Eur J Pharmacol 308(3):311–4. https://doi.org/10.1016/0014-2999(96)00373-1
Reeves JJ, Jones CA, Sheehan MJ, Vardey CJ, Whelan CJ (1997) Adenosine A3 receptors promote degranulation of rat mast cells both in vitro and in vivo. Inflamm Res 46(5):180–184. https://doi.org/10.1007/s000110050169
Shepherd RK, Linden J, Duling BR. 1996Adenosine-induced vasoconstriction in vivo. Role of the mast cell and A3 adenosine receptor. Circ Res 78(4):627–34. https://doi.org/10.1161/01.res.78.4.627.
el-Hashim A, D'Agostino B, Matera MG, Page C. 1996 Characterization of adenosine receptors involved in adenosine-induced bronchoconstriction in allergic rabbits. Br J Pharmacol 119(6):1262–8. https://doi.org/10.1111/j.1476-5381.1996.tb16031.x.
Auchampach JA, Rizvi A, Qiu Y, Tang XL, Maldonado C, Teschner S, Bolli R (1997) Selective activation of A3 adenosine receptors with N6-(3-iodobenzyl)adenosine-5’-N-methyluronamide protects against myocardial stunning and infarction without hemodynamic changes in conscious rabbits. Circ Res 80(6):800–809. https://doi.org/10.1161/01.res.80.6.800
Auchampach JA, Ge ZD, Wan TC, Moore J, Gross GJ (2003) A3 adenosine receptor agonist IB-MECA reduces myocardial ischemia-reperfusion injury in dogs. Am J Physiol Heart Circ Physiol 285(2):H607–H613. https://doi.org/10.1152/ajpheart.01001.2002
van Troostenburg AR, Clark EV, Carey WD, Warrington SJ, Kerns WD, Cohn I, Silverman MH, Bar-Yehuda S, Fong KL, Fishman P (2004) Tolerability, pharmacokinetics and concentration-dependent hemodynamic effects of oral CF101, an A3 adenosine receptor agonist, in healthy young men. Int J Clin Pharmacol Ther 42(10):534–542. https://doi.org/10.5414/cpp42534
Takano H, Bolli R, Black RG Jr, Kodani E, Tang XL, Yang Z, Bhattacharya S, Auchampach JA (2001) A1 or A3 adenosine receptors induce late preconditioning against infarction in conscious rabbits by different mechanisms. Circ Res 88(5):520–528. https://doi.org/10.1161/01.res.88.5.520
Kodani E, Shinmura K, Xuan YT, Takano H, Auchampach JA, Tang XL, Bolli R (2001) Cyclooxygenase-2 does not mediate late preconditioning induced by activation of adenosine A1 or A3 receptors. Am J Physiol Heart Circ Physiol 281(2):H959–H968. https://doi.org/10.1152/ajpheart.2001.281.2.H959
Kodani E, Bolli R, Tang XL, Auchampach JA (2001) Protection of IB-MECA against myocardial stunning in conscious rabbits is not mediated by the A1 adenosine receptor. Basic Res Cardiol 96(5):487–496. https://doi.org/10.1007/s003950170031
Ge ZD, van der Hoeven D, Maas JE, Wan TC, Auchampach JA (2010) A3 adenosine receptor activation during reperfusion reduces infarct size through actions on bone marrow-derived cells. J Mol Cell Cardiol 49(2):280–286. https://doi.org/10.1016/j.yjmcc.2010.01.018
von Lubitz DK, Lin RC, Popik P, Carter MF, Jacobson KA (1994) Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol 263(1–2):59–67. https://doi.org/10.1016/0014-2999(94)90523-1
Fedorova IM, Jacobson MA, Basile A, Jacobson KA (2003) Behavioral characterization of mice lacking the A3 adenosine receptor: sensitivity to hypoxic neurodegeneration. Cell Mol Neurobiol 23(3):431–447. https://doi.org/10.1023/a:1023601007518
Nakashima KI, Iwao K, Inoue T, Haga A, Tsutsumi T, Mochita MI, Fujimoto T, Tanihara H (2018) Stimulation of the adenosine A3 receptor, not the A1 or A2 receptors, promote neurite outgrowth of retinal ganglion cells. Exp Eye Res 170:160–168. https://doi.org/10.1016/j.exer.2018.02.019
Luo C, Yi B, Tao G, Li M, Chen Z, Tang W, Zhang JH, Feng H (2010) Adenosine A3 receptor agonist reduces early brain injury in subarachnoid haemorrhage. NeuroReport 21(13):892–896. https://doi.org/10.1097/WNR.0b013e32833dbd13
Sei Y, von Lubitz DKJE, Abbracchio MP, Ji Xd, Jacobson KA (1997) Adenosine A3 receptor agonist-induced neurotoxicity in rat cerebellar granule neurons. Drug Devel Res 40:267–273
Farr SA, Cuzzocrea S, Esposito E, Campolo M, Niehoff ML, Doyle TM, Salvemini D (2020) Adenosine A3 receptor as a novel therapeutic target to reduce secondary events and improve neurocognitive functions following traumatic brain injury. J Neuroinflammation 17(1):339. https://doi.org/10.1186/s12974-020-02009-7
Bozdemir E, Vigil FA, Chun SH, Espinoza L, Bugay V, Khoury SM, Holstein DM, Stoja A, Lozano D, Tunca C, Sprague SM, Cavazos JE, Brenner R, Liston TE, Shapiro MS, Lechleiter JD (2021) Neuroprotective roles of the adenosine A3 receptor agonist AST-004 in mouse model of traumatic brain injury. Neurotherapeutics 18:2707–2721. https://doi.org/10.1007/s13311-021-01113-7
Liston TE, Hama A, Boltze J, Poe RB, Natsume T, Hayashi I, et al. Adenosine A1R/A3R (adenosine A1 and A3 receptor) agonist AST-004 reduces brain infarction in a nonhuman primate model of stroke. Stroke. 2022;53:238–48. https://www.ahajournals.orghttps://doi.org/10.1161/STROKEAHA.121.036396
Whitehead G, Karcz T, Tosh D, Jung YH, Wen Z, Campbell R, Varun G, Gao ZG, Jacobson KA, Cook D (2022) Effects of purinergic receptor deletion or pharmacologic modulation on pulmonary inflammation in mice. ACS Pharmacology & Translational Science 5:973–984
Cohen S, Fishman P (2019) Targeting the A3 adenosine receptor to treat cytokine release syndrome in cancer immunotherapy. Drug Des Devel Ther 13:491–497. https://doi.org/10.2147/DDDT.S195294
Jeffe F, Stegmann KA, Broelsch F, Manns MP, Cornberg M, Wedemeyer HJ (2009) Adenosine and IFN-{alpha} synergistically increase IFN-gamma production of human NK cells. Leukoc Biol 85(3):452–61. https://doi.org/10.1189/jlb.0108046
Bar-Yehuda S, Luger D, Ochaion A, Cohen S, Patokaa R, Zozulya G, Silver PB, de Morales JM, Caspi RR, Fishman P (2011) Inhibition of experimental auto-immune uveitis by the A3 adenosine receptor agonist CF101. Int J Mol Med 28(5):727–731. https://doi.org/10.3892/ijmm.2011.753
Hein TW, Belardinelli L, Kuo L (1999) Adenosine A2A receptors mediate coronary microvascular dilation to adenosine: role of nitric oxide and ATP-sensitive potassium channels. J Pharmacol Exp Ther 291(2):655–664
Haskó G, Linden J, Cronstein B, Pacher P (2008) Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7(9):759–770. https://doi.org/10.1038/nrd2638
Boros D, Thompson J, Larson DF (2016) Adenosine regulation of the immune response initiated by ischemia reperfusion injury. Perfusion 31(2):103–110. https://doi.org/10.1177/0267659115586579
Cekic C, Linden J (2016) Purinergic regulation of the immune system. Nat Rev Immunol 16(3):177–192. https://doi.org/10.1038/nri.2016.4
Gao Z, Li BS, Day YJ, Linden J (2001) A3 adenosine receptor activation triggers phosphorylation of protein kinase B and protects rat basophilic leukemia 2H3 mast cells from apoptosis. Mol Pharmacol 59(1):76–82. https://doi.org/10.1124/mol.59.1.76
Kim TH, Kim YK, Woo JS (2012) The adenosine A3 receptor agonist Cl-IB-MECA induces cell death through Ca2+/ROS-dependent down regulation of ERK and Akt in A172 human glioma cells. Neurochem Res 37(12):2667–2677. https://doi.org/10.1007/s11064-012-0855-5
Maugeri G, D’Amico AG, Federico C, Saccone S, Giunta S, Cavallaro S, D’Agata V (2019) Involvement of A3 adenosine receptor in neuroblastoma progression via modulation of the hypoxic/angiogenic pathway. J Mol Neurosci 69(1):166–176. https://doi.org/10.1007/s12031-019-01346-4
Ledderose C, Hefti MM, Chen Y, Bao Y, Seier T, Li L, Woehrle T, Zhang J, Junger WG (2016) Adenosine arrests breast cancer cell motility by A3 receptor stimulation. Purinergic Signal 12(4):673–685. https://doi.org/10.1007/s11302-016-9531-6
Ohana G, Bar-Yehuda S, Arich A, Madi L, Dreznick Z, Rath-Wolfson L, Silberman D, Slosman G, Fishman P (2003) Inhibition of primary colon carcinoma growth and liver metastasis by the A3 adenosine receptor agonist CF101. Br J Cancer 89(8):1552–1558. https://doi.org/10.1038/sj.bjc.6601315
Shneyvais V, Jacobson KA, Li A-H, Nawrath H, Zinman T, Isaac A, Shainberg A (2000) Induction of apoptosis in rat cardiac myocytes by A3 adenosine receptor activation and its suppression by isoproterenol. Experimental Cell Res 257:111–126
Kim SG, Ravi RG, Hoffmann CA, Jung YJ, Kim M, Chen A, Jacobson KA (2002) p53-Independent induction of Fas and apoptosis in leukemic cells by an adenosine derivative. Cl-IB-MECA Biochem Pharmacol 63:871–880
Yao Y, Sei Y, Abbracchio MP, Kim YC, Jacobson KA (1997) Adenosine A3 receptor agonists protect HL-60 and U-937 cells from apoptosis induced by A3 antagonists. Biochem Biophys Res Comm 232:317–322
Lu J, Pierron A, Ravid K (2003) An adenosine analogue, IB-MECA, down-regulates estrogen receptor alpha and suppresses human breast cancer cell proliferation. Cancer Res 63(19):6413–6423
Sawynok J, Zarrindast MR, Reid AR, Doak GJ (1997) Adenosine A3 receptor activation produces nociceptive behaviour and edema by release of histamine and 5-hydroxytryptamine. Eur J Pharmacol 333(1):1–7. https://doi.org/10.1016/s0014-2999(97)01110-2
Wu WP, Hao JX, Halldner-Henriksson L, Xu XJ, Jacobson MA, Wiesenfeld-Hallin Z, Fredholm BB (2002) Decreased inflammatory pain due to reduced carrageenan-induced inflammation in mice lacking adenosine A3 receptors. Neuroscience 114(3):523–527. https://doi.org/10.1016/s0306-4522(02)00273-7
Yoon MH, Bae HB, Choi JI, Kim SJ, Chung ST, Kim CM (2006) Roles of adenosine receptor subtypes in the antinociceptive effect of intrathecal adenosine in a rat formalin test. Pharmacology 78(1):21–26. https://doi.org/10.1159/000094762
Chen Z, Janes K, Chen C, Doyle T, Bryant L, Tosh DK, Jacobson KA, Salvemini D (2012) Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J 26(5):1855–1865. https://doi.org/10.1096/fj.11-201541
Janes K, Wahlman C, Little JW, Doyle T, Tosh DK, Jacobson KA, Salvemini D (2015) Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain Behav Immun 44:91–99. https://doi.org/10.1016/j.bbi.2014.08.010
Coppi E, Cherchi F, Fusco I, Failli P, Vona A, Dettori I, Gaviano L, Lucarini E, Jacobson KA, Tosh DK, Salvemini D, Ghelardini C, Pedata F, Di Cesare ML, Pugliese AM (2019) Adenosine A3 receptor activation inhibits pronociceptive N-type Ca2+ currents and cell excitability in dorsal root ganglion neurons. Pain 160(5):1103–1118. https://doi.org/10.1097/j.pain.0000000000001488
Jacobson KA, Giancotti LA, Lauro F, Mufti F, Salvemini D (2020) Treatment of chronic neuropathic pain: purine receptor modulation. Pain 161(7):1425–1441. https://doi.org/10.1097/j.pain.0000000000001857
Durante M, Squillace S, Lauro F, Giancotti LA, Coppi E, Cherchi F, Di Cesare ML, Ghelardini C, Kolar G, Wahlman C, Ope** A, **ao C, Reitman ML, Tosh DK, Hawiger D, Jacobson KA, Salvemini D (2021) Adenosine A3 agonists reverse neuropathic pain via T cell-mediated production of IL-10. J Clin Invest 131(7):e139299. https://doi.org/10.1172/JCI139299
Coppi E, Cherchi F, Lucarini E, Ghelardini C, Pedata F, Jacobson KA, Di Cesare ML, Pugliese AM, Salvemini D (2021) Uncovering the mechanisms of adenosine receptor-mediated pain control: Focus on the A3 receptor subtype. Int J Mol Sci 22(15):7952. https://doi.org/10.3390/ijms22157952
Terayama R, Tabata M, Maruhama K, Iida S (2018) A3 adenosine receptor agonist attenuates neuropathic pain by suppressing activation of microglia and convergence of nociceptive inputs in the spinal dorsal horn. Exp Brain Res 236(12):3203–3213. https://doi.org/10.1007/s00221-018-5377-1
Alles SRA, Smith PA (2018) Etiology and pharmacology of neuropathic pain. Pharmacol Rev 70(2):315–347. https://doi.org/10.1124/pr.117.014399
Mori T, Kuzumaki N, Arima T, Narita M, Tateishi R, Kondo T, Hamada Y, Kuwata H, Kawata M, Yamazaki M, Sugita K, Matsuzawa A, Baba K, Yamauchi T, Higashiyama K, Nonaka M, Miyano K, Uezono Y, Narita M (2017) Usefulness for the combination of G-protein- and β-arrestin-biased ligands of μ-opioid receptors: prevention of antinociceptive tolerance. Mol Pain 13:1744806917740030. https://doi.org/10.1177/1744806917740030
Kvanta A, Seregard S, Sejersen S, Kull B, Fredholm BB (1997) Localization of adenosine receptor messenger RNAs in the rat eye. Exp Eye Res 65(5):595–602. https://doi.org/10.1006/exer.1996.0352
Mitchell CH, Peterson-Yantorno K, Carré DA, McGlinn AM, Coca-Prados M, Stone RA, Civan MM (1999) A3 adenosine receptors regulate Cl- channels of nonpigmented ciliary epithelial cells. Am J Physiol 276(3):C659–C666. https://doi.org/10.1152/ajpcell.1999.276.3.C659
Avila MY, Stone RA, Civan MM (2002) Knockout of A3 adenosine receptors reduces mouse intraocular pressure. Invest Ophthalmol Vis Sci 43(9):3021–3026
Okamura T, Kurogi Y, Hashimoto K, Sato S, Nishikawa H, Kiryu K, Nagao Y (2004) Structure-activity relationships of adenosine A3 receptor ligands: new potential therapy for the treatment of glaucoma. Bioorg Med Chem Lett 14(14):3775–3779. https://doi.org/10.1016/j.bmcl.2004.04.099
Schlötzer-Schrehardt U, Zenkel M, Decking U, Haubs D, Kruse FE, Jünemann A, Coca-Prados M, Naumann GO (2005) Selective upregulation of the A3 adenosine receptor in eyes with pseudoexfoliation syndrome and glaucoma. Invest Ophthalmol Vis Sci 46(6):2023–2034. https://doi.org/10.1167/iovs.04-0915
Zhang M, Hu H, Zhang X, Lu W, Lim J, Eysteinsson T, Jacobson KA, Laties AM, Mitchell CH (2010) The A3 adenosine receptor attenuates the calcium rise triggered by NMDA receptors in retinal ganglion cells. Neurochem Int 56(1):35–41. https://doi.org/10.1016/j.neuint.2009.08.011
Fishman P, Cohen S, Bar-Yehuda S (2013) Targeting the A3 adenosine receptor for glaucoma treatment (review). Mol Med Rep 7(6):1723–1725. https://doi.org/10.3892/mmr.2013.1413
Park CW, Han CT, Sakaguchi Y, Lee J, Youn HY. 2020 Safety evaluation of FM101, an A3 adenosine receptor modulator, in rat, for develo** as therapeutics of glaucoma and hepatitis. EXCLI J 19:187–200. https://doi.org/10.17179/excli2019-2058.
Jacobson KA, Civan MM (2016) Ocular purine receptors as drug targets in the eye. J Ocul Pharmacol Ther 32(8):534–547. https://doi.org/10.1089/jop.2016.0090
Amisten S, Atanes P, Hawkes R, Ruz-Maldonado I, Liu B, Parandeh F, Zhao M, Huang GC, Salehi A, Persaud SJ (2017) A comparative analysis of human and mouse islet G-protein coupled receptor expression. Sci Rep 7:46600. https://doi.org/10.1038/srep46600
Laudadio MA, Psarropoulou C (2004) The A3 adenosine receptor agonist 2-Cl-IB-MECA facilitates epileptiform discharges in the CA3 area of immature rat hippocampal slices. Epilepsy Res 59(2–3):83–94. https://doi.org/10.1016/j.eplepsyres.2004.03.005
Yoshikawa N, Yamada S, Takeuchi C, Kagota S, Shinozuka K, Kunitomo M, Nakamura K (2008) Cordycepin (3’-deoxyadenosine) inhibits the growth of B16-BL6 mouse melanoma cells through the stimulation of adenosine A3 receptor followed by glycogen synthase kinase-3beta activation and cyclin D1 suppression. Naunyn Schmiedebergs Arch Pharmacol 377(4–6):591–595. https://doi.org/10.1007/s00210-007-0218-y
Bar-Yehuda S, Rath-Wolfson L, Del Valle L, Ochaion A, Cohen S, Patoka R, Zozulya G, Barer F, Atar E, Pina-Oviedo S, Perez-Liz G, Castel D, Fishman P (2009) Induction of an anti-inflammatory effect and prevention of cartilage damage in rat knee osteoarthritis by CF101 treatment. Arthritis Rheum 60(10):3061–3071. https://doi.org/10.1002/art.24817
Jacobson KA, Reitman ML (2020) Adenosine-related mechanisms in non-adenosine receptor drugs. Cells 9(4):956. https://doi.org/10.3390/cells9040956
Lasley RD, Narayan P, Jahania MS, Partin EL, Kraft KR, Mentzer RM Jr (1999) Species-dependent hemodynamic effects of adenosine A3-receptor agonists IB-MECA and Cl-IB-MECA. Am J Physiol 276(6):H2076–H2084. https://doi.org/10.1152/ajpheart.1999.276.6.H2076
Sun WC, Moore JN, Hurley DJ, Vandenplas ML, Linden J, Cao Z, Murray TF (2008) Adenosine A2A receptor agonists inhibit lipopolysaccharide-induced production of tumor necrosis factor-alpha by equine monocytes. Vet Immunol Immunopathol 121(1–2):91–100. https://doi.org/10.1016/j.vetimm.2007.08.011
Wittendorp MC, von Frijtag Drabbe Künzel J, IJzerman AP, Boddeke HW, Biber K. (2004) The mouse brain adenosine A1 receptor: functional expression and pharmacology. Eur J Pharmacol 487(1–3):73–9. https://doi.org/10.1016/j.ejphar.2004.01.034
Hill RJ, Oleynek JJ (1997) C F Hoth, M A Kiron, W Weng, R T Wester, W R Tracey, D R Knight, R A Buchholz, S P Kennedy Cloning, expression and pharmacological characterization of rabbit adenosine A1 and A3 receptors. J Pharmacol Exp Ther. 280(1):122–8
DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, Tickner JE, Kennedy SP, Knight DR, Kong J, Oleynek JJ, Tracey WR, Hill RJ (2003) 3′-Aminoadenosine-5′-uronamides: discovery of the first highly selective agonist at the human adenosine A3 receptor. J Med Chem 46:353–355
Tian Y, Marshall M, French BA, Linden J, Yang Z (2015) The infarct-sparing effect of IB-MECA against myocardial ischemia/reperfusion injury in mice is mediated by sequential activation of adenosine A3 and A2A receptors. Basic Res Cardiol 110(2):16. https://doi.org/10.1007/s00395-015-0473-x
Funding
KAJ received support from the NIDDK Intramural Research Program (ZIADK031117) and JAA received funding from NHLBI (R01 grant HL133589).
Author information
Authors and Affiliations
Contributions
ZGG wrote the first draft. ZGG, JAA, and KAJ contributed significantly to the writing and editing. KAJ prepared the figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflicts of interest
Zhan-Guo Gao declares that there is no conflict of interest. John A. Auchampach declares that there is no conflict of interest. Kenneth A. Jacobson declares that there is no conflict of interest.
Ethical approval
Not applicable.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gao, ZG., Auchampach, J.A. & Jacobson, K.A. Species dependence of A3 adenosine receptor pharmacology and function. Purinergic Signalling 19, 523–550 (2023). https://doi.org/10.1007/s11302-022-09910-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-022-09910-1