
Abstract
A growing amount of epidemiological data from multiple countries indicate an increased prevalence of obesity, more importantly central obesity, among hospitalized subjects with COVID-19. This suggests that obesity is a major factor contributing to adverse outcome of the disease. As it is a metabolic disorder with dysregulated immune and endocrine function, it is logical that dysfunctional metabolism contributes to the mechanisms behind obesity being a risk factor for adverse outcome in COVID-19. Emerging data suggest that in obese subjects, (a) the molecular mechanisms of viral entry and spread mediated through ACE2 receptor, a multifunctional host cell protein which links to cellular homeostasis mechanisms, are affected. This includes perturbation of the physiological renin-angiotensin system pathway causing pro-inflammatory and pro-thrombotic challenges (b) existent metabolic overload and ER stress-induced UPR pathway make obese subjects vulnerable to severe COVID-19, (c) host cell response is altered involving reprogramming of metabolism and epigenetic mechanisms involving microRNAs in line with changes in obesity, and (d) adiposopathy with altered endocrine, adipokine, and cytokine profile contributes to altered immune cell metabolism, systemic inflammation, and vascular endothelial dysfunction, exacerbating COVID-19 pathology. In this review, we have examined the available literature on the underlying mechanisms contributing to obesity being a risk for adverse outcome in COVID-19.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There has been a substantial rise in the prevalence of obesity and associated metabolic disorders worldwide during recent years. The World Health Organization has reported over a billion overweight adults worldwide, and over 300 million of these individuals are clinically obese [1]. Being a complex metabolic disorder, obesity is linked to development of several human diseases including cardiovascular diseases (CVD), Type 2 Diabetes (T2D), hypertension, stroke, hepatic steatosis, endometriosis, and certain type of cancers. It is becoming increasingly evident that obesity is also associated with infectious diseases though mechanisms underlying their possible association are not well understood. While the association between obesity and the risk for contracting hospital-borne nosocomial and surgery-site infection is well known, adequate data in support of susceptibility to community-based infections are lacking. In many of the bacterial or viral infections, data on the link between obesity and susceptibility to infection and the outcome of the disease is either limited or controversial [2]. An exception to this is perhaps the H1N1 influenza pandemic in 2009, when a series of epidemiological studies showed that obesity was independently associated with risk for severe disease [3]. Although several studies revealed that obesity was linked with poor outcome in H1N1 infection in terms of hospitalization, Intensive Care Unit (ICU) admission, and death, the relevant pathophysiological mechanisms are not well understood. Some of the obesity-related factors that can affect the course and outcome of the disease may include obesity-related immune dysregulation and impaired immune response, respiratory dysfunction, comorbidities such as diabetes, hypertension, and vascular endothelial dysfunction, and disrupted micro and macrovascular circulation. Unlike bacterial or other microbial infections, viral infection involves taking over of host cell machinery for their multiplication and evasion of host cell immune surveillance, thus, causing havoc to host cell function and leading to disease.
COVID-19 is a viral infectious disease of zoonotic origin, caused by severe acute respiratory syndrome-coronavirus-2 (SARS-CoV2). Coronaviruses (CoVs) are enveloped, positive-sense, single-stranded RNA viruses. They belong to the family Coronaviridae within the order Nidoviridae and sub-order Coronavirinae. The family is further divided into the sub-family Orthocoronavirinae made up of four genera—alpha, beta, gamma, and delta coronavirus. There are seven Human corona viruses (HCoVs) that cause illness in humans and the first of these were discovered in 1966 and 1967. Four of the HCoVs—HCoV-229E, HCoV-OC43, HCoV-NL63, and HKU1—cause mild seasonal respiratory infections. The other three, Severe acute respiratory syndrome coronavirus (SARS-CoV), the Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV2, which are beta-coronaviruses, can infect bronchial epithelial cells, pneumocytes, and cells of the upper respiratory tract. They cause more serious infections which can progress into severe life-threatening disease [4]. SARS-CoV was responsible for an outbreak in Guangdong in 2003 in which 800 out of around 8,000 people died. MERS-CoV caused an outbreak, mainly in Middle Eastern countries in 2012 that affected more than 2500 people killing close to 900 [5].
SARS-CoV2 was reported for the first time in Wuhan, China in December 2019 [6] and has since developed into a pandemic affecting over 200 nations, infecting over 200 million people (289,293,171) of whom more than four million (5,440,643) have lost their life (downloaded on 2nd January, 2022 from Johns Hopkins University Covid resource center). In contrast to SARS and MERS, SARS-CoV2 is characterized by high infectivity, latency, and asymptomatic transmission [7]. A number of epidemiological studies, although heterogeneous in the nature of the material and the reporting of the data, suggest that obesity does adversely affect the severity and outcome of COVID-19. During the initial viral response phase of infection, most individuals are generally asymptomatic and develop only mild symptoms, whereas some progress to severe pulmonary phase of the disease develo** pneumonia with associated symptoms. Some of these patients progress further to a phase of hyper-inflammation and develop acute respiratory distress syndrome (ARDS), sepsis, and multi-organ failure. It is important to understand which of these phases in the progression of the disease is affected in obesity. Does obesity increase susceptibility to infection and its persistence? Does obesity-associated metabolic and immune dysregulation pose risk for progression of the disease and if so, what are the molecular mechanisms? Recent reviews have analyzed the epidemiological data in the context of lessons learnt in previous viral epidemics [8], impact of metabolic and endocrine dysregulation on the susceptibility to serious disease [9, 10], and the impact of SARS-CoV2 infection on organ function relevant in non-communicable diseases and its implications for the obesity epidemic [11]. In this review, we try to analyze the current literature on the role of obesity on COVID-19 pathogenesis focusing on possible mechanisms contributing to the risk for adverse outcome of the infection.
Pathophysiology and clinical course of SARS-CoV2
The SARS-CoV2 genome is about 30 kb long and consists of two flanking, open-reading frames (ORF) and untranslated regions (UTRs) [12]. It encodes at least 27 proteins, including 4 structural proteins (spike (S) protein, envelope (E) protein, membrane (M) protein, and nucleocapsid (N) protein), 16 non-structural proteins (NSP1-11, NSP12-16), and a few accessory proteins (ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b, and ORF14). The ORF1ab at the 5′ end, which encodes the polyprotein precursor for NSPs, constitutes more than two third of the SARS-CoV2 genome. The SARS-CoV2 shares a significant amino acid sequence homology with SARS-CoV. The S-protein that is required for binding to the receptor on host cells is encoded by the S-gene. S-protein-RNA-based vaccines have been introduced against SAR-CoV2 [13]. M-protein is another important structural protein that primarily determines the shape of the virus envelope but is also able to bind to other structural proteins [12, 14]. The N-protein binds with the M protein that stabilizes the N-protein-RNA complex by promoting the completion of viral assembly inside the virion. The envelope or E-protein which is crucial for production and maturation of the virus is the smallest protein in the SARS-CoV structure.
The SARS-CoV2 gains entry into the target cell through the receptor protein, angiotensin-converting enzyme-2 (ACE2) which is present in the heart, lungs, kidneys, and gastrointestinal tract. This is initiated by S-protein attaching to ACE2 on the cell surface, its cleavage by the TMPRSS2 serine protease to expose the fusion peptide, and fusion with the host cell plasma membrane [12, 15, 16]. The virus initiates the replication process inside the host cell by releasing the RNA into the cytoplasm. ORF1ab of the viral RNA is translated into the polyproteins, pp1a and pp1ab which produce a variety of non-structural proteins (NSPs) that form the viral replication/transcription complex (RTC) while the rest of the viral RNA is transcribed into a group of nested subgenomic mRNAs. Viral replication and transcription occur with the help of RTC within the double membrane vesicles (DMV) formed by NSP-mediated reorganization of rough endoplasmic reticulum membranes.
Clinical course of COVID-19
COVID-19 presents with a variety of symptoms such as fever, cough, sore throat, fatigue, headache, myalgia, impaired sense of taste and smell, conjunctivitis, and diarrhea [17, 18]. Severe conditions are associated with pneumonia, fever, and cough and dyspnea. The signs and symptoms of illness associated with COVID-19 vary from person to person and with the severity of the disease. Incubation period is generally for 14 days after exposure though symptoms usually start to develop within 4–5 days [19,20,21,22].
COVID-19 infection can either be asymptomatic, a mild and self-limiting disease or lead to a critical and fatal illness. It is classified into three stages of increasing severity [23]. During the initial viral phase referred to as stage I, predominant symptoms are that of upper respiratory tract infection. Stage II refers to the pulmonary phase of infection characterized by pneumonia and associated symptoms and can be classified into subgroups of pneumonia patients without hypoxia (Stage II A) and those with hypoxia (Stage II B) requiring hospitalization and oxygen supplementation. Stage III refers to the hyper-inflammation phase when patients worsen rapidly and develop Acute Respiratory Distress Syndrome (ARDS) and sepsis, leading to multi-organ failure. It involves inflammation and fluid build-up in the lungs, which limits air-to-blood transfer of oxygen. Such patients generally need invasive mechanical ventilation (IMV) in the intensive care unit (ICU).
Not much information is available on the frequency of asymptomatic infection; it could be as high as 30–40% [24, 25]. Subjects could be symptom-free at diagnosis but turn symptomatic later (pre-symptomatic). Though similar viral loads have been documented in the upper respiratory tract of both symptomatic and asymptomatic cases [26] and in the pre-symptomatic phase, the risk of transmission of virus by asymptomatic people has not been quantified. However, observational and modeling reports suggest that up to 12% of transmission occurs before an index case develops symptoms [27]. It is also observed that while about 80% cases develop only mild disease, about 14% cases have severe disease; 5% of cases become critical and seriously ill and 2 to 3% are fatal. Symptomatic infection predominates in adults with underlying comorbidities [28]. Subjects with underlying comorbidities have been reported to be at increased risk of develo** severe disease following Covid-19 infection. They include individuals with type 1 and type 2 diabetes, hypertension, cancer, chronic lung disease, tuberculosis, chronic renal disease, chronic liver disease and HIV infection. Also at a higher risk are pregnant women and individuals on immunosuppressant therapy [29].
Acute hypoxemic respiratory failure from ARDS is a major finding [25, 30, 31] and the requirement for IMV is high in critically ill patients. Common complications of COVID-related ARDS include acute kidney injury, hepatic injury, cardiac injury including cardiomyopathy, pericarditis, arrhythmia, and sudden cardiac death [28].
Diagnosis and prognosis of the disease have been mostly based on clinical evaluation of the symptoms and radiological imaging and confirmed by laboratory-based RT-PCR tests. CT scan revealed bilateral lung involvement mostly during the intermediate and late stage of the disease and in severe condition revealed ‘white lung’ appearances showing the possible effect of the infection on lung functions [63,64,65,66,67,68,69,70,71,72,73,74]. The Open Safely study reported a greater risk for COVID-19 related death in obese subjects (with a hazard ratio of 1.4 for a BMI between 35 and 39.9, and 1.92 for a BMI over 40) [75].
During the latter half of 2020 and early 2021, additional data have been generated in new case /cohort studies, retrospective, and meta-analysis examining the role of factors such as age, gender, BMI, percentage of obesity more importantly focusing on adiposity, and body composition on the risk for severity of COVID-19 disease. In most of the earlier studies, the impact of obesity on COVID-19 outcome has been generally examined by considering BMI as an index of adiposity (greater or less than 30 kg/m2). A UK-based Biobank study showed a linear relation between BMI and risk of testing positive for COVID-19 [76]. However, a hospital-based study in New York showed a J-shaped relation between BMI and the risk of intubation or death [65]. But these studies carried out mostly in hospital settings, did not give much information regarding the association between adiposity/BMI and the natural course of COVID-19 disease. Two studies comprising patients enrolled in the US Veterans Health administration (VHA) also demonstrated a J-shaped relationship between BMI and risk of adverse outcome in COVID-19 [77, 78]. A general population-based cohort study involving follow-up of 2,524,926 participants conducted in Spain showed that, out of 57,443 individuals who tested positive, 10,562 were hospitalized and 2467 died due to COVID-19. BMI was positively associated with COVID-19 infection and hospitalization and showed a J-shaped relationship with the risk of COVID-related death indicating that both under nutrition and over nutrition (BMI ≤ 18.5 and high BMI) can contribute to COVID-19 -related severe disease [79].
While BMI is widely employed to define overweight and obesity, body composition particularly visceral adiposity (VAT), unlike subcutaneous adiposity, is considered to be a greater risk for obesity-related diseases [80]. Given the role of VAT in increasing risk of pathological conditions in obesity, meta-analysis of epidemiological studies showing computed tomographic analysis of visceral adiposity in COVID-19 patients is particularly relevant. It revealed that COVID-19 patients requiring ICU or IVM-support had increased visceral fat area than those who did not [81, 82]. Watanabe et al. reported that increased subcutaneous fat area was not associated with higher risk for ICU admission [83]. Yang et al. also did not find SFA > 100 mm2, a risk factor for ICU admission [84]. But higher VFA/SFA ratio was associated with a greater risk for ICU admission [85]. Computed tomographic analysis of different fat depots in 165 COVID-19 overweight patients showed an association of severe COVID-19 disease with visceral fat area (VFA) (p = 0.022) but not with subcutaneous fat (p = 0.64); SFA was not altered in patients with mild or severe COVID-19. Further, the ratio of SFA/VFA was significantly low in patients with severe disease.[86]. Not only VFA, but visceral fat thickness as a measure of visceral adiposity is also associated with severe illness in COVID-19 [87]. It was also reported that epicardial adipose tissue volume, as measured by chest CT, was associated with severity of pneumonia and adverse clinical outcomes [88] as well as a predictor of myocardial injury in COVID-19 [89].
In addition to a relation between radiological assessed visceral adiposity and the development of severe COVID-19, anthropometric indicators of abdominal obesity such as waist circumference and waist to hip ratio have also been correlated along BMI with severe outcome of COVID-19. A higher BMI (> 30), higher waist circumference, waist to hip ratio, and waist to height ratio each are positively correlated with increased incidence of covid mortality [90]. Severe cases also showed higher VAT accumulation [82]. Petersen et al. observed that, apart from BMI, VAT and upper abdominal circumference also significantly increased risk of severe COVID [52]. A general population-based cohort study in the United Kingdom showed that hospitalization of COVID-19 patients increased in a linear upward manner with BMI and waist to hip ratio indicating obesity, and central obesity in particular, are risk factors for adverse outcome in COVID-19 [91].
Age and gender are important factors contributing to central obesity and the risk for obesity-related diseases (WHO Report 2008). Results of epidemiological studies on the relation between gender and age on COVID-19 disease severity did not appear to be consistent. While some reports indicate that case fatalities are higher in obese males and increased with age, other reports suggest that obese women are more prone than men [65, 92,93,94]. Recalde et al. reported that the risk of death after hospitalization associated with BMI was higher among females than men [95]. A population-based cohort study in 4,33,995 subjects, (age group 18–79 y) in Spain revealed a similar increase in risk of hospitalization and serious disease due to severe obesity(> 40 BMI) in young adults (< 50y) and aged subjects (65-79y) [96]. Recalde et al. also reported that the incidence of adverse outcome of COVID-19 in obese individuals was higher among the 18–59-year age group compared to older age groups [95]. Further, the risk of hospitalization and disease severity was higher in < 60 age group as compared to > 60y [48, 49, 97, 98] with higher risk in males. On the other hand, in a retrospective study in 200 COVID patients, male gender and increased age were independently associated with disease severity [51]. An analysis of patient data from the QResearch database of general practices in England revealed a positive association between increasing BMI and ICU requirement among COVID-19 patients over the entire BMI range and an increase in risk of hospital admission and death among subjects with a BMI > 23 kg/m2. The increase in risk of hospitalization and ICU requirement with a rise in BMI was greater among subjects without T2D than those with T2D. It was also seen that this increase in risk was higher in subjects with hypertension compared to normotensive subjects and also in subjects with CVD compared to those without CVD [99].
Ethnicity is also considered a determinant for obesity and obesity-related disease. In addition to a higher prevalence of obesity in South Asian, Black, and Arab populations, the obesity-associated cardiometabolic risk at a given BMI in these ethnic communities is also higher than that in the White population. It has been consistently shown that subjects of certain ethnicities are at a higher risk of both contracting COVID-19 as well as an adverse outcome of the disease. Even after considering for ethnicity, it was shown that obesity was associated with adverse outcome as suggested by an increase in hospital admission, ICU/IMV requirement, and mortality. Although this was seen across Chinese, South Asian, and Arab ethnicities, this association was most prominent in populations with Black ethnicity [99,100,101].
The association of obesity and COVID-19 was also seen in children in two case studies in the US. In COVID-19-affected children (2–18 years) with 22–37% obesity, disease severity, hospitalization, and incidence of pneumonia were higher in obese children [102, 103].
Why is obesity a risk for COVID-19 ?—pathways and molecular mechanisms
A number of studies reveal that obesity, more importantly central obesity reflecting expansion of visceral adipose tissue, is critical in leading to adverse outcome in COVID-19 infection. Expansion of VAT causes metabolic dysfunction, endoplasmic reticulum stress, infiltration of immune cells, polarization of macrophages to a pro-inflammatory phenotype, adipocyte cell death, and inflammation. This is also associated with altered expression of adipokines and cytokines causing systemic effects and dysfunction of endocrine and metabolic organs. Such a challenged state of adipose tissue particularly associated with multiple organs such as lungs, vasculature, heart, and kidney may predispose obese subjects to adverse outcomes of COVID-19 infection. It is becoming evident that underlying mechanisms contributing to adverse outcomes for COVID-19 in obesity may include a) pathways of viral entry and spread, b) dysregulated RAS pathway, c) endoplasmic reticulum stress and dysregulated UPR pathway, d) endocrine dysfunction, particularly altered adipokine responses, and e) metabolic reprogramming and altered immune metabolism.
Virus entry and extended shedding in obesity
There is increasing evidence to indicate that ACE2 is the principal host cell receptor that determines the tissue tropism of SARS-CoV2 [15, 16, 104,105,106,107,108]. ACE2 is a single-chain trans-membrane multifunctional protein with an extracellular domain that recognizes the receptor-binding domain (RBD) of the S1 subunit of spike protein [109]. This is followed by S2 subunit-mediated fusion of the virus particle to the host cell membrane and internalization of the virus. The level of ACE2, its relative affinity to viral protein ligands, as well as its organization in the lipid raft structures on the plasma membrane appear to be critical in determining binding, fusion, and internalization of the virus [110]. Another host cell surface factor critical in viral entry is TMPRSS2, a cell surface serine protease that cleaves the viral spike protein into S1 and S2 subunits and primes viral fusion with the host cell membrane.
The initial viral load appears to be one of the determinants of severity of SARS-CoV2 infection and is higher in patients with severe disease than patients with milder forms [111,112,113,114,115]. Analysis of viral RNA in plasma, the respiratory tract, and urine of patients with a wide range of disease severity, including those recovered from COVID-19, showed that disease severity increased with increase in viral load and was predictive of mortality. Further, plasma viral load was also associated with indicators of disease severity such as lower absolute lymphocyte counts, and elevated inflammatory markers CRP and IL-6 [116] which are elevated in obese patients. However, in a hospital-based study, viral load was not seen to be associated with either length of oxygen support or overall survival [117] although clearance rates reported in symptomatic patients were longer than those in asymptomatic subjects [118]. Like influenza A virus infection, SARS-CoV2 also targets lungs and adipose tissue. Longer duration of shedding of influenza A virus has been found in symptomatic obese patients [119], and the viral RNA content in expired aerosol was shown to correlate with BMI of influenza patients [120]. Similarly, COVID-19 patients with obesity could have a higher viral load and longer persistence.
The integral viral load is determined by, among other factors, the relative level of the host cell receptor ACE2. Both ACE2 and TMPRSS2 are expressed in epithelial cells in different human tissues including kidney, liver, heart, lungs, adipose tissue, and GI tissue [121,122,123]. ACE2 overexpressing Hela cells showed greater SARS-CoV2 infection and replication [124]. ACE2 was upregulated both by a diet rich in sucrose or fructose as well as in experimental models of obesity induced by a high-fat diet [125,126,127]. More importantly, the ACE2 expression is increased in lung tissue in experimentally induced obese mice [128]. ACE2 expression was higher in bronchial epithelium of overweight/obese COPD patients compared to patients with BMI < 25 kg/m2 [129]. Elevated expression of both ACE2 and TMPRSS2 has also been shown in obese human subjects [130]. Further, a possible regulation of ACE2 expression by excessive calorie intake was indicated by its decreased expression in subcutaneous adipose tissue of obese subjects who had lost weight following a low-calorie diet [128, 131]. In patients with diabetes [132], kidney disease [133] and non-alcoholic fatty liver [134], who are at greater risk of SARS-CoV2 infection, ACE2 expression is upregulated. It has been suggested that lipid deposits in large airways in lungs make these sites that potential viral reservoirs and its presence in the proximity of alveolar epithelial cells expressing high amounts of ACE2 make obese patients more susceptible to SARS-CoV2 infection [135]. Elevated expression of ACE2 in lower respiratory tract in diet-induced obese male mice, unlike female mice, suggested that a sex-dependent modulation of expression of ACE2 which is significant as incidence of serious SARS-CoV2 infection is higher in males [136]. Although ACE2 is critical in SARS-COV2 infection, other cell surface molecules such as neuropilin [137, 138] and CD147[139] also may function as co-receptors.
Obesity involves hypertrophic and hyperplastic expansion of adipose tissue with enhanced storage of lipids. As pre-adipocytes differentiate into mature adipocytes, significant increase in the expression of ACE2 gene occurs. Apart from mature adipocytes, other resident cells of adipose tissue also express ACE2 [140,141,142] and are known to be targets for multiple viruses [119, 143, 144] including SARS-Cov [145]. Upregulation of ACE2 is apparently mediated through activation of the transcription factor PPARγ as indicated by stimulation of ACE2 expression by PPAR ligands such as thiazolidinedione [126]. Further, while both subcutaneous and visceral adipose tissue express ACE2, its expression was higher in visceral adipose tissue [86]. An association between the activity of transcription factors that regulate expression of genes involved in lipid metabolism such as SREBP1 and PPARγ and ACE2 gene expression in both in vitro as well as in animal models of obesity, suggested a relation between factors regulating lipid metabolism and adiposity, and ACE2 expression [128]. Chronic inflammation is a feature of obesity, and recent reports show that pro-inflammatory cytokines upregulate the expression of ACE2 [146, 147]. Adipose tissue, which is distributed extensively in the body, both under the skin and around different organs—intra-thoracic fat in lungs, epicardial fat in heart, perirenal fat in kidney, and mesenteric fat in intestine—expresses relatively higher levels of ACE2 than lungs [122, 123]. ACE2 expression in adipose tissue makes it a target for SARS-CoV2 infection, and its elevated expression is associated with adverse outcomes of COVID-19 [123]. Elevated expression of ACE2 in adipose tissue in obese/overweight conditions [148] may also lead to greater viral entry and replication, and it may act as a reservoir enhancing viral shedding and spread [144]. Adipose tissue has been reported to be a reservoir for persistence of other viruses [149]; the importance of lipid droplets in virus production [150] is also pertinent in this regard. However, it is still debated whether it is a relative increase in expression of ACE2 gene or ACE2 protein, or an increase in fat mass that results in higher levels of ACE2 in obesity. Although in vitro studies indicated that SARS-CoV2 infects adipocytes and that the virus can persist for longer period of time [130], SARS-CoV2 virus has not been detected in vivo in the adipocyte. But it has been suggested that SARS-CoV2 virus can alter the fate of adipocyte-like cells in lungs [122] and immunohistochemistry for SARS –CoV2 N-protein in autopsy samples showed sporadic positivity in cells in mesocolic and omental fat [151].
Viral replication and mTOR pathway
Another factor determining the integral viral load is the extent of replication and assembly of the virus inside the host cell. The virus uses the machinery of the host cell to replicate its gRNA, transcribe and translate the genes, transport the proteins, assemble and secrete the viral particles[12]. Nearly two third of the viral genome comprising 5’ capped ORF1a and ORF1ab is translated to generate two polyproteins. These are proteolytically cleaved generating 16 different NSPs, including RNA-dependent RNA polymerase (RdRp), which drives transcription of subgenomic RNA and viral genome replication. Virus-induced double membrane vesicles, packed with replication –transcription complex with NSPs and genomic RNA, fuse with ER. Both the genomic and subgenomic viral mRNA acquire 5’cap structure in a process which is mediated by its NSPs particularly NSP14 [12, 152]. Replication and subgenomic RNA synthesis is followed by translation of N –protein mRNA in cytosolic ribosomes whereas mRNAs of S-, E-, and M-proteins are inserted into ER and translated by the ribosomes present in ER. Protein synthesis by 5’cap-dependent translation of viral mRNA occurs by employing host cell machinery involving eukaryotic elongation factors (elFs)[153]. 7-Methyl-GTP cap structure present at the 5’ termini mediates formation of translation-initiation complex which positions the ribosomes near the 5’terminus of the mRNA. It is facilitated by eukaryotic initiation factor 4G (elF4G) acting as a scaffold protein for formation of a protein complex elF4F comprising of initiation factor 4A, an RNA helicase (elF4A) and elF4E,the cap-binding protein[154]. Initiation factor elF 4E is the rate limiting factor and its binding to elF4G is regulated by elF4E–binding protein (4E-BP) causing suppression of translation [155]. Phosphorylation of 4E-BP in response to mitogenic stimuli, dissociates 4E-BP leading to formation of active elF4F complex and initiation of translation. Importance of this pathway in corona virus replication was evident from suppression of human corona virus 229E replication by inhibiting the activity of elF4F complex by blocking elF4E binding to elF4G [156].
The mammalian(mechanistic) target of rapamycin mTOR)-pathway, that modulates activity of elFs, appears to be critical in viral replication and it has been postulated that hyper-activation of this pathway in obesity may contribute to adverse SARS-CoV2 infection[157]. mTOR is a nutrient and energy sensing kinase and it regulates cellular processes like cell survival, proliferation, growth and metabolism in response to nutrient availability and cellular energy levels, by integrating distinct signaling pathways [158, 159]. It is a serine/threonine kinase present as mTOR complex1 (mTORC1) and mTOR complex 2 (mTORC2). mTOR can transmit signals to regulate the expression of major adipogenic transcription factors like PPAR-γ and C/EBP-α family of transcription factors, and thereby stimulate adipogenesis [160]. mTORC1 is a downstream target of PI3K/Akt signaling pathway and is important for regulation of a number of cellular processes including, ribosomal and mitochondrial biogenesis, transcription, and translation [159, 161, 162]. It is activated by increase in ATP, nutrients, growth factors and hormones while absence of growth factors and nutrient deficit cause its inhibition. Activation of mTORC1 leads to phosphorylation and activation of two key effectors-p70 S6 Kinase 1(S6K1) and elF4E binding protein. mTORC1 directly phosphorylates S6K1 on Thr389 leading to its further phosphorylation and activation by phosphoinositide dependent kinase-1 (PDK1). S6K1 phosphorylates and activates elF4B, a positive effector of 5’cap-binding elF4F complex enhancing the translation efficiency. mTORC1 phosphorylates elf4E-BP at multiple sites causing its dissociation from elF4E which can then bind to elF4G and allow translation to occur.
However, dysregulation of mTOR signaling seems to contribute to the disease process in obesity, aging, cancer, and T2D [163,164,165,166,167]. mTORC1 is highly active in multiple tissues during obese and high-fat-fed conditions[168,169,170]. Consistent with this, the activity of S6K1, downstream effector of mTOR, is elevated in human visceral adipose tissue in obesity [171]; S6K1 knockout mice are resistant to obesity [169]. Over-phosphorylation of 4E-BP has also been shown in obesity [172]. Inflammatory mediators such as TNFα, which are upregulated in obesity, activate mTORC1 by activating IκB kinase-β (IKKβ), [173]. Further, insulin resistance in obesity is closely linked to mTOR stimuli [174].
It therefore appears that overactivated mTOR pathway in the obese condition sets a favorable milieu for the SARS-CoV2 viral replication. Transcriptome and proteome study of SARS-CoV2-infected cells showed that inhibition of AkT, upstream of mTOR reduced virus production [175]. Viral infection-associated dysregulation in renin-angiotensin pathway can further activate mTOR/S6K pathway contributing further to pulmonary vasculopathy[176]. SARS-CoV2 infection was shown to increase activity of mTORC1 in kidney epithelial cell line and lung-air interface mucociliary cultures and inhibition of mTORC1 by FDA approved drugs appeared to reduce viral replication. Activation of mTORC1 has also been shown in lung tissue from COVID-19 patients [177]. However, the enzyme catalyzing the formation of 5’-Cap structure is yet to be characterized in SARS-CoV2.
Obesity, endoplasmic reticulum stress, and COVID-19
An important factor that can contribute to SARS-CoV2 replication and disease in obesity is ER stress. Dysregulation of protein folding homeostasis that occurs in response to environmental and cell-intrinsic challenges results in build-up of unfolded proteins in ER lumen causing ER stress. This activates the Unfolded Protein Response (UPR) signaling mediated by three ER resident trans-membrane sensors: PKR-like ER protein kinase (PERK), inositol-requiring protein-1( IRE1) and activating transcription factor-6 (ATF6). These are regulated by an ER-chaperone, glucose-regulatory protein (GRP78), which dissociates from these receptors during ER stress leading to their activation. UPR is an adaptive mechanism which initially aims at rebalancing protein folding homeostasis by shutdown of the cellular protein synthesis, enhances ER-chaperone expression and mediates misfolded protein degradation through ER-associated degradation (ERAD) pathway. But, if the cells fail to recover when the load of misfolded proteins exceeds, such as during viral infection, UPR triggers apoptotic signaling by activating (a) C/EBP homologous protein ( CHOP) (b) c-Jun N-terminal kinase( JNK) pathway and (c) ER-associated caspases, eventually resulting in activation of caspase-3. ER stress-related inflammation and apoptosis in various cells are associated with the pathogenesis/progression of several diseases [178,179,180,181].
Infection by several viruses including SARS-CoV caused ER stress [182,183,184]. Corona virus appears to induce ER stress in host cell by (a) excessive synthesis of viral proteins, their post translational modification (PTM) and folding, (b) restructuring of the ER membrane while forming double membrane vesicles for replication, and (c) the exhaustion of the ER membrane due to continued formation of the virion [182]. Though one of the mechanisms to overcome ER stress is shutdown of global protein synthesis [183], the viruses have evolved mechanisms to counteract this and ensure viral protein translation. For instance, viral NSP1, apart from its role in suppressing host cell immune response by IFN inhibition, inhibits host cell translation by blocking mRNA binding to 40S subunit of ribosomes, and promotes mRNA degradation [185, 186] but the presence of 5’leader sequence in viral mRNA prevents its degradation, allowing viral protein translation [187, 188]. This appears to be a general phenomenon in coronaviruses. SARS-CoV2 NSP 16 disrupts host mRNA splicing by binding to mRNA recognition domains of snRNAs and NSP 8 and NSP 9 disrupt protein trafficking by binding to signal recognition particle in HEK293T cells infected with SARS-CoV2. Further, NSP1 is also known to bind in the mRNA entry channel of ribosome causing translational inhibition of host cell proteins [189]. Recent proteome data in SARS-CoV2-infected cells also showed suppression of host cell protein production and dysregulation of ER proteostasis pathways, and increase in serum levels of GRP78 and CHOP in COVID-19 patients indicating ER stress and suppression of IFN production[108, 189,190,191].
Several types of viruses like Zika virus[192], Coxsacki virus [193], Dengue virus [194], Japanese encephalitis virus [195] take advantage of the chaperone property of GRP78 to bind to viral proteins for their entry and replication within the host cells. Though GRP78 is primarily an ER protein aiding protein folding and vesicular transport to Golgi, it has also been shown on the cell surface, probably due to its mis-sorting or due to saturation of its binding to the KDEL receptor for its reverse transport to ER [196]. It may act as a co-receptor for the viral entry. Structural analysis and molecular docking suggested binding of S-protein of SARS-CoV2 to GRP78 [197]. The nascent S-protein, M-protein, and E-protein of SARS-CoV and SARS-CoV2, undergo post translational modification,particularly glycosylation and folding in the ER. Accumulation of these nascent viral proteins causes ER stress. In vitro and in vivo studies have suggested that different types of cells including endothelial cells, alveolar epithelial cells, cardiomyocytes are under ER stress in subjects with comorbid conditions such as diabetes and obesity. Circulating GRP78 levels, due to shedding from cell surface increased in subjects with diabetes and obesity and correlated with CRP levels [198]. Both metabolic stress and chronic inflammation occurring in obesity appear to influence the level and distribution of GRP78. Treatment with pharmacological chaperones that alleviate ER stress suppressed NF-kB activity and inflammation in obese mice suggesting that ER stress contributes to the chronic inflammation occurring in obesity [199]. It, therefore, appears that ER stress associated with metabolic stress and inflammation in obesity, by virtue of increase in GRP 78, might contribute to increased viral entry. However, direct evidence supporting a role for GRP78 in SARS-CoV2 infection is lacking, although serum levels of GRP78 were found to be higher in patients with COVID-19 compared to those with pneumonia, and healthy controls [191]. Signaling pathways of UPR and inflammation are linked by mechanisms like the generation of ROS, calcium efflux from ER, activation of NF-κB by PERK, NF-κB and MAPK activation by IRE1, and induction of the acute-phase response. Further, Ang-II that accumulates (as discussed later) during SARS-CoV2 infection, acts on ER through AT1R to increase ER stress and downstream inflammatory signaling through NF-κB; it also induces TGFβ-mediated apoptosis and fibrosis [200]. The systemic inflammation of adipocytes in obesity is primarily mediated through ER dysfunction. UPR signaling is also implicated in vascular inflammation and possibly endothelial cell dysfunction [201, 202] and such ER stress-mediated pre-activation make obese subjects vulnerable to severe COVID-19. Further, loss of ACE2-mediated protection of ER stress through suppression of apoptosis on viral infection exacerbates ER stress [203, 204].
Dysregulation of Renin-angiotensin system and COVID-19-related respiratory dysfunction
As indicated before, the most common complication of COVID-19 is altered respiratory function associated with infection of the lung progressing to SARS. Obesity may increase the risk of develo** respiratory dysfunction by different mechanisms including pulmonary restriction and imbalance between ventilation and perfusion. Accumulated fat within the thorax and abdominal cavity may mechanically affect both chest wall and lung compliance. Restriction of diaphragmatic mobility and chest wall movement reduces functional residual capacity [205, 206]. One of the major molecular pathways that regulate pulmonary function is the renin-angiotensin system (RAS) which is critical for maintenance of blood pressure, electrolyte and fluid balance and affects the functions of several organs including heart, lungs, kidney, liver, blood vessels and adipose tissue [207, 208]. The classical RAS pathway consists of the enzyme renin which proteolytically cleaves angiotensinogen to Ang1; it is then converted to Ang-II by angiotensin-converting enzyme (ACE1) [209]. Ang-II, through its cell surface receptor (AT1R) regulates vasoconstriction, inflammation and oxidative stress. It is further cleaved by ACE2 to Ang(1–7), which binds to Mas receptor and exerts vasodilation, vasoprotection, anti-proliferative and anti-inflammatory effects. Balance between ACE/Ang-II/AT1 receptor and ACE2/Ang(1–7)/Mas receptor axis contributes to cellular homeostasis and vascular function. ACE2 also serves to regulate the kallikrein/kinin system that generates, by sequential proteolysis, ligands for bradykinin-II and bradykinin-I receptors which play a role in blood pressure regulation, inflammation and coagulation. Further, by inactivating BI receptor ligands, ACE2 protects against pulmonary edema, whereas decrease in ACE2 increases pulmonary vascular permeability, edema, hypertension, inflammation, ARDS and cardiac failure [210,211,212,213,214].
As ACE2 is a multifunctional molecule actively involved in pathways critical to cellular homeostasis, hijacking this host molecule by SARS-CoV2 can adversely affect host cell function. Evidence in support of hyper-activated RAS pathway as one of the potential mechanisms contributing to adverse outcome of COVID-19 in obese subjects is accumulating. RAS pathway is active in lungs, vascular tissue, heart, adipose tissue, liver and kidney [215, 216] and the presence of significant amount of ACE2 in lungs, vasculature and adipose tissue of obese subjects allows entry of SARS-CoV2 virus [217, 218]. Significantly elevated levels of angiotensinogen in obese subjects feed more amounts of Ang-II into the RASpathway [219,220,221] and it will accumulate if sufficient ACE2 is not available as happens in SARS-CoV and SARS-CoV2 infection [206 214]. Serum levels of Ang-II are raised in COVID-19 patients [222] and relate positively with viral load and lung injury [111, 223, 224], indicating dysfunction/reduced action of ACE2. Accumulation of Ang-II causes pulmonary dysfunction as suggested by earlier studies in experimentally induced acute lung injury where Ang-II has been shown to cause pulmonary edema and inflammation [225] Further, it has pro-coagulant effects as infusion of Ang-II caused platelet activation [226]. Binding of SARS-CoV2 to cell surface ACE2 through the viral spike protein, followed by cell entry is associated with decrease in ACE2 [104, 227, 228]. This may be due to cleavage of its ectodomain by ADAM17, the cell surface metalloprotease that also activates TNFα by its proteolytic cleavage [229]. Presence of cleavage sites in the ectodomain and endodomain suggests a possible cleavage of ACE2 by TMPRSS2 as well [230]. Plasma ACE2, a result of proteolytic shedding of cellular ACE2, was higher in COVID-19 patients than in healthy controls [231]. Further, in SARS-CoV2-infected cells in culture, there was a significant reduction in the expression of both TMPRSS2 and ACE2 [232] and the levels of total ACE2 [190]. Though it is not clear how cell surface ACE2 is downregulated in SARS-CoV2 infection, cleavage of ACE2 by proteases as indicated by elevated levels of plasma form of ACE2 and a shutdown of host gene expression, by downregulation of translation and degradation of host mRNAs [233] are two possible mechanisms. Further, a feed-forward effect of AT1R activation by accumulating ANG-II could transcriptionally downregulate ACE2 expression [234]. SARS-CoV1 infection has also been associated with a decrease in ACE2 [227, 233, 235]. Modulation of RASpathway, particularly ACE2 expression by metabolic regulators including hormones such as insulin [236] and glucagon-like peptide-1 receptor agonist [237] suggest a role for host’s metabolic state in regulating viral entry and response to infection.
The dysregulation of the RAS pathway and its implications in the development of severe disease in obese subjects infected with SARS-CoV2 is illustrated in Fig. 1. A decrease in ACE2 can affect the equilibrium between pro-inflammatory Ang-II and anti-inflammatory Ang(1–7). Accumulation of Ang-II triggers receptor-mediated JAK-STAT pathway while decrease in Ang(1–7) shuts down Mas receptor signaling causing upregulation of pro-inflammatory factors and downregulation of anti-inflammatory factors. Decrease in ACE2 and consequent accumulation of ANG-II on SARS-COV2 infection in obese subjects can have effects beyond RAS pathway contributing further to pulmonary vasculopathy. For instance, accumulation of ANG-II can heighten activation of mTOR/S6K pathway and further impact insulin responsiveness and cause endothelial dysfunction [160, 238, 239]. Activation of mTORC1, as discussed before, can increase SARS-CoV2 replication. Ang-II, acting through G-protein coupled ATR1 increases ROS production by stimulating Nox family NADPH-oxidase and cause lung endothelial dysfunction. It also disturbs mitochondrial function and modifies cell metabolism [200]. Further, ER stress-induced inflammation and apoptosis of alveolar epithelial cells is regulated by Ang-II/Ang(1–7) system [203]. Decrease in ACE2/Ang(1–7) on SARS–Cov2 infection of the lung epithelium can lead to loss of the protective effect of Ang(1–7) against ER stress-induced inflammation and apoptosis as shown earlier in experimentally induced ER stress in lung epithelium and microvascular endothelial cell [204].

Dysregulation of RAAS pathway in Covid-19 subjects with obesity. A. Angiotensinogen is converted by Renin to Ang I which is converted by ACE 1 to Ang-II that acts through AT1R to exert vasoconstrictive and inflammatory effects. Ang-II is alternately cleaved to Ang 1–7 which can act through the Mas-R and exert a vasoprotective, anti-inflammatory and anti-thrombotic effect. Balance between the Ang-II-AT1R and Ang 1–7 – Mas-R axes maintains normal cellular homeostasis. B. Increased ATN in obesity increases amount of Ang-II in the RAAS. ACE2-mediated entry of SARS-CoV2 leads to decreased ACE2 availability and imbalance between Ang-II-AT1R and Ang 1–7 – Mas-R axes resulting in increased Ang-II and a shift toward Ang–II-AT1R-mediated effects, leading to increased inflammation, thrombosis and cellular dysfunction. Thick arrows-increased, Thin arrows-decreased. Red-Effect of SARS-CoV2  . Gold-Effect of Obesity
. Gold-Effect of Obesity
Metabolic reprogramming in host cell on SARS-CoV2 infection
Viruses appear to have developed suitable strategies to reprogramme host cell metabolism to their advantage for replication and survival; specific host cell metabolic pathways of carbohydrate, lipids, amino acids and nucleotides are differentially affected by viral infection. Viral infections such as influenza virus have been reported to induce a shift into glycolytic metabolism of glucose and inhibition of glycolysis reduced severity of infection suggesting that virus-induced shift into glycolytic phenotype was critical for its survival [240,241,242]. Reprogramming of host cell metabolism as an adaptive mechanism to potentiate host immunity is also likely. SARS-CoV2 infection also appears to cause diverse effects in host cell metabolism, though detailed information is not available yet. In vitro studies in monocytes supplemented with glucose or monocytes isolated from obese/diabetic patients showed that increase in glucose resulted in increase in viral load indicating that glucose load favored SARS-COV2 infection. SARS-CoV2 infection leads to stimulation of glycolysis along with an increase in the glycolytic capacity in monocytes, and inhibition of glycolysis resulted in reduced viral replication and cytokine production suggesting that glycolysis is required to sustain COV2 infection in monocytes [243]. Alteration in whole body metabolism was indicated by plasma metabolomics and lipidomic analysis of COVID-19 patients of varying disease severity; decrease in TCA cycle intermediates such as malic acid indicated altered energy metabolism. There was a gradual decrease in carbamoyl phosphate, an intermediate of the urea cycle, with increase in fatality, indicating hepatic dysfunction. Both GMP and carbamoyl phosphate were significantly lower in fatal cases than mild ones [244]. Metabolomic analysis of plasma samples of COVID-19 patients revealed alteration in pathways of metabolism of amino acids, lipids, and energy metabolism. A meta-analysis of six such studies revealed a decrease in TCA cycle and propionate pathway as well as perturbation of porphyrin metabolism pathway [298]. Role of exosomal miRNA in mediating cell–cell interaction and influencing host cell defense was evident from the disruption of lung epithelial cell integrity and mitochondrial function by exosomes containing miRNA 23a-27a-24 cluster secreted by alveolar macrophage infected with HIV protein [299]. The importance of host cell miRNA in suppression of SARS-CoV replication and immune evasion has also been demonstrated [300]. In this context, investigations into the possible involvement of miRNAs in SARS-CoV2 disease are also underway. Both ACE2 mRNA and protein expression in cardiomyocytes were downregulated by miR-200c [363, 364]. Its expression is decreased by inflammatory cytokines TNFα and IL-6, which are induced as fat accumulates in adipose tissue [365]. Decrease in adiponectin occurring in obesity and in insulin-resistant conditions [366] may, thus, exaggerate inflammatory response and dysregulation of vascular endothelial homeostasis [356]. Further, adiponectin levels are lower in males who are at increased risk for COVID-19 than females [367]; similarly, its levels are low in certain ethnic groups [368, 369], who are at increased risk for COVID-19. It has been postulated that decrease in adiponectin in obesity may contribute to respiratory failure in COVID-19 [10]. In a recent study, decrease in plasma levels of adiponectin was observed in 12 COVID-19 patients with respiratory failure compared to non-COVID patients with respiratory failure [370]. It appears that leptin and adiponectin have opposite effects on vascular endothelium, metabolism, immune response, and inflammation, and an inverse pattern of expression with leptin showing an increase and adiponectin decreasing in obesity [364]; the ratio of these two adipokines in circulation might be important in the pathophysiology of COVID-19.
Vascular endothelium and thrombotic risk
It is also evident that dysfunctional adipose tissue contributes to thrombotic risk, which can contribute to adverse outcome in COVID-19 patients with obesity. Apart from pneumonia-related respiratory dysfunction affecting lungs, a clinical feature of COVID-19 in several cases is pulmonary thromboembolism and thrombotic microangiopathy involving endothelial system. Laboratory investigations and imaging studies suggested hyper-inflammation and thrombotic phenomena as important characteristics of severe cases of COVID-19 and SARS-CoV2 may predispose patients to thrombotic disease[371]. Diffuse vascular endothelial inflammation, associated with apoptosis, which is reported to make vascular endothelial cells pro-coagulant [372], suggested that vascular endothelium is a target for SARS-CoV2 [373] and could cause impaired microcirculation. Risk for thrombosis is high in obesity and is associated with a shift to pro-thrombotic state with dysfunctional endothelium [374], activated platelets, and decreased fibrinolytic activity [375,376,377]. Decrease in fibrinolysis appears to be due to obesity-associated insulin resistance; by modulating the transcription factor Egr-1, insulin is suggested to regulate levels of tissue factor and plasminogen activation inhibitor (PAI-1) [378]. Increased fat mass-associated elevation of circulating PAI-1 can inhibit plasmin-mediated fibrinolysis. Overall coagulation potential and decreased fibrinolysis (as reflected in overall homeostatic potential) have been demonstrated to increase with increase in BMI [379]. Platelet activation in obesity was indicated by higher excretion of 11-dehydroTBX2, a metabolite of thromboxane and a marker of platelet activation, by obese subjects and its reduction to normal levels on weight loss [380]. Further, insulin-induced anti-aggregation of platelets in non-obese subjects was neutralized in obese insulin-resistant subjects indicating that the abnormal metabolic state accompanying insulin resistance, and obesity alters platelet activity [381]. Elevated levels of von Willebrand Factor, TF, factor VII, Factor VIII, and fibrinogen in circulation indicated a hypercoagulable state in obesity [377, 382]. Moreover, C-reactive protein, of which plasma levels are increased in obesity [383] and severe COVID-19 [384], also exerts a pro-thrombotic effect [385]. It induces TF and PAI-1 and enhances monocyte-endothelial cell interaction by inducing expression of endothelial cell adhesion molecules [386].
Dysregulation of several molecular pathways in obesity can impact endothelial function. For instance, alteration in eNOS activity, increase in pro-inflammatory cytokines and circulating free fatty acids (FFA), and decrease in protective adipokines in obesity contribute to endothelial dysregulation. FFAs induce endothelial dysfunction by several mechanisms including disruption of calcium signaling-mediated NO production, oxidant stress and inflammatory signaling, RAS activation-dependent elevation of endothelin leading to vasoconstriction, and activation of apoptotic pathways [387, 388]. A relation between FFA-induced endothelial dysfunction and RAS was indicated by prevention of FFA effect by inhibition of RAS [389]. FFAs appear to exert this effect by activation of leukocytes through Ang-II production in mononuclear and polynuclear cells and consequent enhanced adhesion of leukocytes to endothelium [390]. Increase in endothelial permeability in obesity permits passage of the virus across the endothelium to infect pericytes or pneumocytes expressing the ACE2 receptor. Plasma levels of VEGF that increase endothelial permeability are increased in obese subjects [391]. Further, increase in FFA can increase endothelial permeability through Nlrp3 inflammasome activation coupled with a decrease in tight junction proteins ZO-1/ZO-2[392, 393]. Plasma metabolomic analysis showed significant increase in FFA correlating with markers of inflammation such as IL-6 and CRP and severity of COVID-19 [394]. FFA-induced immune and endothelial dysfunction, and hyper-inflammation may also make obese subjects vulnerable to adverse outcome in COVID-19.
Recent reports have implicated auto-antibodies in pathogenesis of complications of COVID-19. This was indicated by detection of increased levels of antibodies against interferon-1 (both IFNω and IFNα) in patients admitted with serious COVID-19 [395]. It also appears that the auto-antibodies can contribute to an increased pro-thrombotic state. This was suggested by a higher titer of anti-phospholipid antibodies (aPLs) in serum of patients with severe COVID-19 and an increased ability for IgG antibodies purified from these serum samples to induce venous thromboembolism in an experimental mouse model [396]. The levels of these aPLs were associated with higher platelet count, severe respiratory disease, and low glomerular filtration rate. The antibodies detected were against Cardiolipin, β2 Glycoprotein I, and phosphatidyl serine/prothrombin. A recent meta-analysis also indicated a rise in aPL prevalence in critically ill COVID-19 patients compared to non-critically ill patients but could not demonstrate any association between the presence of aPLs and requirement of IMV, mortality, D-dimer levels, or development of venous thromboembolism [397]. Obesity has been shown to be associated with increased production of auto-antibodies against intracellular proteins in different organs. Moreover, it has also been demonstrated that obese adipose tissue contributes to secretion of these auto-antibodies [398]. Parallels between the coagulopathy seen in conditions like Anti-phospholipid antibody syndrome and that seen in serious COVID-19, and the increase in circulating auto-antibodies seen in obesity suggest that an altered auto-immune response could be another possible link between obesity and adverse outcome in COVID-19.
Neutrophil activation
The role of peripheral blood leukocytes, particularly neutrophils, in the development of obesity and related diseases is becoming increasingly evident [399,400,401]. Increase in neutrophil level and its activation indicated by increased expression of elastase and myeloperoxidase, were reported in obese male subjects[402]. High plasma levels of adiponectin were associated with reduced production of the chemokine CXCL8 by neutrophils and the neutrophil activation in obese subjects appeared to be a consequence of decrease in adiponectin [403]. An increase in neutrophils in BAL [404], elevated levels of unique markers of neutrophil activation, and their correlation with disease severity [38] indicated that neutrophil activation is critical for pathogenesis of COVID-19 complications.
One of the mechanisms by which neutrophils exert their effect is through formation of Neutrophilic Extracellular Traps (NETs) in a process called NETosis [405]. This involves release of de-condensed nuclear chromatin, associated with histones and neutrophilic antimicrobial granular proteins, in the form of a reticular scaffold which traps invading pathogens within the DNA fibers. This limits spread of infective agent and recruits antimicrobial factors to the infection site. However, NET action is non-specific and can result in injury to surrounding tissue and worsen the inflammatory response.
The inflammatory state in obesity is also characterized by increase in NETosis [405]. A higher amount of NET formation was observed in plasma of a group of subjects with morbid obesity compared to controls [406]. In experimental mouse models of obesity, there was increased NET formation in the adipose tissue [407].
Viral infections such as influenza A [408], Respiratory Syncytial virus [409], and Chikungunya [410] induce NETosis. Mice on a high-fat diet, infected with influenza, showed higher NETosis than mock-infected controls [411]. In patients hospitalized with COVID-19, there was an increase in markers of NETosis, such as MPO-DNA, in patients requiring mechanical ventilation. Further, sera from COVID-19 patients triggered more NET formation in neutrophils, in vitro, than that from controls [412]. Increase in aPLs in COVID-19 patients was associated with neutrophil hyperactivity and release of NETs, and purified IgG fractions from serum of these patients promoted NETosis in neutrophils isolated from healthy subjects [396]. NETs have been reported to increase in BAL fluid of patients with ARDS [413] as well as those with respiratory failure following acute exacerbation of COPD [414]. Histochemical analysis of autopsy samples of lungs of COVID-19 patients showed the presence of neutrophilic plugs [151]. NETosis has also been implicated in various conditions characterized by arterial and venous thrombosis [415,416,417]. It, therefore, appears that increased NETosis due to acute inflammation in COVID-19 might further aggravate the pre-existing NETosis in the chronically inflamed obese state, thus, priming it for a more severe outcome.
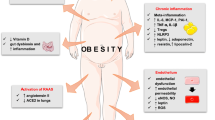
It appears that dysregulation of the metabolic and endocrine functions of adipose tissue, compounded by inflammation, leads to local and systemic effects that affect functioning of multiple organs, making obese patients vulnerable to a more adverse outcome in SARS-CoV2 infection (Fig. 2). Increase of adipose tissue mass and elevated levels of ACE2 can make it a reservoir for the virus. Besides affecting virus shedding, the pre-existing pro-inflammatory state is aggravated by enhanced acute production of inflammatory cytokines. Higher levels of angiotensinogen and downregulation of ACE2 following infection result in loss of protective effect of ACE2. The resulting dysregulation of the RAAS pathway and ANG-II accumulation further worsen the inflammatory state. Obesity-associated insulin resistance and hyperglycemia are aggravated by SARS-CoV2-induced hyperglycemia consequent to virus-targeting pancreas, leading to further metabolic dysfunction. Dysregulation of endocrine function, particularly a rise in pro-inflammatory leptin and reduction in protective adiponectin, causes systemic effects that affect vascular endothelial function, induce oxidant imbalance, dysregulate immune metabolism, and impair immune response, making obese subjects more prone to severe COVID-19. Alterations in the components of the complement pathway which are mostly of adipose tissue origin and, associated with both adiposity and insulin resistance, can make obese subjects susceptible to microthrombosis in COVID-19. Obesity-associated metabolic and endocrine imbalance can also contribute to dysregulated platelet function and altered fibrinolytic system leading to increased risk for thrombotic events in COVID-19 patients. Several of these changes in adipose tissue function that occur in obesity and have implications for adverse outcome for COVID patients with obesity, could be reversed on calorie restriction-induced weight loss or by surgical intervention suggesting the possibility of adopting weight reduction strategies to reduce risk for severe COVID-19 disease.

Challenged adipose tissue and exacerbation of COVID-19. A. Leptin, through its receptor-associated JAK/ STAT-dependent and independent pathways in central and peripheral tissues, regulates energy homeostasis, glucose and lipid metabolism, and immune function. It stimulates polarization of CD4 T-cells to a pro-inflammatory Th1 rather than anti-inflammatory Th2 type, activation of monocytes and macrophages, NK cell activation, the production of pro-inflammatory cytokines, and neutrophil chemotaxis. Adiponectin acts through its two receptors, mediated via AMPK or PPARα, to promote fatty acid oxidation and inhibit lipogenesis, suppress mTOR and IKK-NF-kB-PTEN signaling, and improve insulin signaling. Adipose tissue produces pro-inflammatory cytokines of which levels depend on the relative levels of pro-inflammatory leptin and anti-inflammatory adiponectin. Adipose tissue also contributes to the RAAS pathway. B. Obesity is characterized by metabolic dysfunction, increase in inflammation, ER stress, immune impairment, RAAS dysregulation, and an increased thrombogenic state. Altered adipokine and cytokine production can result in systemic effects adversely affecting organ function. Increased leptin action causes increased immune cell-mediated inflammation with increased vascular permeability, neutrophil activation with neutrophilic extracellular traps, increased pro-inflammatory, and decreased anti-inflammatory cytokines and increased polarization of macrophages to a pro-inflammatory M1 type from an anti-inflammatory M2 type. It also results in depressed innate and adaptive immune response with reduced interferon response and decreased dendritic cell activity. Dysregulated adipokine production and Insulin resistance affect immune cell metabolism and diminishes immune response. Increase in pro-inflammatory cytokines, decrease in adiponectin, and increased ER stress lead to endothelial dysfunction. Decreased adiponectin also leads to platelet activation. Increased tissue factor and PAI-1 from adipose tissue lead to activation of coagulation cascade and decreased fibrinolysis, respectively. Anti-fibrinolytic effect, hypercoagulable state, and activated platelet with endothelial dysfunction lead to a highly thrombotic state. This challenged system is further compromised by SARS-CoV2 entry that exacerbates inflammation and ER stress. It dysregulates RAAS, leading to loss of protective effect of ACE2 and accumulation of Ang-II, causing further ER stress and inflammation leading to more endothelial dysfunction. Impaired immune response, increased inflammation, and activated thrombotic state can increase severity of COVID-19 in obesity. Red effect of SARS-CoV2  . Gold effect of Obesity
. Gold effect of Obesity
Conclusion
A series of epidemiological studies revealed obesity, particularly central obesity, to be an independent risk factor contributing to higher morbidity and mortality among SARS-CoV2-infected patients. Data from clinical samples of infected patients and studies of cells infected with SARS-CoV2 virus suggest that the underlying mechanisms contributing to adverse outcomes of COVID-19 in subjects with obesity include (a) molecular mechanisms enhancing viral entry and spread and (b) dysregulation of host cell homeostasis adversely affecting functions of key organs which remain critically challenged in obese subjects.
Increased expression of host cell proteins, particularly ACE2, and overactivated mTOR can lead to increased viral entry and spread. Aggravated ER stress, dysregulated RAS pathway, and reprogrammed host cell metabolism may contribute to an adverse outcome.
Disproportionate expansion of adipose tissue, more importantly that of VAT, results in dysfunctional, metabolically challenged and pro-inflammatory adipose tissue leading to distorted cross-talk and loss of homeostasis. The resulting imbalance between pro- and anti-inflammatory cytokines and adipokines causes local and systemic inflammation and an altered immune response due to dysregulated immune cell metabolism. Altered redox balance, decrease in protective adipokines such as adiponectin, and increase in free fatty acids induce vascular endothelial damage. This adiposopathy increases the risk for adverse outcome of COVID-19 in patients with obesity. Adipose tissue as a reservoir would increase the latency of the virus which might become susceptible to mutation. Appropriate in vivo model-based studies would be required to obtain further insights.
Host-cell response involves differential expression of several genes including those relating to various metabolic and signaling pathways, and a number of miRNAs that target genes regulating cellular processes that contribute to progression and severity of the disease. However, not much information on the targeting of SARS-CoV2 genes by host miRNAs, or sequestering of host miRNAs by viral mRNAs causing loss of regulation of host genes is available. Whether cell/tissue tropism of SARS-CoV2 depends on miRNAs, apart from ACE2, as has been suggested for liver tropism of HCV by miR-122 [418] is also relevant.
Several molecular and signaling pathways dysregulated in SARS-CoV2 infection align with parallel changes in these molecular pathways in obesity, to exacerbate the pathological process and cause severe disease outcome. Some of these molecular pathways are so critical that targeting them could have immense therapeutic potential. Since mTOR pathway is critical in viral replication and elevated in obesity, it is a potential therapeutic target. FDA approved mTOR inhibitors which showed suppression of viral replication in cell-based studies could be clinically tested for their therapeutic potential. ANG-II/RAS pathway is another important therapeutically potent molecular system that contributes significantly to severe disease in COVID-19 patients with obesity. Based on experimental studies on angiotensin receptor blockers (ARB) and ACE inhibitors, concern has been expressed regarding a possible compensatory upregulation of ACE2 that could increase viral load and lung injury. Further, it is also debated whether use of such inhibitors should be continued in CVD patients infected with SARS-CoV2 [419]. As elevated levels of serum ANG-II are correlated with viral load, assessment of serum ANG-II may be done before considering the use of ARB or ACE inhibitors. Yet another molecular pathway involved in acute inflammation and associated complication is the interleukin and JAK/STAT pathway. IL-6 is one of the key upregulated interleukins in SARS-CoV2 infection aligning with its elevated levels in pre-existing chronic inflammation in obesity. Antibodies against IL-6 receptor (Sarilumab and Tocilizumab) and IL-6 (Siltuximab) are two types of IL-6 inhibitors approved by FDA and the NIH panel on COVID-19 treatment guidelines has recommended Tocilizumab along with corticosteroids in certain hospitalized cases with severe disease.
Several of the derangements in metabolism, inflammation and immune cell metabolism and associated molecular pathways, with implications for severe COVID-19, in obesity could be reversed by weight reduction either by diet and life style-based caloric restriction or surgical intervention. Such obesity reduction approaches could be effective in reducing risk for adverse outcome in COVID-19 infections and could even reduce risk for post COVID-19 diseases.
Availability of data and material
Not applicable.
References
Ritchie H, Roser M (2017) "Obesity". Published online at OurWorldInData.org. Retrieved from: 'https://ourworldindata.org/obesity' [Online Resource]
Huttunen R, Syrjänen J (2013) Obesity and the risk and outcome of infection. Int J Obes 37:333–340. https://doi.org/10.1038/ijo.2012.62
Honce R, Schultz-Cherry S (2019) Impact of obesity on Influenza A virus pathogenesis, immune response, and evolution. Front Immunol 10:1071. https://doi.org/10.3389/fimmu.2019.01071
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses (2020) The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5(4):536–544. https://doi.org/10.1038/s41564-020-0695-z
https://www.niaid.nih.gov/diseases-conditions/covid-19. Last Accessed Nov 2021
Zhou P, Yang XL, Wang XG et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273. https://doi.org/10.1038/s41586-020-2012-7
Guy RK, DiPaola RS, Romanelli F et al (2020) Rapid repurposing of drugs for COVID-19. Science 368(6493):829–830. https://doi.org/10.1126/science.abb9332
Bhattacharya I, Ghayor C, Pérez Dominguez A et al (2020) From Influenza Virus to Novel Corona Virus (SARS-CoV-2)—the contribution of obesity. Front Endocrinol (Lausanne) 11:556962. https://doi.org/10.3389/fendo.2020.556962
Drucker DJ (2021) Diabetes, obesity, metabolism, and SARS-CoV-2 infection: the end of the beginning. Cell Metab 33:479–498. https://doi.org/10.1016/j.cmet.2021.01.016
Lockhart SM, O’Rahilly S (2020) When two pandemics meet: why is obesity associated with increased COVID-19 mortality? Med (N Y) 1:33–42. https://doi.org/10.1016/j.medj.2020.06.005
Stefan N, Birkenfeld AL, Schulze MB (2021) Global pandemics interconnected—obesity, impaired metabolic health and COVID-19. Nat Rev/ Endocrinol 17:135–149. https://doi.org/10.1038/s41574-020-00462-1
V’kovski P, Kratzel A, Steiner S et al (2021) Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol 19(3):155–170. https://doi.org/10.1038/s41579-020-00468-6
Voysey M, Clemens SAC, Madhi SA et al (2021) Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 397:10269, 99–111. https://doi.org/10.1016/S0140-6736(20)32661-1
Wang M-Y, Zhao R, Gao L-J et al (2020) SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front Cell Infect Microbiol 10:587269. https://doi.org/10.3389/fcimb.2020.587269
Hoffmann M, Kleine-Weber H, Schroeder S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280. https://doi.org/10.1016/j.cell.2020.02.052
Wang Q, Zhang Y, Wu L et al (2020) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181(4):894–904. https://doi.org/10.1016/j.cell.2020.03.045
Song Y, Liu P, Shi XL et al (2020) SARS-CoV-2 induced diarrhoea as onset symptom in patient with COVID-19. Gut 69:1143–44. https://doi.org/10.1136/gutjnl-2020-320891
Giacomelli A, Pezzati L, Conti F et al (2020) Self-reported olfactory and taste disorders in SARS-CoV-2 patients: a cross-sectional study. Clin Infect Dis 71:889–890. https://doi.org/10.1093/cid/ciaa330
Guan WJ, Ni ZY, Hu Y (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382:1708–1720. https://doi.org/10.1056/NEJMoa2002032
Chan JF, Yuan S, Kok KH, et al (2020) A Familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395(10223):514–523. https://doi.org/10.1016/50140-6736(20)30154-9
Lauer SA, Grantz KH, Bi Q et al (2020) The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann Intern Med 172:577–582. https://doi.org/10.7326/M20-0504
Vetter P, Vu DL, L’Huillier AG et al (2020) Clinical features of COVID-19. BMJ 369:m1470. https://doi.org/10.1136/bmj.m1470
Wiesbauer F, Jacobs S (2020) COVID-19 disease progression [Internet]. Available from: https://www.medmastery.com/guide/covid-19-clinical-guide/covid-19-disease-progression. Last Accessed Feb 2021
Oran DP, Topol E (2020) Prevalence of asymptomatic SARS-CoV-2 infection: a narrative review. Ann Intern Med 173(5):362–367. https://doi.org/10.7326/M20-3012
Lavezzo E, Franchin E, Ciavarella C et al (2020) Suppression of a SARS-CoV-2 outbreak in the Italian municipality of Vo’. Nature 584(7821):425–429
Hoehl S, Rabenau H, Berger A et al (2020) Evidence of SARS-CoV-2 infection in returning travelers from Wuhan, China. N Engl J Med 382:1278–1280. https://doi.org/10.1056/NEJMc2001899
Wei WE, Li Z, Chiew CJ et al (2020) Presymptomatic transmission of SARS-CoV-2—Singapore, January 23-March 16, 2020. MMWR 69(14):411–415. https://doi.org/10.15585/mmwr.mm6914e1
McIntosh K (2021) Coronavirus disease-2019 (COVID-19): Epidemiology, virology, and prevention [Internet] Available from https://www.uptodate.com/contents/covid-19-clinical-features. Last Accessed Feb 2021
https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html. Last Accessed Nov 2021
Cummings MJ, Baldwin MR, Abrams D et al (2020) Epidemiology, clinical course and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet 395(10239):1763–1770. https://doi.org/10.1016/S0140-6736(20)31189-2
Suleyman G, Fadel RA, Malette KM et al (2020) Clinical characteristics and morbidity associated with coronavirus disease 2019 in a series of patients in metropolitan detroit. JAMA Netw Open 3(6):e2012270. https://doi.org/10.1001/jamanetworkopen.2020.12270
**e Y, Wang Z, Liao H et al (2020) Epidemiologic, clinical, and laboratory findings of the COVID-19 in the current pandemic: systematic review and meta-analysis. BMC Infect Dis 20:640. https://doi.org/10.1186/s12879-020-05371-2
Zayet S, Kadiane-Oussou NJ, Lepiller Q et al (2020) Clinical features of COVID-19 and influenza: a comparative study on Nord Franche-Comte cluster. Microbes Infect 22(9):481–488. https://doi.org/10.1016/j.micinf2020.05.016
Ellinghaus D, Degenhardt F, Bujanda L et al (2020) Genome wide association study of Severe COVID-19 with respiratory failure. N Engl J Med 383:1522–1534. https://doi.org/10.1056/NEJMoa2020283
Chen T, Wu D, Chen H et al (2020) Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ 368:m1091. https://doi.org/10.1136/bmj.m1091
Zhang Z-L, Hou Y-L, Li D-T, Lic F-Z (2020) Laboratory findings of COVID-19: a systematic review and meta-analysis. Scand J Clin Lab Invest. https://doi.org/10.1080/00365513.2020.1768587
Wölfel R, Corman VM, Guggemos W et al (2020) Virological assessment of hospitalized patients with COVID19. Nature 581:465–469. https://doi.org/10.1038/s41586-020-2196-x
Meizlish ML, Pine AB, Bishai JD et al (2021) A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv 5:1164–1177. https://doi.org/10.1182/bloodadvances.2020003568
Cai Q, Chen F, Wang T et al (2020) Obesity and COVID-19 severity in a designated hospital in Shenzhen, China. Diabetes Care 43:1392–1398. https://doi.org/10.2337/dc20-0576
Huang R, Zhu L, Xue L et al (2020) Clinical findings of patients with coronavirus disease 2019 in Jiangsu province, China: a retrospective, multi-center study. PLoS Negl Trop Dis 14:e0008280. https://doi.org/10.1371/journal.pntd.0008280
Halvatsiotis P, Kotanidou A, Tzannis K et al (2020) Demographic and clinical features of critically Ill patients with COVID-19 in Greece: the burden of diabetes and obesity. Diabetes Res Clin Pract 166:108331. https://doi.org/10.1016/j.diabres.2020.108331
Hajifathalian K, Kumar S, Newberry C et al (2020) Obesity is associated with worse outcomes in COVID-19: Analysis of Early Data From New York City. Obes (Silver Spring) 28:1606–1612. https://doi.org/10.1002/oby.22923
Hernández-Garduño E (2020) Obesity is the comorbidity more strongly associated for COVID-19 in Mexico. A case-control study. Obes Res Clin Pract 14:375–379. https://doi.org/10.1016/j.orcp.2020.06.001
Chiumello D, Pozzi T, Storti E et al (2020) Body Mass Index and ARDS severity in patients with and without SARS-CoV-2 infection. Br J Anaesth 125:e376–e377. https://doi.org/10.1016/j.bja.2020.07.006
Carrillo-Vega MF, Salinas-Escudero G, Garcia-Peña C (2020) Early estimation of the risk factors for hospitalisation and mortality by COVID-19 in Mexico. PLoS ONE 5(9):e0238905. https://doi.org/10.1371/journal.pone.0238905
Bello-Chavolla OY, Bahena-López JP, Antonio-Villa NE (2020) Predicting mortality due to SARS-CoV-2: a mechanistic score relating obesity and diabetes to COVID-19 outcomes in Mexico. J Clin Endocrinol Metab 105:dgaa346. https://doi.org/10.1210/clinem/dgaa346
Busetto L, Bettini S, Fabris R et al (2020) Obesity and COVID-19: an Italian snapshot. Obesity 28:1600–1605. https://doi.org/10.1002/oby.22918
Ebinger JE, Achamallah N, Ji H et al (2020) Pre-existing traits associated with COVID-19 illness severity. PLoS ONE 2020(15):e0236240
Lighter J, Phillips M, Hochman S et al (2020) Obesity in patients younger than 60 years is a risk factor for COVID-19 hospital admission. Clin Infect Dis 71:896–897. https://doi.org/10.1093/cid/ciaa415
Müssig K (2020) Obesity favors a severe course of COVID-19. Info Diabetol 14:12–13. https://doi.org/10.1007/s15034-020-2135-9
Palaiodimos L, Kokkinidis DG, Li W et al (2020) Severe obesity, increasing age and male sex are independently associated with worse in-hospital outcomes, and higher in-hospital mortality, in a cohort of patients with COVID-19 in the Bronx, New York. Metabolism 108:154262. https://doi.org/10.1016/j.metabol.2020.154262
Petersen A, Bressem K, Albrecht J et al (2020) The role of visceral adiposity in the severity of COVID-19: Highlights from a unicenter cross-sectional pilot study in Germany. Metab Clin Exp 110:154317. https://doi.org/10.1016/j.metabol.2020.154317
Simonnet A, Chetboun M, Poissy J et al (2020) High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Requiring Invasive Mechanical Ventilation. Obesity (Silver Spring) 28:1195–1199
Squalli J (2020) Evaluating the determinants of COVID-19 mortality: a cross-country study. medRxiv 2020
Urra JM, Cabrera CM, Porras L et al (2020) Selective CD8 cell reduction by SARS-CoV-2 is associated with a worse prognosis and systemic inflammation in COVID-19 patients. Clin Immunol 217:108486. https://doi.org/10.1016/j.clim.2020.108486
Docherty AB, Harrison EM, Green CA et al (2020) Features of 20, 133 UK patients in hospital with COVID-19 using the ISARICWHO Clinical Characterisation Protocol: prospective observational cohort study. BMJ 369:m1985. https://doi.org/10.1136/bmj.m1985
Caci G, Albini A, Malerba M et al (2020) COVID-19 and obesity: dangerous liaisons. J Clin Med 9:2511. https://doi.org/10.3390/jcm9082511
De Siqueira JVV, Almeida LG, Zica BO et al (2020) Impact of obesity on hospitalizations and mortality, due to COVID-19: a systematic review. Obesity Res Clin Pract 14:398–403. https://doi.org/10.1016/j.orcp.2020.07.005
Ritter A, Kries N-N, Louwen F, Yuan J (2020) Obesity and COVID-19; molecular mechanisms Linking both pandemics. Int J Mol Sci 21:5793. https://doi.org/10.3390/ijms21165793
Rychter AM, Zawada A, Ratajczak AE et al (2020) Should patients with obesity be more afraid of COVID-19? Obes Rev 21:e13083. https://doi.org/10.1111/obr.13083
Caussy C, Pattou F, Wallet F et al (2020) Prevalence of obesity among adult inpatients with COVID-19 in France. Lancet Diabetes Endocrinol 8:562–564. https://doi.org/10.1016/S2213-8587(20)30160-1
Kalligeros M, Shehadeh F, Mylona EK et al (2020) Association of obesity with disease severity among patients with covid19. Obesity 28:1200–1204. https://doi.org/10.1002/oby.22859
Hu X, Pan X, Wei W et al (2020) Clinical epidemiological analyses of overweight/obesity and abnormal liver function contributing to prolonged hospitalization in patients infected with COVID-19. Int J Obes. 44:1784–1789. https://doi.org/10.1038/s41366-020-0634-3
Hamer M, Kivimaki M, Gale CR et al (2020) Lifestyle risk factors, inflammatory mechanisms and covid19 hospitalisation; a community based cohort study of 387109 adults in UK. Brain Behav Immunity 87:184–187. https://doi.org/10.1016/j.bbi.2020.05.059
Anderson MR, Geleris J, Anderson DR et al (2020) Body Mass Index and risk for intubation or death in SARS-CoV-2 infection: a retrospective cohort study. Ann Intern Med 173:782–790. https://doi.org/10.7326/M20-3214
Al-Sabah S, Al-Haddad M, Al-Youha S et al (2020) COVID-19: Impact of obesity and diabetes on disease severity. Clin Obes 10:e12414. https://doi.org/10.1111/cob.12414
Goyal P, Ringel JB, Rajan M et al (2020) Obesity and COVID-19 in New York City: a retrospective cohort study. Ann Intern Med 173:855–858. https://doi.org/10.7326/M20-2730
Kass DA, Duggal P, Cingolani O (2020) Obesity could shift severe COVID-19 disease to younger ages. Lancet 395(10236):1544–1545. https://doi.org/10.1016/S0140-6736(20)31024-2
de Lusignan S, Doward J, Correa A et al (2020) Risk factors for SARS CoV2 among patients in the Oxford Royal college of General Pactioner. Lancet Infect Dis 20:1034–1042. https://doi.org/10.1016/s1473-3099(20)30371-6
Malik P, Patel U, Patel, K et al (2020) Obesity a predictor of outcomes of COVID-19 hospitalised patients—a systematic review and meta analysis. Med Virol. Doi;: https://doi.org/10.1002/jmv.26555
Nakeshbandi M, Maini R, Daniel P et al (2020) The impact of obesity on COVID-19 complications: a retrospective cohort study. Int J Obes 44:1832–1837. https://doi.org/10.1038/s41366-020-0648-x
Petrilli CM, Jones SA, Yang J et al (2020) Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ 369:m1966. https://doi.org/10.1136/bmj.m1966
Popkin BM, Shufa D, Green WD et al (2020) Individuals with obesity and COVID-19; a global perspective on the epidemiology and biological relationships. Obesity Rev 21:e13128. https://doi.org/10.1111/obr.13128
Al Heialy S, Hachim MY, Hachim IY et al (2021) Combination of obesity and co-morbidities leads to unfavorable outcomes in COVID-19 patients. Saudi J Biol Sci 28:1445–1450
Williamson EJ, Walker AJ, Bhaskaran K et al (2020) Factors associated with COVID-19-related death using OpenSAFELY. Nature 584:430–436. https://doi.org/10.1038/s41586-020-2521-4
Yates T, Razieh C, Zaccardi F et al (2020) Obesity and risk of COVID-19: analysis of UK biobank. Prim Care Diabetes 14:566–567. https://doi.org/10.1016/j.pcd.2020.05.011
Breland JY, Wong MS, Steers WN et al (2021).BMI and risk for severe COVID-19 among veterans health administration patients. Obesity (SilverSpring) 29:825–828. https://doi.org/10.1002/oby.23121.
Eastment MC, Berry K, Locke E et al (2020) BMI and outcomes of SARS-CoV-2 among US Veterans. Obesity (Silver Spring) 29:900–908. https://doi.org/10.1002/oby.23111
Recalde M, Pistillo A, Fernandez-Bertolin S et al (2020a) BMI and risk of COVID-19 diagnosis, hospitalisation and death; a population based multi state cohort analysis including 2524926people in catalonia spain. medRxiv preprint. https://doi.org/10.101101/2020.11.25.20237776
Neeland IJ, Ross R, Després J-P et al (2019) Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: a position statement. Lancet Diabetes Endocrinol 7(9):715–725. https://doi.org/10.1016/S2213-8587(19)30084-1
Földi M, Farkas N, Kiss S et al (2021) Visceral adiposity elevates the risk of critical condition in COVID-19: a systematic review and meta-analysis. Obesity 29:521–528. https://doi.org/10.1002/oby.23096
Huang Y, Yao L, Huang Y-M et al (2020) Obesity in patients with COVID-19: a systematic review and meta-analysis. Metabolism 113:154378. https://doi.org/10.1016/j.metabol.2020.154378
Watanabe M, Caruso D, Tuccinardi D et al (2020) Visceral fat shows the strongest association with the need of intensive care in patients with COVID-19. Metabolism 111:154319. https://doi.org/10.1016/j.metabol.2020.154319
Yang Y, Ding L, Zou X et al (2020) Visceral adiposity and high intramuscular fat deposition independently predict critical illness in patients with SARS-CoV-2. Obesity (Silver Spring) 28:2040–2048. https://doi.org/10.1002/oby.22971
Chandarana H, Dane B, Mikheev A et al (2020) Visceral adipose tissue in patients with COVID-19:risk stratification for severity. Abdom Radiol (NY). https://doi.org/10.1007/s00261-020-02693-2
Favre G, Legueult K, Pradier C et al (2021) Visceral fat is associated to the severity of COVID-19. Metabolism 115:154440. https://doi.org/10.1016/j.metabol.2020.154440
Battisti S, Pedone C, Napoli N et al (2020) Computed tomography highlights increased visceral adiposity associated with critical illness in COVID-19. Diabetes Care 43:e129–e130. https://doi.org/10.2337/dc20-1333
Grodecki K, Lin A, Razipour A et al (2021) Epicardial adipose tissue is associated with extent of pneumonia and adverse outcomes in patients with COVID-19. Metabolism 115:154436. https://doi.org/10.1016/j.metabol.2020.154436
Wei ZY, Qiao R, Chen J et al (2021) Pre-existing health conditions and epicardial adipose tissue volume: potential risk factors for myocardial injury in COVID-19 patients. Front Cardiovasc Med 7:585220. https://doi.org/10.3389/fcvm.2020.585220
Peters SAE, MacMahon S, Woodward M (2021) Obesity as a risk factor for COVID-19 mortality in women and men in the UK biobank: comparisons with influenza/pneumonia and coronary heart disease. Diabetes Obes Metab 23:258–262. https://doi.org/10.1111/dom.14199
Hamer M, Gale CR, Kivimaki M et al (2020) Overweight, obesity and risk of hospitalisation for covid19; a community based cohort study of adults in the UK. Proc Natl Acad Sci USA 117:21011–21013. https://doi.org/10.1073/pnas.2011086117
Tartof SY, Qian L, Hong V et al (2020) Obesity and mortality among patients diagnosed with COVID-19: results from an integrated health care organization. Ann Intern Med 173:773–781. https://doi.org/10.7326/M20-3742
Sattar N, McInnes IB, McMurray JV (2020) Obesity is a risk factor for severe COVID-19 infection: multiple potential mechanisms. Circulation 142(1):4–6. https://doi.org/10.1161/CIRCULATIONAHA.120.04765920
Hendren NS, de Lemos JA, Ayers C et al (2021) Association of body mass index and age with morbidity and mortality in patients hospitalized with COVID-19. Circulation 143:135–144. https://doi.org/10.1161/CIRCULATIONAHA.120.051936
Recalde M, Roel E, Pistillo A et al (2020b) Characteristics and outcomes of 627 044 COVID-19 patients with and without obesity in the United States, Spain, and the United Kingdom. https://doi.org/10.1101/2020.09.02.20185173 Medrxiv (b)
Fresán U, Guevara M, Elía F et al (2021) Independent role of severe obesity as a risk factor for COVID-19 hospitalization: a Spanish Population-Based Cohort Study. Obesity 29:29–37. https://doi.org/10.1002/oby.23029
Klang E, Kassim G, Soffer S et al (2020) Severe obesity as an independent risk factor for COVID-19 mortality in hospitalized patients younger than 50. Obesity (Silver Spring) 28:1595–1599. https://doi.org/10.1002/oby.22913
Peña JE, Rascón-Pacheco RA, Ascencio-Montiel IJ et al (2020) Hypertension, diabetes and obesity, major risk factors for death in patients with COVID-19 in Mexico. Arch Med Res 16(S0188–4409):32243–32248. https://doi.org/10.1016/j.arcmed.2020.12.002
Gao M, Piernas C, Astbury NM et al (2021) Associations between body-mass index and COVID-19 severity in 6·9 million people in England: a prospective, community-based, cohort study. Lancet 9(6):350–359. https://doi.org/10.1016/S2213-8587(21)00089-9
Yates T, Zaccardi F, Islam N et al (2021) Obesity, ethnicity, and risk of critical care, mechanical ventilation, and mortality in patients admitted to hospital with COVID-19: analysis of the ISARIC CCP-UK Cohort. Obesity (Silver Spring, Md.) 29(7):1223–1230. https://doi.org/10.1002/oby.23178
Coleman P, Barber TM, van Rens T et al (2021) COVID-19 outcomes in minority ethnic groups: do obesity and metabolic risk play a role? Curr Obesity Rep. https://doi.org/10.1007/s13679-021-00459-5
Garg S, Kim L, Whitaker M et al (2020) Hospitalization rates and characteristics of patients hospitalized with laboratory-confirmed coronavirus disease 2019—COVID-NET, 14 States, March 1–30, 2020. CDC Report Morb Mortal Wkly Rep (MMWR) 69:458–464
Zachariah P, Johnson CL, Halabi KC et al (2020) Epidemiology, clinical features, and disease severity in patients with coronavirus disease (COVID-19) in a Children’s Hospital in New York City, New York. JAMA Pediatrics. https://doi.org/10.1001/jamapediatrics.2020.2430
Walls AC, Park Y-J, Tortorici MA et al (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181(2):281-292.e6. https://doi.org/10.1016/j.cell.2020.02.058
Tai W, He L, Zhang X et al (2020) Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol 17:613–620. https://doi.org/10.1038/s41423-020-0400-4
Ou X, Liu Y, Lei X, Li P et al (2020) Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 11:1620. https://doi.org/10.1038/s41467-020-15562-9
Lei C, Qian K, Li T et al (2020) Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat Commun 11:2070. https://doi.org/10.1038/s41467-020-16048-4
Blanco-Melo D, Nilsson-Payant BE, Liu WC et al (2020) Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181(5):1036–1045. https://doi.org/10.1016/j.cell.2020.04.026
Yan R, Zhang Y, Li Y et al (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367(6485):1444–1448. https://doi.org/10.1126/science.abb2762
Cuervo NZ, Grandvaux N (2020) ACE2: evidence of role as entry receptor for SARS-CoV-2 and implications in comorbidities. eLife 9:e61390. https://doi.org/10.7554/eLife.61390
Liu R, Yi S, Zhang J et al (2020) Viral load dynamics in sputum and nasopharyngeal swab in patients with COVID-19. J Dent Res 99:1239–1244. https://doi.org/10.1177/0022034520946251
Liu Y, Yan L-M, Wan L et al (2020) Viral dynamics in mild and severe cases of COVID-19. Lancet Infect Dis 20:656–657. https://doi.org/10.1016/S1473-3099(20)30232-2
Magleby R, Westblade LF, Trzebucki A et al (2020) Impact of SARS-CoV-2 viral load on risk of intubation and mortality among hospitalized patients with coronavirus disease 2019. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa851
Maltezou HC, Raftopoulos V, Vorou R et al (2021) Association between upper respiratory tract viral load, comorbidities, disease severity and outcome of patients with SARS-CoV-2 infection. J Infect Dis 223:1132–1138. https://doi.org/10.1093/infdis/jiaa804
Tsukagoshi H, Shinoda D, Saito M et al (2021) Relationships between viral load and the clinical course of COVID-19. Viruses 13(2):304. https://doi.org/10.3390/v13020304
Fajnzylber J, Regan J, Coxen K et al (2020) SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat Commun 11:5493. https://doi.org/10.1038/s41467-020-19057-5|
Argyropoulos KV, Serrano A, Hu J et al (2020) Association of Initial Viral Load in Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) patients with outcome and symptoms. Am J Pathol 190:1881–1887. https://doi.org/10.1016/j.ajpath.2020.07.001
Uhm J-S, Ahn JY, Hyun JH et al (2020) Patterns of viral clearance in the natural course of asymptomatic COVID-19: Comparison with symptomatic non-severe COVID-19 Int. J Infec dis 99:279–285
Maier HE, Lopez R, Sanchez N et al (2018) Obesity increases the duration of influenza A virus shedding in adults. J Infect Dis 218:1378–1382. https://doi.org/10.1093/infdis/jiy370
Yan J, Grantham M, Pantelic J et al (2018) EMIT Consortium. Infectious virus in exhaled breath of symptomatic seasonal influenza cases from a college community. Proc Natl Acad Sci USA 115:1081–1086. https://doi.org/10.1073/pnas.1716561115
Patel VB, Clarke N, Wang Z et al (2014) Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol 66:167–176. https://doi.org/10.1016/j.yjmcc.2013.11.017
Kruglikov IL, Scherer PE (2020) The role of adipocytes and adipocyte-like cells in the severity of COVID-19 infections. Obesity 28:1187–1190. https://doi.org/10.1002/oby.22856
Al-Benna S (2020) Association of high level gene expression of ACE2 in adipose tissue with mortality of COVID-19 infection in obese patients. Obes Med 19:100283. https://doi.org/10.1016/j.obmed.2020.100283
Zhou H, Chen X, Hu T et al (2020) A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr Biol 30(11):2196–2203. https://doi.org/10.1016/j.cub.2020.05.023
Coelho MS, Lopes KL, Freitas RA (2010) High sucrose intake in rats is associated with increased ACE2 and angiotensin-(1–7) levels in the adipose tissue. Regul Pept 162:61–67. https://doi.org/10.1016/j.regpep.2010.03.008
Zhang W, Xu Y-Z, Liu B et al (2014) Pioglitazone upregulates angiotensin converting enzyme 2 expression in insulin-sensitive tissues in rats with high-fat diet-induced nonalcoholic steatohepatitis. Sci World J 2014:id603409. https://doi.org/10.1155/2014/603409
Hernández-Díazcouder A, Romero-Nava R, Carbó R et al (2019) High fructose intake and adipogenesis. Int J Mol Sci 20:2787. https://doi.org/10.3390/ijms20112787
Al Heialy S, Hachim MY, Senok A et al (2020) Regulation of angiotensin- converting enzyme 2 in obesity: implications for COVID-19. Front Physiol 11:555039. https://doi.org/10.3389/fphys.2020.555039
Higham A, Singh D (2020) Increased ACE2 expression in bronchial epithelium of COPD patients who are overweight. Obesity 28:1586–1589. https://doi.org/10.1002/oby.22907
Kruglikov IL, Shah M, Scherer PE (2020) Obesity and diabetes as comorbidities for COVID-19: Underlying mechanisms and the role of viral-bacterial interactions. Elife 9:e61330. https://doi.org/10.7554/eLife.61330
Li L, Spranger L, Soll D et al (2020) Metabolic impact of weight loss induced reduction of adipose ACE-2—potential implication in COVID-19 infections? Metabolism 113:154401. https://doi.org/10.1016/j.metabol.2020.154401
Rao S, Lau A, So H-C (2020) Exploring diseases/traits and blood proteins causally related to expression of ACE2, the putative receptor of SARS-CoV-2: a mendelian randomization analysis highlights tentative relevance of diabetes-related traits. Diabetes Care 43:1416–1426. https://doi.org/10.2337/dc20-0643
Gilbert RE, Caldwell L, Misra PS et al (2021) Overexpression of the Severe Acute Respiratory Syndrome Coronavirus-2 Receptor, Angiotensin-Converting Enzyme 2, in Diabetic Kidney Disease: Implications for Kidney Injury in Novel Coronavirus Disease 2019. Can J Diabetes 45:162–166. https://doi.org/10.1016/j.jcjd.2020.07.003
Soldo J, Heni M, Königsrainer A et al (2020) Increased hepatic ACE2 expression in NAFL and diabetes—a risk for COVID-19 patients? Diabetes Care 43:134–136. https://doi.org/10.2337/dc20-1458
Pasquarelli-do-Nascimento G, Braz-de-Melo HA, Faria SS et al (2020) Hypercoagulopathy and adipose tissue exacerbated inflammation may explain higher mortality in COVID-19 patients with obesity. Front Endocrinol 11:530. https://doi.org/10.3389/fendo.2020.00530
Sarver DC, Wong GW (2021) Obesity alters Ace2 and Tmprss2 expression in lung, trachea, and esophagus in a sex-dependent manner: Implications for COVID-19. Biochem Biophys Res Commun 538:92–96. https://doi.org/10.1016/j.bbrc.2020.10.066
Cantuti-Castelvetri L, Ojha R, Pedro LD et al (2020) Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370:856–860. https://doi.org/10.1126/science.abd2985
Daly JL, Simonetti B, Klein K et al (2020) Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 370:861–865. https://doi.org/10.1126/science.abd3072
Wang K, Chen W, Zhang Z et al (2020) CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther 5(1):283. https://doi.org/10.1038/s41392-020-00426-x
Gupte M, Boustany-Kari CM, Bharadwaj K et al (2008) ACE2 is expressed in mouse adipocytes and regulated by a high-fat diet. Am J Physiol Regul Integr Comp Physiol 295:R781–R788. https://doi.org/10.1152/ajpregu.00183.2008
Uhlen M, Oksvold P, Fagerberg L et al (2010) Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 28:1248–1250. https://doi.org/10.1038/nbt1210-1248
Gupte M, Thatcher SE, Boustany-Kari CM et al (2012) Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol 32:1392–1399. https://doi.org/10.1161/ATVBAHA.112.248559
Ponterio E, Cangemi R, Mariani S et al (2015) Adenovirus 36 DNA in human adipose tissue. Int J Obes 39:1761–1764. https://doi.org/10.1038/ijo.2015.163
Ryan PM, Caplice NM (2020) Is adipose tissue a reservoir for viral spread, immune activation, and cytokine amplification in Coronavirus disease 2019? Obesity 28:1191–1194. https://doi.org/10.1002/oby.22843
Gu J, Korteweg C (2007) Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol 170:1136–1147. https://doi.org/10.2353/ajpath.2007.061088
Hennighausen L, Lee HK (2020) Activation of the SARS-CoV-2 receptor Ace2 by cytokines through JAK-STAT-dependent enhancers during pregnancy. Cell Rep 32:108199. https://doi.org/10.1016/j.celrep.2020.108199
Sajuthi SP, DeFord P, Li Y et al (2020) Type 2 and interferon inflammation regulate SARS-CoV-2 entry factor expression in the airway epithelium. Nat Commun 11:5139. https://doi.org/10.1038/s41467-020-18781-2
Patel VB, Basu R, Oudit GY (2016) ACE2/Ang 1–7 axis: a critical regulator of epicardial adipose tissue inflammation and cardiac dysfunction in obesity. Adipocyte 5:306–11. https://doi.org/10.1080/21623945.2015.1131881
Bourgeois C, Gorwood J, Barrail-Tran A et al (2019) Specific biological features of adipose tissue, and their impact on HIV persistence. Front Microbiol 10:2837. https://doi.org/10.3389/fmicb.2019.02837
Miyanari Y, Atsuzawa K, Usuda N et al (2007) The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9(9):1089–1097. https://doi.org/10.1038/ncb1631
Schurink B, Roos E, Radonic T et al (2020) Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe 1(7):e290–e299. https://doi.org/10.1016/S2666-5247(20)30144-0
Chen Y, Cai H, Pan J et al (2009) Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc Natl Acad Sci USA 106:3484–3489. https://doi.org/10.1073/pnas.0808790106
Nakagawa K, Lokugamage KG, Makino S (2016) Viral and cellular mRNA translation in coronavirus-infected cells. Adv Virus Res 96:165–192. https://doi.org/10.1016/bs.aivir.2016.08.001
Pain VM (1996) Initiation of protein synthesis in eukaryotic cells European. J Biochem 236:747–771. https://doi.org/10.1111/j.1432-1033.1996.00747.x
Pause A, Belsham GJ, Gingras AC et al (1994) Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5’-cap function. Nature 371(65):762–7. https://doi.org/10.1038/371762a0
Cencic R, Desforges M, Hall DR et al (2011) Blocking eIF4E-eIF4G interaction as a strategy to impair coronavirus replication. J Virol 85:6381–6389. https://doi.org/10.1128/JVI.00078-11
Bolourian A, Mojtahedi Z (2020) Obesity and COVID-19: The mTOR pathway as a possible culprit. Obesity Rev 21:1–3. https://doi.org/10.1111/obr.13084
Gulati P, Thomas G (2007) Nutrient sensing in the mTOR/S6K1 signalling pathway. Biochem Soc Trans 35:236–238. https://doi.org/10.1042/BST0350236
Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168:960–976. https://doi.org/10.1016/j.cell.2017.02.004
Jia G, Aroor AR, Martinez-Lemus LA et al (2014) Overnutrition, mTOR signaling, and cardiovascular diseases. Am J Physiol Regul Integr Comp Physiol 307:R1198–R1206. https://doi.org/10.1152/ajpregu.00262.2014
Dunlop EA, Tee AR (2009) Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cell Signal 21:827–835. https://doi.org/10.1016/j.cellsig.2009.01.012
Laplante M, Sabatini DM (2009) mTOR signaling at a glance. J Cell Sci 122:3589–3594
Dann SG, Selvaraj A, Thomas G (2007) mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med 13:252–259. https://doi.org/10.1016/j.molmed.2007.04.002
Dowling RJO, Topisirovic I, Fonseca BD et al (2010) Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta 1804:433–439. https://doi.org/10.1016/j.bbapap.2009.12.001
Fasolo A, Sessa C (2012) Targeting mTOR pathways in human malignancies. Curr Pharm Des 18:2766–2777. https://doi.org/10.2174/138161212800626210
Cornu M, Albert V, Hall MN (2013) mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev 23:53–62. https://doi.org/10.1016/j.gde.2012.12.005
Cai H, Dong L, Liu F (2016) Recent advances in adipose mTOR signaling and function: therapeutic prospects. Trends Pharmacol Sci 37:303–317. https://doi.org/10.1016/j.tips.2015.11.011
Khamzina L, Veilleux A, Bergeron S et al (2005) Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 146:1473–1481. https://doi.org/10.1210/en.2004-0921
Tavares MR, Pavan IC, Amaral CL et al (2015) The S6K protein family in health and disease. Life Sci 131:1–10. https://doi.org/10.1016/j.lfs.2015.03.001
Catalán V, Gómez-Ambrosi J, Rodríguez A et al (2015) Expression of S6K1 in human visceral adipose tissue is upregulated in obesity and related to insulin resistance and inflammation. Acta Diabetol 52:257–266. https://doi.org/10.1007/s00592-014-0632-9
Mao Z, Zhang W (2018) Role of mTOR in glucose and lipid metabolism. Int J Mol Sci 19(7):2043. https://doi.org/10.3390/ijms19072043
Lastres-Becker I, Nonis D, Eich F, Klinkenberg M et al (2016) Mammalian ataxin-2 modulates translation control at the pre-initiation complex via PI3K/mTOR and is induced by starvation. Biochim Biophys Acta. 1862:1558–69. https://doi.org/10.1016/j.bbadis.2016.05.017
Lee D-F, Kuo H-P, Chen C-T et al (2007) IKKβ suppression ofTSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130:440–455. https://doi.org/10.1016/j.cell.2007.05.058
Yoo M-S (2017) The role of mammalian target of rapamycin (mTOR) in insulin signaling. Nutrients 9(11):1176. https://doi.org/10.3390/nu9111176
Appelberg S, Gupta S, Svensson Akusjärvi S et al (2020) Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg Microbes Infect 9:1748–1760. https://doi.org/10.1080/22221751.2020.1799723
Kim JA, Jang HJ, Martinez-Lemus LA et al (2012) Activation of mTOR/p70S6 kinase by ANG II inhibits insulin-stimulated endothelial nitric oxide synthase and vasodilation. Am J Physiol Endocrinol Metab 302:E201–E208. https://doi.org/10.1152/ajpendo.00497.2011
Mullen PJ, Garcia G, Purkayastha A et al (2021) SARS-CoV-2 infection rewires host cell metabolism and is potentially susceptible to mTORC1 inhibition. Nat Commun 12:1876. https://doi.org/10.1038/s41467-021-22166-4
Szegezdi E, Logue SE, Gorman AM et al (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7:880–885. https://doi.org/10.1038/sj.embor.7400779
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529. https://doi.org/10.1038/nrm2199
Tabas I, Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13:184–190. https://doi.org/10.1038/ncb0311-184
Almanza A, Carlesso A, Chintha C et al (2019) Endoplasmic reticulum stress signalling—from basic mechanisms to clinical applications. FEBS J 286:241–278. https://doi.org/10.1111/febs.14608
Fung TF, Liu DX (2014) Coronavirus infection ER stress, apoptosis and innate immunity. Front Microbiol 5:296. https://doi.org/10.3389/fmicb.2014.00296
Fung TS, Liao Y, Liu DX (2016) Regulation of stress responses and translational control by Coronavirus. Viruses 8:184. https://doi.org/10.3390/v8070184
Fung TF, Liu DX (2019) The ER stress sensor IRE1 and MAP kinase ERK modulate autophagy induction in cells infected with coronavirus infectious bronchitis virus. Virology 533:34–44. https://doi.org/10.1016/j.virol.2019.05.00
Schubert K, Karousis ED, Jomaa A et al (2020) SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol 27:959–966. https://doi.org/10.1038/s41594-020-0511-8.Erratum.In:NatStructMolBiol.202027:1094
Thomas M, Buschauer R, Ameismeier M et al (2020) Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 369(6508):1249–1255. https://doi.org/10.1126/science.abc8665
Huang C, Lokugamage KG, Rozovics JM et al (2011) SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog 7(12):e1002433. https://doi.org/10.1371/journal.ppat.1002433
Tanaka T, Kamitani W, DeDiego ML et al (2012) Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J Virol 86(20):11128–11137. https://doi.org/10.1128/JVI.01700-12
Banerjee AK, Blanco MR, Bruce EA et al (2020) SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183:1325–1339. https://doi.org/10.1016/j.cell.2020.10.004
Bojkova D, Klann K, Koch B et al (2020) Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583:469–472. https://doi.org/10.1038/s41586-020-2332-7
Köseler A, Sabirli R, Gören T et al (2020) Endoplasmic reticulum stress markers in SARS-COV-2 infection and pneumonia: case-control study. In Vivo 34 (3 suppl):1645–1650
Ojha CR, Rodriguez M, Lapierre J et al (2018) Complementary mechanisms potentially involved in the pathology of Zika Virus. Front Immunol 9:2340. https://doi.org/10.3389/fimmu.2018.02340
Triantafilou K, Triantafilou M (2003) Lipid raft microdomains: key sites for Coxsackievirus A9 infectious cycle. Virology 317:128–135. https://doi.org/10.1016/j.virol.2003.08.036
**dadamrongwech S, Thepparit C, Smith DR (2004) Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch Virol 149(5):915–927. https://doi.org/10.1007/s00705-003-0263-x
Nain M, Mukherjee S, Karmakar SP et al (2017) GRP78 is an important host factor for Japanese Encephalitis Virus Entry and replication in mammalian cells. J Virol 91(6):e02274-e2316. https://doi.org/10.1128/JVI.02274-16
Capitani M, Sallese M (2009) The KDEL receptor: new functions for an old protein. FEBS Lett 583:3863–3871. https://doi.org/10.1016/j.febslet.2009.10.053
Allam L, Ghrifi F, Mohammed H et al (2020) Targeting the GRP78-dependant SARS-CoV-2 cell entry by peptides and small molecules. Bioinform Biol Insights 14:1177932220965505. https://doi.org/10.1177/1177932220965505
Girona J, Rodríguez-Borjabad C, Ibarretxe D et al (2019) The circulating GRP78/BiP is a marker of metabolic diseases and atherosclerosis: bringing endoplasmic reticulum stress into the clinical scenario. J Clin Med 8:1793. https://doi.org/10.3390/jcm8111793
Chen Y, Wu Z, Zhao S et al (2016) Chemical chaperones reduce ER stress and adipose tissue inflammation in high fat diet-induced mouse model of obesity. Sci Rep 6:27486. https://doi.org/10.1038/srep27486obesity
Forrester SJ, Booz GW, Sigmund CD et al (2018) Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev 298:1627–1738. https://doi.org/10.1152/physrev.00038.2017
Zhang K, Kaufman RJ (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454(7203):455–462. https://doi.org/10.1038/nature07203
Cao SS, Luo KL, Shi L (2016) Endoplasmic reticulum stress interacts with inflammation in human diseases. J Cell Physiol 231:288–294. https://doi.org/10.1002/jcp.25098
Uhal BD, Nguyen H, Dang M et al (2013) Abrogation of ER stress-induced apoptosis of alveolar epithelial cells by angiotensin 1–7. Am J Physiol Lung Cell Mol Physiol 305(1):L33–L41. https://doi.org/10.1152/ajplung.00001.2013
Zhang M, Gao Y, Zhao W et al (2018) ACE-2/ANG1-7 ameliorates ER stress-induced apoptosis in seawater aspiration-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 315(6):L1015–L1027. https://doi.org/10.1152/ajplung.00163.2018
Peters U, Dixon AE (2018) The effect of obesity on lung function. Exp Rev Resp Med 12:755–767. https://doi.org/10.1080/17476348.2018.1506331
Dietz W, Santos-Burgoa C (2020) Obesity and its implications for COVID-19 mortality. Obesity 28:1005. https://doi.org/10.1002/oby.22818
Kuba K, Imai Y, Penninger JM (2006) Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol 6:271–276. https://doi.org/10.1016/j.coph.2006.03.001
Fleming I (2006) Signaling by the angiotensin-converting enzyme. Circ Res 98(7):887–896. https://doi.org/10.1161/01.RES.0000217340.40936.53
Tikellis C, Thomas MC (2012) Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept. https://doi.org/10.1155/2012/256294
Muñoz-Durango N, Fuentes CA, Castillo AE et al (2016) Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int J Mol Sci 17(7):797. https://doi.org/10.3390/ijms17070797
Colafella KMM, Bovée DM, Danser AHJ (2019) The renin-angiotensin-aldosterone system and its therapeutic targets. Exp Eye Res 186:107680. https://doi.org/10.1016/j.exer.2019.05.020
Wang K, Gheblawi M, Oudit GY (2020) Angiotensin converting enzyme-2: a double-edged-sword. Circulation 142:426–428. https://doi.org/10.1161/CIRCULATIONAHA.120.047049
Chung MK, Karnik S, Saef J et al (2020) SARS-CoV-2 and ACE2:The biology and clinical data settling the ARB and ACEI controversy. EBioMedicine (The Lancet) 58:102907. https://doi.org/10.1016/j.ebiom.2020.102907
Van deVeerdonk FL, Netea MG, van Deuren M et al (2020) Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. ELife 9:e57555. https://doi.org/10.7554/eLife.57555
Jugdutt BI (2015) Expanding saga of the renin-angiotensin system: the Angiotensin II counter-regulatory AT2 receptor pathway. Circulation 131(16):1380–1383. https://doi.org/10.1161/CIRCULATIONAHA.115.016328
Patel S, Rauf A, Khan H et al (2017) Renin-angiotensin-aldosterone (RAAS): the ubiquitous system for homeostasis and pathologies. Biomed Pharmacother 94:317–325. https://doi.org/10.1016/j.biopha.2017.07.091
Hamming I, Timens W, Bulthuis MLC et al (2003) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203:631–637. https://doi.org/10.1002/path.1570
Hikmet F, Méar L, Edvinsson A et al (2020) The protein expression profile of ACE2 in human tissues. Mol Syst Biol 16(7):e9610. https://doi.org/10.15252/msb.20209610
Cooper R, McFarlane-Anderson N, Bennett FI et al (1997) ACE, angiotensinogen and obesity: a potential pathway leading to hypertension. J Hum Hypertens 11:107–111. https://doi.org/10.1038/sj.jhh.1000391
Segura J, Ruilope LM (2007) Obesity, essential hypertension and renin-angiotensin system. Public Health Nutr 10:1151–1155. https://doi.org/10.1017/S136898000700064X
Cabandugama PK, Gardner MJ, Sowers JR (2017) The renin angiotensin aldosterone system in obesity and hypertension: roles in the cardiorenal metabolic syndrome. Med Clin N Am 101:129–137. https://doi.org/10.1016/j.mcna.2016.08.009
Wu Z, Hu R, Zhang C et al (2020) Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit Care 24(1):290. https://doi.org/10.1186/s13054-020-03015-0
Yu X, Sun S, Shi Y et al (2020) SARS-CoV-2 viral load in sputum correlates with risk of COVID-19 progression. Critical Care. 24:Article No. 170. https://doi.org/10.1186/s13054-020-02893-8
Liu Y, Yang Y, Zhang C et al (2020) Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci 63:364–374. https://doi.org/10.1007/s11427-020-1643-8
Deng J, Wang DX, Deng W, Li CY, Tong J (2012) The effect of endogenous angiotensin II on alveolar fluid clearance in rats with acute lung injury. Can Respir J 19(5):311–318. https://doi.org/10.1155/2012/951025
Larsson PT, Schwieler JH, Wallén NH (2000) Platelet activation during angiotensin II infusion in healthy volunteers. Blood Coagul Fibrinolysis 11:61–69
Kuba K, Imai Y, Rao S et al (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 11:875–879. https://doi.org/10.1038/nm1267
Gheblawi M, Wang K, Viveiros A et al (2020) Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ Res 126:1456–1474. https://doi.org/10.1161/CIRCRESAHA.120.317015t
Zipeto D, Palmeira JdF, Argañaraz GA et al (2020) ACE2/ADAM17/TMPRSS2 interplay may be the main risk factor for COVID-19. Front Immunol 11:576745. https://doi.org/10.3389/fimmu.2020.576745
Heurich A, Hofmann-Winkler H, Gierer S et al (2014) TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol 88:1293–307. https://doi.org/10.1128/JVI.02202-13
Patel SK, Juno JA, Lee WS et al (2021) Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: implications for COVID-19 pathogenesis and consequences. Eur Respir J. https://doi.org/10.1183/13993003.03730-2020
Sun J, Ye F, Wu A et al (2020) Comparative transcriptome analysis reveals the intensive early stage responses of host cells to SARS-CoV-2 infection. Front Microbiol 11:593857. https://doi.org/10.3389/fmicb.2020.593857
Perrotta F, Matera MG, Cazzola M et al (2020) Severe respiratory SARS-CoV2 infection: Does ACE2 receptor matter? Respir Med 168:105996. https://doi.org/10.1016/j.rmed.2020.105996
Ferrario CM, Jessup J, Chappell MC et al (2005) Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 111:2605–2610. https://doi.org/10.1161/CIRCULATIONAHA.104.510461
Li W, Moore M, Vasilieva N et al (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. https://doi.org/10.1038/nature02145
Salem ES, Grobe N, Elased KM (2014) Insulin treatment attenuates renal ADAM17 and ACE2 shedding in diabetic Akita mice. Am J Phy Renal physiology 306:F629–F639. https://doi.org/10.1152/ajprenal.00516.2013
Romaní-Pérez M, Outeiriño-Iglesias V, Moya CM et al (2015) Activation of the GLP-1 receptor by liraglutide increases ACE2 expression, reversing right ventricle hypertrophy, and improving the production of SP-A and SP-B in the lungs of type 1 diabetes rats. Endocrinology 156:3559–69. https://doi.org/10.1210/en.2014-1685
Whaley-Connell AT, Habibi J, Nistala R et al (2012) Mineralocorticoid receptor-dependent proximal tubule injury is mediated by a redox-sensitive mTOR/S6K1 pathway. Am J Nephrol 35:90–100
Shimada S, Yang C, Kumar V et al (2020) Angiotensin II activates mTORC1 pathway in the kidneys through a pressor-independent mechanism. FASEB J 34:11. https://doi.org/10.1096/fasebj.2020.34.s1.02210
Ritter JB, Wahl AS, Freund S et al (2010) Metabolic effects of influenza virus infection in cultured animal cells: Intra- and extracellular metabolite profiling BMC Sys. Biol 4:61. https://doi.org/10.1186/1752-0509-4-61
Ohno M, Sekiya T, Nomura N et al (2020) Influenza virus infection affects insulin signaling, fatty acid-metabolizing enzyme expressions, and the tricarboxylic acid cycle in mice. Sci Rep 10:10879. https://doi.org/10.1038/s41598-020-67879-6
Keshvarz M, Solaymani-Mohammadi F, Namdari H (2020) Metabolic host response and therapeutic approaches to influenza infection. Cell Mol Biol Lett 25:15. https://doi.org/10.1186/s11658-020-00211-2
Codo AC, Davanzo GG, Monteiro LB et al (2020) Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab 32(3):437–446. https://doi.org/10.1016/j.cmet.2020.07.007.Erratum.In:CellMetab.32:498-499
Wu D, Shu T, Yang X et al (2020) Plasma metabolomic and lipidomic alterations associated with COVID-19. Natl Sci Rev. https://doi.org/10.1093/nsr/nwaa086
Pang Z, Zhou G, Chong J, **a J (2021) Comprehensive meta-analysis of COVID-19 global metabolomics datasets. Metabolites 11:44. https://doi.org/10.3390/metabo11010044
Zhu L, She ZG, Cheng X et al (2020) Association of blood glucose control and outcomes in patients with COVID-19 and pre-existing type 2 diabetes. Cell Metab 31(6):1068–1077. https://doi.org/10.1016/j.cmet.2020.04.021
Šestan M, Marinović S, Kavazović I et al (2018) Virus-induced interferon-γ causes insulin resistance in skeletal muscle and derails glycemic control in obesity. Immunity 49:164–177. https://doi.org/10.1016/j.immuni.2018.05.005
Harper SA, Bradley JS, Englund JA, et al. (2009) Seasonal influenza in adults and children–diagnosis, treatment, chemoprophylaxis, and institutional outbreak management: clinical practice guidelines of the Infectious Diseases Society of America. External icon Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 48(8):1003–1032. https://doi.org/10.1086/598513.
Chen J, Chunhua WC, Wang X et al (2020) The impact of COVID-19 on blood glucose: a systematic review and meta-analysis. Front Endocrinol 11:574. https://doi.org/10.3389/fendo.2020.574541541
Carrasco-Sánchez FJ, López-Carmona MD, Martínez-Marcos FJ et al (2021) Admission hyperglycaemia as a predictor of mortality in patients hospitalized with COVID-19 regardless of diabetes status: data from the Spanish SEMI-COVID-19 Registry. Ann Med 53:103–116. https://doi.org/10.1080/07853890.2020.1836566
Müller JA, Groß R, Conzelmann C et al (2021) SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nat Metab 3(2):149–165. https://doi.org/10.1038/s42255-021-00347-1
Willson C, Watanabe M, Tsuji-Hosokawa A et al (2019) Pulmonary vascular dysfunction in metabolic syndrome. J Physiol 597:1121–1141. https://doi.org/10.1113/JP275856
Song J-W, Lam SM, Fan X et al (2020) Omics-driven systems interrogation of metabolic dysregulation in COVID-19 pathogenesis. Cell Metab 32:188–202. https://doi.org/10.1016/j.cmet.2020.06.016
Moolamalaa STR, Chauhan R, Priyakumar UD et al (2020) Host metabolic reprogramming in response to SARS-Cov-2. bioRxiv. https://doi.org/10.1101/2020.08.02.232645
Cariou PB, Hadjadj S, Wargny M et al (2020) Phenotypic characteristics and prognosis of inpatients with COVID-19 and diabetes: the CORONADO study. Diabetologia 63:1500–1515. https://doi.org/10.1007/s00125-020-05180-x
Bramante CT, Ingraham NE, Murray TA et al (2020) Observational study of metformin and risk of mortality in patients hospitalized with COVID-19. Preprint. medRxiv. 2020.06.19.20135095. https://doi.org/10.1101/2020.06.19.20135095
Singh AK, Singh R (2020) Is metformin ahead in the race as a repurposed host-directed therapy for patients with diabetes and COVID-19? Diabetes Res Clin Pract 165:108268. https://doi.org/10.1016/j.diabres.2020.108268
de las Heras N, Giménez VMM, Ferder L et al (2020) Implications of oxidative stress and potential role of mitochondrial dysfunction in COVID-19: therapeutic effects of Vitamin D. Antioxidants 9:897. https://doi.org/10.3390/antiox9090897
Shen B, Yi X, Sun Y et al (2020) Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 182(1):59-72.e15. https://doi.org/10.1016/j.cell.2020.05.032
Weiler Miralles CS, Wollinger LM, Marin D et al (2015) Waist-to-height ratio (WHtR) and triglyceride to HDL-C ratio (TG/HDL-c) as predictors of cardiometabolic risk. Nutr Hosp 31:2115–2121. https://doi.org/10.3305/nh.2015.31.5.7773
Marfia G, Navone S, Guarnaccia L et al (2021) Decreased serum level of sphingosine-1-phosphate: a novel predictor of clinical severity in COVID-19. EMBO Mol Med 13:424. https://doi.org/10.15252/emmm.202013424
Chew WS, Torta F, Ji S et al (2019) Large-scale lipidomics identifies associations between plasma sphingolipids and T2DM incidence. JCI Insight 5:e126925. https://doi.org/10.1172/jci.insight.126925
O’Rourke RW, Lumeng CN (2021) Pathways to severe COVID-19 for people with obesity. Obesity 29:645–653. https://doi.org/10.1002/oby.23099
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. https://doi.org/10.1016/j.cell.2009.01.002
Ghildiyal M, Zamore PD (2009) Small silencing RNAs:an expanding universe. Nat Rev Genet 10:94–108. https://doi.org/10.1038/nrg2504
Ha M, Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15:509–524. https://doi.org/10.1038/nrm3838
Doench JG, Sharp PA (2004) Specificity of microRNA target selection in translational repression. Genes Dev 18:504–511. https://doi.org/10.1101/gad.1184404
Grimson A, Farh KK-H, Johnston WK et al (2007) MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27:91–105. https://doi.org/10.1016/j.molcel.2007.06.017airing
Rupaimoole R, Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 16:203–222. https://doi.org/10.1038/nrd.2016.246
Iacomino G, Siani A (2017) Role of microRNAs in obesity and obesity-related diseases. Genes Nutr 12:23. https://doi.org/10.1186/s12263-017-0577-z
Landrier J-F, Derghal A, Mounien L (2019) MicroRNAs in obesity and related metabolic disorders. Cells 8:859. https://doi.org/10.3390/cells8080859
Zhang J, Li S, Li L et al (2015) Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteom Bioinform 13:17–24. https://doi.org/10.1016/j.gpb.2015.02.001
Desdín-Micó G, Mittelbrunn M (2017) Role of exosomes in the protection of cellular homeostasis. Cell Adh Migr 11:127–134. https://doi.org/10.1080/19336918.2016.1251000
Bhome R, Vecchio FD, Lee G-H et al (2018) Exosomal microRNAs (exomiRs): small molecules with a big role in cancer. Cancer Lett 420:228–235. https://doi.org/10.1016/j.canlet.2018.02.002
Fernández-Hernando C, Ramírez CM, Goedeke L et al (2013) MicroRNAs in metabolic disease. Arterioscler Thromb Vasc Biol 33:178–185. https://doi.org/10.1161/ATVBAHA.112.300144
Deiuliis JA (2016) MicroRNAs as regulators of metabolic disease: pathophysiologic significance and emerging role as biomarkers and therapeutics. Int J Obes 40:88–101. https://doi.org/10.1038/ijo.2015.170
Jiang Q, Wang Y, Hao Y et al (2009) miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 37:D98-104. https://doi.org/10.1093/nar/gkn714
Dumortier O, Hinault C, Obberghen EV (2013) MicroRNAs and metabolism crosstalk in energy homeostasis. Cell Metab 18:312–324. https://doi.org/10.1016/j.cmet.2013.06.004
Heneghan HM, Miller N, McAnena OJ et al (2011) Differential miRNA expression in omental adipose tissue and in the circulation of obese patients identifies novel metabolic biomarkers. J Clin Endocrinol Metab 96:E846–E850. https://doi.org/10.1210/jc.2010-2701
Kristensen MM, Davidsen PK, Vigelso A et al (2017) miRNAs in human subcutaneous adipose tissue: effects of weight loss induced by hypocaloric diet and exercise. Obesity (Silver Spring) 25:572–580. https://doi.org/10.1002/oby.21765
Zhu H, Shyh-Chang N, Segrè AV et al (2011) The Lin28/let-7 axis regulates glucose metabolism. Cell 147:81–94. https://doi.org/10.1016/j.cell.2011.08.033
Sun T, Fu M, Bookout AL et al (2009) MicroRNA let-7 regulates 3T3-L1 adipogenesis. Mol Endocrinol 23:925–931. https://doi.org/10.1210/me.2008-0298
Fu X, Dong B, Tian Y et al (2015) MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J Clin Invest 125:2497–2509. https://doi.org/10.1172/JCI75438
Ahn J, Lee H, Jung CH et al (2013) MicroRNA-146b promotes adipogenesis by suppressing the SIRT1-FOXO1 cascade. EMBO Mol Med 5:1602–1612. https://doi.org/10.1002/emmm.201302647
Karkeni E, Astier J, Tourniaire F et al (2016) Obesity-associated inflammation induces microRNA-155 expression in adipocytes and adipose tissue: outcome on adipocyte function. J Clin Endocrinol Metab 101:1615–1626. https://doi.org/10.1210/jc.2015-3410
Wang P, Hou J, Lin L et al (2010) Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol 185(10):6226–6233. https://doi.org/10.4049/jimmunol.1000491
Mittelbrunn M, Gutiérrez-Vázquez C, Villarroya-Beltri C et al (2011) Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun 2:282. https://doi.org/10.1038/ncomms1285
Vickers KC, Palmisano BT, Shoucri BM et al (2011) MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol 13(4):423–433. https://doi.org/10.1038/ncb2210
Karolina DS, Tavintharan S, Armugam A et al (2012) Circulating miRNA profiles in patients with metabolic syndrome. J Clin Endocrinol Metab 97:E2271–E2276. https://doi.org/10.1210/jc.2012-1996
Zampetaki A, Kiechl S, Drozdov I et al (2010) Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res 107(6):810–817. https://doi.org/10.1161/CIRCRESAHA.110.226357
Zhang T, Lv C, Li L et al (2013) Plasma miR-126 is a potential biomarker for early prediction of type 2 diabetes mellitus in susceptible individuals. Biomed Res Int 2013:761617. https://doi.org/10.1155/2013/761617
Ortega FJ, Mercader JM, Catalán V et al (2013) Targeting the circulating microRNA signature of obesity. Clin Chem 59(5):781–792. https://doi.org/10.1373/clinchem.2012.195776
Thomou T, Mori MA, Dreyfuss JM et al (2017) Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542:450–455. https://doi.org/10.1038/nature21365
Ying W, Riopel M, Bandyopadhyay G et al (2017) Adipose tissue macrophage-derived exosomal miRNAs can modulate in vivo and in vitro insulin sensitivity. Cell 171(2):372–384. https://doi.org/10.1016/j.cell.2017.08.035
Abu-Izneid T, Al Hajri N, Mohammed Ibrahim A et al (2020) Micro-RNAs in the regulation of immune response against SARS COV-2 and other viral infections. J Adv Res. https://doi.org/10.1016/j.jare.2020.11.013
Fulzele S, Sahay B, Yusufu I et al (2020) COVID-19 virulence in aged patients might be impacted by the host cellular microRNAs abundance/profile. Aging Dis 11:509–522. https://doi.org/10.14336/AD.2020.0428
Trobaugh DW, Klimstra WB (2017) MicroRNA regulation of RNA virus replication and pathogenesis. Trends Mol Med 23(1):80–93. https://doi.org/10.1016/j.molmed.2016.11.003
Ma Y, Wang C, Xue M et al (2018) The coronavirus transmissible gastroenteritis virus evades the Type I interferon response through IRE1α-mediated manipulation of the microRNA miR-30a-5p/SOCS1/3 axis. J Virol 92(22):e00728–e00731. https://doi.org/10.1128/JVI.00728-188
Yuan Z, Petree JR, Lee FE-H et al (2019) Macrophages exposed to HIV viral protein disrupt lung epithelial cell integrity and mitochondrial bioenergetics via exosomal microRNA shuttling. Cell Death Dis 10(8):580. https://doi.org/10.1038/s41419-019-1803-y
Mallick B, Ghosh Z, Chakrabarti J (2009) MicroRNome analysis unravels the molecular basis of SARS infection in bronchoalveolar stem cells. PLoS ONE 4:e7837. https://doi.org/10.1371/journal.pone.0007837
Lu D, Chatterjee S, **ao K et al (2020) MicroRNAs targeting the SARS-CoV-2 entry receptor ACE2 in cardiomyocytes. J Mol Cell Cardiol 148:46–49. https://doi.org/10.1016/j.yjmcc.2020.08.017
Matarese A, Gambardella J, Sardu C et al (2020) Santulli G miR-98 regulates TMPRSS2 expression in human endothelial cells: key implications for COVID-19. Biomedicines 8(11):462. https://doi.org/10.3390/biomedicines8110462
Chow JT-S, Salmena L (2020) Prediction and Analysis of SARS-CoV-2-Targeting MicroRNA in Human Lung Epithelium. Genes 11:1002. https://doi.org/10.3390/genes11091002
Khan MAAK, Sany MRU, Islam MS et al (2020) Epigenetic regulator miRNA pattern differences among SARS-CoV, SARS-CoV-2, and SARS-CoV-2-world-wide isolates delineated the mystery behind the epic pathogenicity and distinct clinical characteristics of pandemic COVID-19. Front Genet 11:765. https://doi.org/10.3389/fgene.2020.00765
Wyler E, Mösbauer K, Franke V et al (2021) Transcriptomic profiling of SARS-CoV-2 infected human cell lines identifies HSP90 as target for COVID-19 therapy. iScience 24(3):102151. https://doi.org/10.1016/j.isci.2021.102151
Soni DK, Cabrera-Luque J, Kar S, et al (2020) Suppression of miR-155 attenuates lung cytokine storm induced by SARS-CoV-2 infection in human ACE2-transgenic mice. bioRxiv. https://doi.org/10.1101/2020.12.17.423130
Li C, Hu X, Li L et al (2020) Differential microRNA expression in the peripheral blood from human patients with COVID-19. J Clin Lab Anal 34:e23590. https://doi.org/10.1002/jcla.23590.020
Sabbatinelli J, Giuliani A, Matacchione G et al (2021) Decreased serum levels of the inflammaging marker miR-146a are associated with clinical non-response to tocilizumab in COVID-19 patients. Mech Ageing Dev 193:111413. https://doi.org/10.1016/j.mad.2020.111413
Mirzaei R, Mahdavi F, Badrzadeh F et al (2021) The emerging role of microRNAs in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Int Immunopharmacol 90:107204. https://doi.org/10.1016/j.intimp.2020.107204
Ortega FJ, Mercader JM, Moreno-Navarrete JM et al (2015) Surgery-Induced weight loss is associated with the downregulation of genes targeted by micrornas in adipose tissue. J Clin Endocrinol And Metabol 100:E1467–E1476. https://doi.org/10.1210/jc.2015-2357
Tsubota A, Mogushi K, Aizaki H et al (2014) Involvement of MAP3K8 and miR-17-5p in poor virologic response to interferon-based combination therapy for chronic hepatitis C. PLoS ONE 9(5):e97078. https://doi.org/10.1371/journal.pone.0097078
Nahand JS, Karimzadeh MR, Nezamnia M et al (2020) The role of miR-146a in viral infection. IUBMB Life 72(3):343–360. https://doi.org/10.1002/iub.2222
Roos J, Dahlhaus M, Funcke JB et al (2021) miR-146a regulates insulin sensitivity via NPR3. Cell Mol Life Sci 78:2987–3003. https://doi.org/10.1007/s00018-020-03699-1
Sanada T, Sano T, Sotomaru Y et al (2020) Anti-inflammatory effects of miRNA-146a induced in adipose and periodontal tissues. Biochem Biophys Rep 22:100757. https://doi.org/10.1016/j.bbrep.2020.100757
Desjarlais M, Wirth M, Lahaie I et al (2020) Nutraceutical targeting of inflammation-modulating microRNAs in severe forms of COVID-19: a novel approach to prevent the cytokine storm. Front Pharmacol 11:602999. https://doi.org/10.3389/fphar.2020.602999
Roganović JR (2020) MicroRNA-146a and -155, upregulated by periodontitis and type 2 diabetes in oral fluids, are predicted to regulate SARS-CoV-2 oral receptors genes. J Periodontol. https://doi.org/10.1002/JPER.20-0623
Cheng HS, Njock MS, Khyzha N et al (2014) Noncoding RNAs regulate NF-κB signaling to modulate blood vessel inflammation. Front Genet 5:422. https://doi.org/10.3389/fgene.2014.00422
Markopoulos GS, Roupakia E, Tokamani M et al (2018) Roles of NF-κB signaling in the regulation of miRNAs impacting on inflammation in cancer. Biomedicines 6(2):40. https://doi.org/10.3390/biomedicines6020040
Chang C-C, Wu M-J, Yang J-Y et al (2015) Leptin-STAT3-G9a signaling promotes obesity-mediated breast cancer progression. Cancer Res 75:2375–2386. https://doi.org/10.1158/0008-5472.CAN-14-3076
Liu Q, Du J, Yu X et al (2017) miRNA-200c-3p is crucial in acute respiratory distress syndrome. Cell Discov 3:17021. https://doi.org/10.1038/celldisc.2017.21
Foley NH, OˈNeill LA. (2012) miR-107: A Toll-like receptor-regulated miRNA dysregulated in obesity and type II diabetes. J Leuko Biol 92:521–527. https://doi.org/10.1189/jlb.0312160
Xu Y, Du J, Zhang P et al (2018) MicroRNA-125a-5p mediates 3T3-L1 preadipocyte proliferation and differentiation. Molecules 23:E317. https://doi.org/10.3390/molecules23020317
Brovkina O, Nikitin A, Khodyrev D et al (2019) Role of microRNAs in the regulation of subcutaneous white adipose tissue in individuals with obesity and without type 2 diabetes. Front Endocrinol 10:840. https://doi.org/10.3389/fendo.2019.00840
Gomes JLP, Tiago Fernandes T, Soci UPR et al (2017) Obesity downregulates microRNA-126 inducing capillary rarefaction in skeletal muscle: effects of aerobic exercise training. Oxid Med Cell Longev Article ID 2415246. https://doi.org/10.1155/2017/2415246
Hijmans JG, Diehl KJ, Bammert TD et al (2018) Influence of overweight and obesity on circulating inflammation-related microRNA. Microrna 7(2):148–154. https://doi.org/10.2174/2211536607666180402120806
Garg A, Seeliger B, Derda AA et al (2021) Circulating cardiovascular microRNAs in critically ill COVID-19 patients. Eur J Heart Fail 23(3):468–475. https://doi.org/10.1002/ejhf.2096
Seeger T, Fischer A, Muhly-Reinholz M et al (2014) Long-term inhibition of miR-21 leads to reduction of obesity in db/db mice. Obesity 22:2352–2360. https://doi.org/10.1002/oby.20852
Guglielmi V, D’Adamo M, Menghini MR et al (2017) MicroRNA 21 is up-regulated in adipose tissue of obese diabetic subjects. Nutr Healthy Aging. https://doi.org/10.3233/NHA-160020
Kershaw EE, Flier JS (2004) Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89:2548–2556. https://doi.org/10.1210/jc.2004-0395
Coelho M, Oliveira T, Fernandes R (2013) Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci 9:191–200. https://doi.org/10.5114/aoms.2013.33181
Weisberg SP, McCann D, Desai M et al (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112(12):1796–1808. https://doi.org/10.1172/JCI19246
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867. https://doi.org/10.1038/nature05485
Mancuso P (2010) Obesity and lung inflammation. J Appl Physiol 108(3):722–728. https://doi.org/10.1152/japplphysiol.00781.2009
Ubags ND, Stapleton RD, Vernooy JH et al (2016) Hyperleptinemia is associated with impaired pulmonary host defense. JCI insight 1(8):e82101
Sanchez-Margalet V, Martin-Romero C (2001) Human leptin signaling in human peripheral blood mononuclear cells: activation of the JAK-STAT pathway. Cell Immunol 211:30–36. https://doi.org/10.1006/cimm.2001.1815
Matarese G, Moschos S, Christos S (2005) Leptin in immunology. J Immunol 174:3137–42. https://doi.org/10.4049/jimmunol.174.6.3137
MacIver NJ, Michalek RD, Rathmell JC (2013) Metabolic regulation of T LYMPHOCYTES. Annu Rev Immunol 31:259–328. https://doi.org/10.1146/annurev-immunol-032712-095956
Martinez-Martinez E, Jurado-Lopez R, Valero-Munoz M et al (2014) Leptin induces cardiac fibrosis through galectin-3, mTOR and oxidative stress: potential role in obesity. J Hypertens 32:1104–1114. https://doi.org/10.1097/HJH.0000000000000149
Saucillo DC, Gerriets VA, Sheng J (2014) Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol. 192:136–144. https://doi.org/10.4049/jimmunol.1301158
Rebello CJ, Kirwan JP, Greenway FL (2020) Obesity, the most common comorbidity in SARS-CoV-2: is leptin the link? Int J Obes 44:1810–1817. https://doi.org/10.1038/s41366-020-0640-5
Jain M, Budinger GRS, Lo A et al (2011) Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-γ. Am J Respir Crit Care Med 183:1490–1498. https://doi.org/10.1164/rccm.201009-1409OC
Zhang AJ, To KK, Li C et al (2013) Leptin mediates the pathogenesis of severe 2009 pandemic influenza A (H1N1) infection associated with cytokine dysregulation in mice with diet-induced obesity. J Infectious Dis 207:1270–1280. https://doi.org/10.1093/infdis/jit031
Lee SM, Choi HJ, Oh CH et al (2014) Leptin increases TNF-α expression and production through phospholipase D1 in raw 2647 cells. PLoS ONE 9:e102373. https://doi.org/10.1371/journal.pone.0102373
Akhtar LN, Qin H, Muldowney MT et al (2010) Suppressor of cytokine signaling 3 inhibits antiviral IFN-beta signaling to enhance HIV-1 replication in macrophages. J Immunol (Baltimore, M.D.:1950) 185(4):2393–2404. https://doi.org/10.4049/jimmunol.0903563
Michaud F, Coulombe F, Gaudreault E et al (2010) Epstein-Barr virus interferes with the amplification of IFNalpha secretion by activating suppressor of cytokine signaling 3 in primary human monocytes. PLoS ONE 5(7):e11908. https://doi.org/10.1371/journal.pone.0011908
Wang J, Xu Y, Zhang X et al (2021) Leptin correlates with monocytes activation and severe condition in COVID-19 patients. J Leukoc Biol. https://doi.org/10.1002/JLB.5HI1020-704R
Kordonowy LL, Burg E, Lenox CC et al (2012) Obesity is associated with neutrophil dysfunction and attenuation of murine acute lung injury. Am J Respir Cell Mol Biol 47:120–127. https://doi.org/10.1165/rcmb.2011-0334OC
Teran-Cabanillas E, Montalvo-Corral M, Caire-Juvera G et al (2013) Decreased interferon-α and interferon-β production in obesity and expression of suppressor of cytokine signaling. Nutrition 29(1):207–212. https://doi.org/10.1016/j.nut.2012.04.019
Terán-Cabanillas E, Hernández J (2017) Role of leptin and SOCS3 in inhibiting the Type I interferon response during obesity. Inflammation 40(1):58–67. https://doi.org/10.1007/s10753-016-0452-x
Gammone MA, D’Orazio N (2021) Review: obesity and COVID-19: a detrimental intersection. Front Endocrinol 12:652639. https://doi.org/10.3389/fendo.2021.652639
Alti D, Sambamurthy C, Kalangi SK (2018) Emergence of leptin in infection and immunity: scope and challenges in vaccines formulation. Front Cell Infect Microbiol 8:147. https://doi.org/10.3389/fcimb.2018.00147
Hadjadj J, Yatim N, Barnabei L et al (2020) Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369(6504):718–724. https://doi.org/10.1126/science.abc6027
Zhang Q, Bastard P, Liu Z et al (2020) Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (NY) 370(6515):eabd4570. https://doi.org/10.1126/science.abd4570
De Rosa A, Ludovica Monaco M, Capasso M et al (2013) Adiponectin oligomers as potential indicators of adipose tissue improvement in obese subjects. Eur J Endocrinol 169:37–43. https://doi.org/10.1530/EJE-12-1039
Ouchi N, Walsh K (2007) Adiponectin as an anti-inflammatory factor. Clin Chim Acta 380:24–30. https://doi.org/10.1016/j.cca.2007.01.026
Antoniades C, Antonopoulos AS, Tousoulis D et al (2009) Adiponectin: from obesity to cardiovascular disease. Obes Rev 10:269–279. https://doi.org/10.1111/j.1467-789X.2009.00571.x
Rojas E, Rodríguez-Molina D, Bolli P et al (2014) The role of adiponectin in endothelial dysfunction and hypertension. Curr Hypertens Rep 16:463. https://doi.org/10.1007/s11906-014-0463-7
Montagnani M, Funahashi T, Shimomura I et al (2003) Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem 278:45021–45026. https://doi.org/10.1074/jbc.M307878200
Antonopoulos AS, Margaritis M, Coutinho P et al (2015) Adiponectin as a link between type 2 diabetes and vascular NADPH oxidase activity in the human arterial wall: the regulatory role of perivascular adipose tissue. Diabetes 64:2207–2219. https://doi.org/10.2337/db14-1011
Ouchi N, Kihara S, Arita Y et al (1999) Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation 100:2473–2476. https://doi.org/10.1161/01.cir.100.25.2473
Ouedraogo R, Gong Y, Berzins B et al (2007) Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J Clin Invest 117(6):1718–1726. https://doi.org/10.1172/JCI29623
Ye R, Scherer PE (2013) Adiponectin, driver or passenger on the road to insulin sensitivity? Mol Metab 2(3):133–141. https://doi.org/10.1016/j.molmet.2013.04.001
Kasahara DI, Kim HY, Williams AS et al (2012) Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: role of IL-17A. J Immunol 188:4558–4567. https://doi.org/10.4049/jimmunol.1102363
Shah D, Romero F, Duong M et al (2015) Obesity-induced adipokine imbalance impairs mouse pulmonary vascular endothelial function and primes the lung for injury. Sci Rep 5:11362. https://doi.org/10.1038/srep11362
Simons PJ, van den Pangaart PS, Aerts JM et al (2007) Pro-inflammatory delipidizing cytokines reduce adiponectin secretion from human adipocytes without affecting adiponectin oligomerization. J Endocrinol 192(2):289–299. https://doi.org/10.1677/JOE-06-0047.Erratum.In:JEndocrinol.2007192(3):683
Nigro E, Scudiero O, Monaco ML et al (2014) New insight into adiponectin role in obesity and obesity-related diseases. Biomed Res Int. https://doi.org/10.1155/2014/658913
Duncan BB, Schmidt MI, Pankow JS et al (2004) Adiponectin and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes 53:2473–2478. https://doi.org/10.2337/diabetes.53.9.2473
Abate N, Chandalia M, Snell PG, Grundy SM (2004) Adipose tissue metabolites and insulin resistance in non-diabetic Asian Indian men. J Clin Endocrinol Metab 89:2750–2755. https://doi.org/10.1210/jc.2003-031843
Bush NC, Darnell BE, Oster RA et al (2005) Adiponectin is lower among African Americans and is independently related to insulin sensitivity in children and adolescents. Diabetes 54:2772–2778. https://doi.org/10.2337/diabetes.54.9.2772
Kearns SM, Ahern KW, Patrie JT et al (2021) Reduced adiponectin levels in patients with COVID- 19 acute respiratory failure: a case- control study. Physiol Rep 9:e14843. https://doi.org/10.14814/phy2.14843
Spiezia L, Boscolo A, Poletto F et al (2020) COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb Haemost 120:998–1000. https://doi.org/10.1055/s-0040-1710018
Bombeli T, Karsan A, Tait JF et al (1997) Apoptotic vascular endothelial cells become procoagulant. Blood 89:2429–2442
Varga Z, Flammer AJ, Steiger P et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395(10234):1417–1418. https://doi.org/10.1016/S0140-6736(20)30937-5
Jung F, Krüger-Genge A, Franke RP et al (2020) COVID-19 and the endothelium. Clin Hemorheol Microcirc 75:7–11. https://doi.org/10.3233/CH-209007
Mertens I, Van Gaal LF (2002) Obesity, haemostasis and the fibrinolytic system. Obes Rev 3:85–101. https://doi.org/10.1046/j.1467-789x.2002.00056.x
Anfossi G, Russo I, Trovati M (2009) Platelet dysfunction in central obesity. Nutr Metab Cardiovasc Dis 19:440–449. https://doi.org/10.1016/j.numecd.2009.01.006
Vilahur G, Ben-Aicha S, Badimon L (2017) New insights into the role of adipose tissue in thrombosis. Cardiovasc Res 113(9):1046–1054. https://doi.org/10.1093/cvr/cvx086
Aljada A, Ghanim H, Mohanty P et al (2002) Insulin inhibits the pro-inflammatory transcription factor early growth response gene-1 (Egr)-1 expression in mononuclear cells (MNC) and reduces plasma tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) concentrations. J Clin Endocrinol Metab 87:1419–1422. https://doi.org/10.1210/jcem.87.3.8462
Rakusa MJ, Jensterle M, Božič-Mijovski M et al (2017) Increased coagulation and decreased fibrinolysis as measured with overall hemostatic potential are dependent on BMI and not associated with PCOS. Metab Syndr Relat Disord 15:194–198. https://doi.org/10.1089/met.2016.0148
Basili S, Pacini G, Guagnano MT et al (2006) Insulin resistance as a determinant of platelet activation in obese women. J Am Coll Cardiol 48:2531–2538. https://doi.org/10.1016/j.jacc.2006.08.040
Trovati M, Mularoni EM, Burzacca S et al (1995) Impaired insulin-induced platelet antiaggregating effect in obesity and in obese NIDDM patients. Diabetes 44(11):1318–1322. https://doi.org/10.2337/diab.44.11.1318
Sjöholm A, Nyström T (2005) Endothelial inflammation in insulin resistance. Lancet 365(9459):610–612. https://doi.org/10.1016/S0140-6736(05)17912-4
Sudhakar M, Silambanan S, Chandran AS et al (2018) C-Reactive Protein (CRP) and leptin receptor in obesity: binding of monomeric CRP to leptin receptor. Front Immunol 9:1167. https://doi.org/10.3389/fimmu.2018.01167
Mostaghim A, Sinha P, Bielick C et al (2020) Clinical outcomes and inflammatory marker levels in patients with COVID-19 and obesity at an inner-city safety net hospital. PLoS ONE 15:e0243888. https://doi.org/10.1371/journal.pone.0243888
Molins B, Peña E, de la Torre R et al (2011) Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc Res 92(2):328–337. https://doi.org/10.1093/cvr/cvr226
Grad E, Danenberg HD (2013) C-reactive protein and atherothrombosis: cause or effect? Blood Rev 27:23–29. https://doi.org/10.1016/j.blre.2012.12.001
Boden G (2008) Obesity and free fatty acids. Endocrinol Metab Clin N Am. 37:635–46, viii–ix. https://doi.org/10.1016/j.ecl.2008.06.007
Ghosh A, Gao L, Thakur A et al (2017) Role of free fatty acids in endothelial dysfunction. J Biomed Sci 24:50. https://doi.org/10.1186/s12929-017-0357-5
Watanabe S, Tagawa T, Yamakawa K et al (2005) Inhibition of the renin-angiotensin system prevents free fatty acid-induced acute endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol 25(11):2376–2380. https://doi.org/10.1161/01.ATV.0000187465.55507.85
Azekoshi Y, Yasu T, Watanabe S et al (2010) Free fatty acid causes leukocyte activation and resultant endothelial dysfunction through enhanced angiotensin II production in mononuclear and polymorphonuclear cells. Hypertension 56:136–142. https://doi.org/10.1161/HYPERTENSIONAHA.110.153056
Sudhakar M, Silambanan S, Prabhakaran AA et al (2021) Angiogenic potential, circulating angiogenic factors and insulin resistance in subjects with obesity. Ind J Clin Biochem 36:43–50. https://doi.org/10.1007/s12291-019-0816-8
Legrand-Poels S, Esser N, Lhomme L et al (2014) Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes Biochem Pharmacol 92:131–41. https://doi.org/10.1016/j.bcp.2014.08.013
Wang L, Chen Y, Li X et al (2016) Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget 7(45):73229–73241. https://doi.org/10.18632/oncotarget.12302
Thomas T, Stefanoni D, Reisz JA et al (2020) COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 5(14):e140327. https://doi.org/10.1172/jci.insight.140327
Bastard P, Rosen LB, Zhang Q et al (2020) Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (New York, NY) 370(6515):eabd4585. https://doi.org/10.1126/science.abd4585
Zuo Y, Estes SK, Ali RA et al (2020) Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl Med 12(570):3876. https://doi.org/10.1126/scitranslmed.abd3876
Taha M, Samavati L (2021) Antiphospholipid antibodies in COVID-19: a meta-analysis and systematic review. RMD Open 7(2):e001580. https://doi.org/10.1136/rmdopen-2021-001580
Frasca D, Diaz A, Romero M et al (2020) Identification and characterization of adipose tissue-derived human antibodies with “Anti-self” specificity. Front Immunol 11:392. https://doi.org/10.3389/fimmu.2020.00392
Burg ND, Pillinger MH (2001) The neutrophil: function and regulation in innate and humoral immunity. Clin Immunol 99:7–17. https://doi.org/10.1006/clim.2001.5007
Talukdar S, Oh DY, Bandyopadhyay G et al (2012) Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18(9):1407–1412. https://doi.org/10.1038/nm.2885
Brotfain E, Hadad N, Shapira Y et al (2015) Neutrophil functions in morbidly obese subjects. Clin Exp Immunol 181:156–163. https://doi.org/10.1111/cei.12631
Xu X, Su S, Wang X et al (2015) Obesity is associated with more activated neutrophils in African American male youth. Int J Obes (Lond) 39(1):26–32. https://doi.org/10.1038/ijo.2014.194
Trellakis S, Rydleuskaya A, Fischer C et al (2012) Low adiponectin, high levels of apoptosis and increased peripheral blood neutrophil activity in healthy obese subjects. Obes Facts 5(3):305–318. https://doi.org/10.1159/000339452
Liao M, Liu Y, Yuan J et al (2020) Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med 26:842–844. https://doi.org/10.1038/s41591-020-0901-9
Mutua V, Gershwin LJ (2020) A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. https://doi.org/10.1007/s12016-020-08804-7
D’Abbondanza M, Martorelli EE, Ricci MA et al (2019) Increased plasmatic NETs by-products in patients in severe obesity. Sci Rep 9:14678. https://doi.org/10.1038/s41598-019-51220-x
Braster Q, Silvestre Roig C, Hartwig H et al (2016) Inhibition of NET release fails to reduce adipose tissue inflammation in mice. PLoS ONE 11:e0163922. https://doi.org/10.1371/journal.pone.0163922
Tripathi S, Verma A, Kim EJ et al (2014) LL-37 modulates human neutrophil responses to influenza A virus. J Leukoc Biol 96(5):931–938. https://doi.org/10.1189/jlb.4A1113-604RR
Muraro SP, De Souza GF, Gallo SW et al (2018) Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci Rep 8:14166. https://doi.org/10.1038/s41598-018-32576-y
Hiroki CH, Toller-Kawahisa JE, Fumagalli MJ et al (2020) Neutrophil extracellular traps effectively control acute Chikungunya virus infection. Front Immunol 10:3108. https://doi.org/10.3389/fimmu.2019.03108
Moorthy AN, Tan KB, Wang S et al (2016) Effect of high-fat diet on the formation of pulmonary neutrophil extracellular traps during Influenza Pneumonia in BALB/c mice. Front Immunol 7:289. https://doi.org/10.3389/fimmu.2016.00289
Zuo Y, Yalavarthi S, Shi H et al (2020) Neutrophil extracellular traps in COVID-19. JCI Insight 5(11):e138999. https://doi.org/10.1172/jci.insight.138999
Mikacenic C, Moore R, Dmyterko V et al (2018) Neutrophil extracellular traps (NETs) are increased in the alveolar spaces of patients with ventilator-associated pneumonia. Crit Care 22:358. https://doi.org/10.1186/s13054-018-2290-8
Grabcanovic-Musija F, Obermayer A, Stoiber W (2015) Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res 16:59. https://doi.org/10.1186/s12931-015-0221-7
Fuchs TA, Brill A, Duerschmied D et al (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci (U S A) 107:15880–15885. https://doi.org/10.1073/pnas.1005743107
Brill A, Fuchs TA, Savchenko AS et al (2012) Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost 10:136–144. https://doi.org/10.1111/j.1538-7836.2011.04544.x
Borissoff JI, Joosen IA, Versteylen MO et al (2013) Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol 33:2032–2040. https://doi.org/10.1161/ATVBAHA.113.301627
Luna JM, Scheel TK, Danino T et al (2015) Hepatitis C virus RNA functionally sequesters miR-122. Cell 160:1099–1110. https://doi.org/10.1016/j.cell.2015.02.025
Kai H, Kai M (2020) Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens Res 43:648–654. https://doi.org/10.1038/s41440-020-0455-8
Kokkinopoulou I, Maratou E, Mitrou P et al (2019) Decreased expression of microRNAs targeting type-2 diabetes susceptibility genes in peripheral blood of patients and predisposed individuals. Endocrine 66:226–239. https://doi.org/10.1007/s12020-019-02062-0
Kuryłowicz, A., Zofia Wicik,Z., Magdalena Owczarz,M.,et al (2018) NGS reveals molecular pathways affected by obesity and weight loss-related changes in miRNA levels in adipose tissue. Int. J. Mol. Sci. 19, 66; doi:https://doi.org/10.3390/ijms19010066
Acknowledgements
The authors thank Dr Ganesh Venkatraman, Department of Human Genetics, SRIHER (DU) for a critical reading of the manuscript and helpful comments.
Funding
Financial assistance from Indian Council for Medical Research, New Delhi to M.S in the form of a research associateship No. 3/1/2(16)/OBS/2019-NCD-II is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
MS was responsible for article conception, review of literature, and writing of the first draft. All authors contributed to editing and revising of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sudhakar, M., Winfred, S.B., Meiyazhagan, G. et al. Mechanisms contributing to adverse outcomes of COVID-19 in obesity. Mol Cell Biochem 477, 1155–1193 (2022). https://doi.org/10.1007/s11010-022-04356-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-022-04356-w