
Abstract
Context
In the replication of SARS-CoV-2, the main protease (Mpro/3CLpro) is significant. It is conserved in a number of novel coronavirus variations, and no known human proteases share its cleavage sites. Therefore, 3CLpro is an ideal target. In the report, we screened five potential inhibitors (1543, 2308, 3717, 5606, and 9000) of SARS-CoV-2 Mpro through a workflow. The calculation of MM-GBSA binding free energy showed that three of the five potential inhibitors (1543, 2308, 5606) had similar inhibitor effects to X77 against Mpro of SARS-CoV-2. In conclusion, the manuscript lays the groundwork for the design of Mpro inhibitors.
Methods
In the virtual screening phase, we used structure-based virtual screening (Qvina2.1) and ligand-based virtual screening (AncPhore). In the molecular dynamic simulation part, we used the Amber14SB + GAFF force field to perform molecular dynamic simulation of the complex for 100 ns (Gromacs2021.5) and performed MM-GBSA binding free energy calculation according to the simulation trajectory.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It has been about 2.5 years since the first case of novel coronavirus pneumonia was reported. So far, there have been millions of cases of COVID-19, including a lot of deaths, reported to WHO [1]. Although this is not the first time humans have been hit by coronavirus, no other epidemic has been as widespread as this one [2, 3]. The virus that caused this outbreak was named SARS-CoV-2 by WHO [4]. In short, the novel coronavirus pandemic has affected health, economy, culture, and politics around the world [5, 6]. Therefore, we have to accelerate our speed to develop drugs to prevail over the pandemic.
SARS-CoV-2 which appears round or oval in shape is a β coronavirus with a coating. The viral genome contains about 30,000 nucleotides, encodes two polyproteins and four structural proteins including spike protein, envelope protein, membrane protein, and nucleocapside protein, and interspersed with several auxiliary proteins from 5′ to 3′ [7, 8]. Mpro (nsp5) and PLpro (a domain in nsp3) cleave two polyproteins to create 16 non-structural proteins, which are crucial for SARS-CoV-2 replication and immunological escape [9,10,11,12].
Because of how close its function is to that of the 3C protease found in small RNA viruses, Mpro is also called 3C-like protease. Mpro has a catalytic Cys-His binary structure, unlike other chymotrypsin-like and many serine or cysteine proteases. It consists of 306 amino acids, and most of the cleaved sites of this protease are identified as leu-gln-|- (ser, ala, gly). The mass of this protease is determined by mass spectrometry to 33,797.0 Da [13]. The Mpro structure is divided into 3 regions, region 1 (8-101) and region 2 (102-184) have an inverse parallel β structure containing 3 β barrels each, region 3 (201-303) contains 5 α spirals, and region 2 and region 3 are connected by a long ring structure (185-200). The dimer form of Mpro is active, the monomer is inactivated, and region 3 plays a crucial role in the dimerization process [14]. It has 96% sequence homology with SARS-CoV. But there are two mutation sites T285A and I286L in SARS-CoV-2, which makes the three regions closer, which eventually leads to an increase in catalytic turnover [15, 16].
The protease’s substrate binding pocket lies between regions 1 and 2, and its active position is made up of 4 pockets [17, 18] (Fig. 1). The Mpro of SARS-CoV-2 has also been found to have a novel role, demonstrating that the protease may directly cleave proteins including NLRP12 and TAB1 involved in the host’s normal immune response. The elevated cytokines and improved inflammatory response in patients may be explained by NSP5-mediated cleavage of NLRP12 and TAB1 [19].

Pocket of Mpro
Mpro is a desirable target for the exploitation of antiviral medicines due to its functional significance and the lack of closely similar human homologues [20].
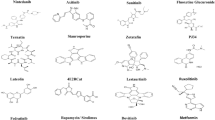
So far, the FDA has approved a new oral coronavirus drug, Paxlovid, consisting of Mpro inhibitor nirmatrelvir and cytochrome P450 enzyme CYP3A4 inhibitor ritonavir [21]. In addition, S-217622 developed by Japan’s Shionogi & Co., Ltd has also conducted clinical trials. The drug has strong metabolic stability, high oral availability, and low clearance, and it can turn into a relatively expectant heavy-weight drug against SARS-CoV-2 [22]. Some non-covalent inhibitors are listed here (Table 1) [23,24,25,26].
Based on the severity of the transmission of new coronavirus, we still need to find drugs to fight or live with COVID-19, and this paper found potential non-covalent potential inhibitors, which lays a foundation for the design of novel non-covalent inhibitors of Mpro.
Results
Sequence alignment and protein structure alignment
First, we compared the amino acid sequences of the main protease of five variants of SARS-CoV-2 and found that the amino acid sequence identity was 99.8% [28] (Fig. 2). Among them, one mutation is K90R, while another mutation is H132P. We superposed the three crystal structures and highlighted their active pockets and found that the small changes in the active pockets are almost negligible (Fig. 3). We selected the structures of these two mutants (PDB: 7U29 [24], 7TVX [29]) and primitive Mpro (7RNK [30]) on the RCSB PDB and compared them. Pairwise structure alignment [31] shows that RMSD between 7RNK and 7U29 (K90R) is 0.5 and TM score is 0.99. The two values are between 7RNK and 7TVX (P132H) of 0.35 and 0.98, respectively. These results reflect to some extent that the Mpro of multiple variants of SARS-CoV-2 is highly conservative.

Amino acid sequence alignment

Superposition of the three crystal structures and minor differences in the active pocket
Drug-like screening and structure-based virtual screening
We screened 5365 drug-like compounds from 15,033 compounds (L5610 and L5600 from TargetMol), and the drug-like standards were partially explained by ADMETlab 2.0 [32]. Prior to the structure-based virtual screening, we used the PDB structure 6LU7 and 6W79 to verify the effectiveness of virtual screening [33]. By docking X77 and Mpro in 6LU7 for many times, we found that Qvina2.1 [20, 34] was reliable in evaluating the non-covalent binding between small molecules and proteins. The RMSD between X77 in crystal structure and highest scoring conformation produced by docking is 1.275 at − 8.3 kcal/mol, which explains the credibility of Qvina2.1 (Fig. 4).

X77 and Mpro docking by Qvina2.1 (cyan: crystal structure, green: docking structure)
We screened 901 compounds from 5365 drug-like compounds with docking score less than − 8.0 kcal/mol by using Qvina2.1. The top eight compounds with the highest docking score are provided. The presented parameters include the docking score, MW, logP, nHA, nHD, and TPSA (Table 2). The compound number that corresponds to the compound name attached to the supplementary materials (Table S1).
Pharmacophore modeling and pharmacophore screening
Since there are several known active Mpro inhibitors, we selected several crystal structures from the RCSB PDB (PDB: 6lze, 6m0k, 6wtj, 6y2f, 7jyc, 7k0g, 7k0h, 7k40, 7lbn, 7ltj, 7lzv, 7m04, 7tdu, 7tfr, 7tll [35,36,37,38,39,40,41,42,43]) to define the pharmacophore characteristics of the inhibitors, including hydrogen bond donors (HD) and acceptors (HA), aromatic rings (AR), and covalent bond (CV) features (Fig. 5). Almost all inhibitors contain hydrogen bond donors, hydrogen bond receptors, and covalent bonds, differing in their number. 7ltj contains a special inhibitor that contains an aromatic ring that has a Π-Π stacking interaction with Mpro of SARS-CoV-2. In addition to providing the characteristics of the pharmacophore, the pharmacophore model also provides the diversity of the pharmacophore position.

Pharmacophore modeling by AncPhore
At the same time, we used AncPhore to generate the low-energy conformations of 901 compounds selected by Qvina2.1. Each compound corresponds to ten low-energy conformations at most. The top ten compounds of APScore [44] were selected from the low-energy conformation database of these compounds (Table 3). And they have a high degree of matching with the pharmacophore characteristics of 7ltj, which corresponds to the Mcule-5948770040 (take the 9000 as an example) (Fig. 6). 7ltj’s high match rate may be due to AncPhore’s determination that Mcule-5948770040 has relatively few interactions with Mpro.

The 9000 compound matched with Mcule-5948770040
Molecular dynamic simulation
RMSD
From the eutectic structure of Mpro and its noncovalent inhibitor X77 (PDB: 6w79 [27]), the chemical structure of X77 was extracted and processed and then docked with Mpro (PDB: 6lu7). Then, the initial conformation with the highest Qvina2.1 score was selected as the control group for molecular dynamic simulation. The size of RMSD reflects structural deviation of study object. We extracted the RMSD of each complex (Fig. 7a) relative to the Mpro protein backbone and the RMSD (Fig. 7b) of the Mpro protein backbone itself from the 100-ns MD simulation trajectory. We also extracted the RMSD of each ligand (Fig. 8) relative to the ligand in the initial conformation of the complex to explore whether the ligand is stable in its initial position. In all complexes, we found that the RMSD trend of the complex was the same as the RMSD of the Mpro backbone itself, so we only discuss the change of the RMSD trend of the complex and ligand here.


a The RMSD of each complex relative to the Mpro backbone in the 100-ns MD. b The RMSD of the Mpro relative to itself in the 100-ns MD

The RMSD of ligands relative to ligands in the 100-ns MD
The RMSD of the complexes of Mpro with eight compounds (2246, 2308, 2933, 3717, 5606, 481, 2432, 2908) remained at a stable level during the simulation of 100 ns. For 1047 (45 ns), 4368 (55 ns), 2490 (22 ns), 3083 (40 ns), 5125 (15 ns), and 9000 (65 ns), all of them keep low RMSD at first, then rise at a certain node, and keep stable in the following time range (the time node after rising and stabilizing is in parentheses). The RMSD of Mpro-783 complex fluctuates in the first 50 ns and returns to stable in the last 50 ns. Mpro-1179 complex has low RMSD between 60 and 80 ns, and it is stable in other time. The RMSD of Mpro-1543 complex increased steadily in the first 50 ns and remained stable in the first 50–85 ns and then decreased slightly. The most interesting complex is Mpro-9328, whose RMSD has a sharp rising and falling trend between 32 and 35 ns. This reflects the instability of the complex over a short period of time, but does not affect its subsequent RMSD which remains stable for a longer period of time. Finally, the RMSD of the control group Mpro-X77 increased within 35–65 ns but remained low and stable for the rest of the time.
As can be seen from that RMSD of the ligand, almost all of the compounds are not maintained in the initial conformation. Some compounds (1047, 2308, 481) maintain one conformation initially and then cross the energy barrier to maintain another low energy conformation. Some compounds (4368, 5125, 3083, X77) were initially unstable and subsequently maintained a stable conformation for a long period of time. Most compounds (2246, 2933, 783, 1179, 2432, 1543, 5606, 9328, 2908, 9000, 3717) are in a state of fluctuation, but some (1179, 3717, 5606, 2908) of these compounds can be seen in the final period of conformational stability. The RMSD of these compounds is likely to be normal, no matter how to change, because many highly flexible small molecules do not have a single stable conformation, and they have a large number of low-energy conformations. And that potential energy barrier between the conformations is low and can be easily crossed.
Root mean square fluctuation analysis
RMSF is the average value of atomic position change over time, which can characterize the flexibility and motion intensity of protein amino acids in the whole simulation process. According to the RMSF extraction results (Fig. 9), it can be seen that the compound 9328 disturbs the protein residues to a greater extent, which is also consistent with the RMSD extraction results of the complex. The effects of other compounds on protein residue flexibility were similar to those of X77.

The RMSF of Mpro amino acid residues in the 100-ns MD
H_Bond number
Hydrogen bonds is crucial in maintaining the stability of the complex. The number of hydrogen bonds (Fig. 10) formed between compounds and Mpro during the whole simulation process was counted. The results showed that the number of H_Bonds formed between compounds 2308, 4368, 5606, 3083, and 1543 and Mpro was higher or equal to that between X77 and Mpro. The amount of hydrogen bond with Mpro for 3717 and 2490 increased from 30 and 60 ns, respectively, and that amount of hydrogen bond with Mpro for 783 and 1047 decreased from 30 and 50 ns respectively. The remaining compounds form fewer hydrogen bonds with Mpro.

H_Bond number between ligands and Mpro in the 100-ns MD
FEL (free energy landscape)
The free energy landscape is a graph that shows the change of the free energy of the complexes. The free energy topography map is drawn when the RMSD of the complex is stable, that is, when the molecule is stable, generally it has only one energy well, so as to find the characteristic structure of the complex. Therefore, we select the simulation track after 50 ns of each complex here and take Rg (Rg is the gyration radius of the composite, which can characterize the change of the loose degree of the composite structure in the simulation process) as the abscissa and RMSD as the ordinate to draw the free energy topography map (Fig. 11).

FEL of all compounds and corresponding characteristic conformation (red: initial conformation in simulation, cyan: characteristic low-energy conformation corresponding to time)
A rise from blue to red indicates that the energy is increasing, while the low-energy conformation tends to be stable. We extracted the low-energy conformations indicated in each free energy landscape map separately. The complexes of compounds 1047, 5606, and 9328 with Mpro suggest two low-energy conformations, while the other complexes have only one. However, all compounds are similar to X77 in that binding to Mpro results in a similar state of inhibition of the complex.
MM-GBSA
We also used the trajectory after 50 ns of simulation when calculating the ligand and Mpro’s binding free energy. The binding energies (Table 4) of 1503, 2308, and 5606 with Mpro are slightly lower than those of X77 with Mpro, indicating that the interaction between these three compounds and Mpro is slightly stronger than that between X77 and Mpro. The fact that the binding energies of 3717, 9000, and X77 are slightly higher than those of X77 suggests that these two compounds have a slightly weaker ability to bind to Mpro, while the other complexes’ binding free energies are reasonable. It is worth noting that the electrostatic interaction between 1047 and 481 and Mpro is positive because the docked small molecule has a negative charge, and the protein itself also has a negative charge, so it is in a mutually repelling state in the gas phase. It is also because these two compounds are negatively charged that their polar solvation energy is greater, so it shows a more negative value in ΔEGB. Compounds 2246 and 9000 are positively charged after docking, which can explain the energy decomposition term for these two compounds.
Interaction between Mpro and its potential inhibitors
According to the MD analysis results and MM-GBSA calculation results, we found five potential inhibitors of Mpro, which are 1543, 2308, 3717, 5606, and 9000, respectively. Then, we analyzed the interaction between these five compounds and Mpro (Fig. 12). In the Mpro-1543 complex, Glu166 forms a hydrogen bond with the sulfur-oxygen bond of the compound, Gln192 forms a hydrogen bond with the hydroxyl group of 1543, and the catalytic residue His 41 forms a Π-Π stacking with one of the benzene rings of 1543, as shown in the figure below. In the Mpro-2308 complex, the amide group in 2308 plays an important role in forming hydrogen bonds with multiple amino acids of the main protease. In the Mpro-3717 complex, the sulfur-oxygen bond acts as a hydrogen bond acceptor, forming hydrogen bonds with Gly143, Ser144, His163, and His172 of the main protease, in addition to which the sulfur in the sulfur-oxygen bond forms a Π-sulfur interaction with the benzene ring in His172. In the Mpro-5606 complex, Π-alkyl interactions are more common. In the Mpro-9000 complex, the more specific interaction is the halogen bond, mainly due to the presence of fluorine in the compound, with Thr26 acting as an electron donor.

Interaction between Mpro and potential inhibitors
Discussion
In this study, we demonstrated the conservation of Mpro and preliminarily screened 18 compounds according to the structure of the known inhibitors of Mpro. Through further molecular dynamic simulation and MM-GBSA binding free energy calculation, we found five potential inhibitors of Mpro and analyzed the interaction between the potential inhibitors and Mpro. These five compounds have more diverse interactions with Mpro than previously discovered non-covalent inhibitors, due to the diversity of groups in the compounds themselves. Compared with other works of the same type, the same point is that we have carried out the whole process of virtual screening of SARS-CoV-2 Mpro, and the difference may be that the screening methods we choose are different. Some work uses classic QSAR screening, and other work involves the design of small molecules from scratch. In the MD simulation stage, the force field and the software selected in various works are also different. It must be admitted that GROMACS is still well suited to simulating biological systems. Due to limitations, we have not been able to verify the enzyme activation experiments of the screened potential inhibitors. But computational experiments still provide us with a subtle perspective to observe the characteristics of potential inhibitors.
Three compounds (1047, 3717, 5606) have a sulfur-oxygen bond and thus provide hydrogen bond acceptors and sulfur elements. Compound 9000 has a trifluoromethyl group and a cyano group, which is very similar to nirmatrelvir, but the cyano group seems to have only weak van der Waals forces with the main protease due to its position. And we found that the chemical structure of the positive control X77 occupies almost the entire active pocket of SARS-CoV-2 Mpro. The two sulfur-containing residues of Mpro, Met49, and Cys145 form Π-S bond with the aromatic ring of X77, and the N and O in X77 mainly act as hydrogen bond receptors. The potential inhibitors we identified, while not completely occupying the active pocket of the enzyme, provide interactions outside the active pocket. Overall, the structures of these compounds provide some ideas for drug design.
Materials and methods
Compounds and database
The database are from TargetMol and the compound files are from ChemDiv.
Sequence alignment
Download the nucleotide sequences of SARS-CoV-2 variants on NCBI. These variants include B.1.1.7 (α), B.1.351 (β), P.1 (γ), B.1.617.2 (δ), BA.1 (o), BA.1.1 (o), BA.2 (o), and BA.3 (o). Convert the nucleotide sequence of each variant strain into an amino acid sequence using the ExPASy translate tool. DNAMAN was used for amino acid sequence alignment.
Drug-like screening
In that aspect of analogue screen, we used the predicted physicochemical properties in the sdf file of compounds and referred to the partial analogue standard of ADMETlab 2.0, i. e. 100 < MW < 600, 0 < nHA < 12, 0 < nHD < 7, 0 < TPSA < 140, and 0 < logP < 3.
Preparation for docking and virtual screening
We chose chain A of 6lu7 as the receptor. Download 6LU7 PDB structure from the RCSB PDB website, pretreat it with PyMOL to remove water molecules, chain B and N3 ligands, and then use AutoDock Tools to hydrogenate the protein, calculate the charge, and set the atomic type. Convert the small molecule from L5610 and L5600 library of TargetMol into a 3D structure with Open Babel. AutoDock Tools’ prepare_ ligand4.py script was used to deal with small molecules. The detailed information about the grid box was shown as center (x = − 10.351, y = 13.549, z = 69.904.) and size (x = 22.5, y = 28.5, z = 26.25).
When everything was ready, we validated the docking model using the processed receptors and ligands. We wrote a script for virtual screening using Qvina2.1 with energy_range = 3, num_modes = 9, and exhaustiveness = 24.
AncPhore screening based on Mpro inhibitor structures
We used AncPhore [27] to generate 10 conformations of 901 small molecules screened by Qvina2.1. At the same time, we downloaded several co-crystal structures of Mpro and its inhibitors from RCSB PDB, including 6lze, 6m0k, 6wtj, 6y2f, 7jyc, 7k0g, 7k0h, 7k40, 7lbn, 7ltj, 7lzv, 7m04, 7tdu, 7tfr, and 7tll. The PDB number corresponds to the inhibitor name attached to the supplementary materials (Table S2). We treated the complex with PyMOL to save the protein as pdb and the ligand as mol2, then generated individual pharmacophore models using AncPhore, and then combined these pharmacophore models to screen the small molecule database.
Molecular dynamic simulation and post-MD analysis
In the MD simulation phase, the force field used was Amber14SB + GAFF. RESP2 charge of small molecules was calculated using Multiwfn in combination with ORCA [26, 45,46,47]. GROMACS (5.1.2) was used to set up initial parameters within TIP3P water model dodecahedral box with an edge distance of 1 nm. The complexes were neutralized with 0.15 M Nacl. We then performed energy minimization utilizing the steepest plummet technique to restrict the energy to 100 kJ/mol/nm [48].
In the NVT phase, the temperature rises from 0 to 300 K in 100 ps and continues for 100 ps. Hydrogen atoms are confined by LINCS. Temperature coupling algorithm is V-rescale without pressure coupling. Cutoff scheme is Verlet, and periodic boundary conditions are used in all directions. Subsequently, we perform npt for 500 ps, the pressure control mode is Berendsen, and no initial velocity is generated [49]. In the formal MD stage, the time is set to 100 ns. And the voltage control mode is changed to Parrinello-Rahman, and the temperature control mode remains unchanged.
We extracted RMSD of protein, small molecule and complex, RMSF of amino acid of protein, a number of hydrogen bonds formed between small molecule and protein, rdf of 50 ns after simulation and radius of gyration of protein, combined Rg (deleting xyz direction information) and RMSD information of complex, and generated sham file by using sham command of gmx. The xpm2png.py script was used to generate that free energy topography, and the shamlog and bindex files were used to find the conformation information of the complex at the lowest energy point.
Binding free energy calculation
We installed gmx_MMPBSA [50] and generated an input file for calculating MMGBSA with a start time of 50 ns and an end time of 100 ns, sampling 5000 frames, sampling every 10 frames. All other settings are default.
In summary, the following is how the binding free energy is calculated:
Data analysis
GraphPad Prism 9.0 is used to process the data, and PyMOL and Discovery Studio Visualizer are used to process the view.
Data availability
The data that supports the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy.
References
WHO coronavirus disease (COVID-19) dashboard. Available online: https://covid19.who.int/ (accessed on 19 July 2022)
Zhu ZX, Lian XH, Su XS et al (2020) From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir Res 21(1):224. https://doi.org/10.1186/s12931-020-01479-w
Chen B, Tian EK, He B et al (2020) Overview of lethal human coronaviruses. Signal Transduct Target Ther 5(1):89. https://doi.org/10.1038/s41392-020-0190-2
Gorbalenya AE, Baker SC, Baric RS et al (2020) The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5(4):536–544. https://doi.org/10.1038/s41564-020-0695-z
Bump JB, Baum F, Sakornsin M et al (2021) Political economy of covid-19: extractive, regressive, competitive. Bmj-Brit Med J 372:n73. https://doi.org/10.1136/bmj.n73
Clemente-Suarez VJ, Ramos-Campo DJ, Mielgo-Ayuso J et al (2021) Nutrition in the actual COVID-19 pandemic. A narrative review. Nutrients 13(6):1924. https://doi.org/10.3390/nu13061924
Chan JF-W, Kok K-H, Zhu Z et al (2020) Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 9(1):221–236. https://doi.org/10.1080/22221751.2020.1719902
Wu C-r, Yin W-c, Jiang Y et al (2022) Structure genomics of SARS-CoV-2 and its Omicron variant: drug design templates for COVID-19. Acta Pharmacol Sin. https://doi.org/10.1038/s41401-021-00851-w
Yoshimoto FK (2020) The proteins of severe acute respiratory syndrome coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J 39(3):198–216. https://doi.org/10.1007/s10930-020-09901-4
Klemm T, Ebert G, Galleja DJ et al (2020) Mechanism and inhibition of the papain-like protease PLpro of SARS-CoV-2. EMBO J 39(18): e106275. https://www.ncbi.nlm.nih.gov/pubmed/32845033
Baez-Santos YM, St John SE, Mesecar (2015) The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115: 21–38. https://www.ncbi.nlm.nih.gov/pubmed/25554382
Lv Z, Cano KE, Jia L et al (2021) Targeting SARS-CoV-2 proteases for COVID-19 antiviral development. Front Chem 9: 819165. https://www.ncbi.nlm.nih.gov/pubmed/35186898
Kneller DW, Phillips G, Weiss KL et al (2020) Unusual zwitterionic catalytic site of SARS–CoV-2 main protease revealed by neutron crystallography. J Biol Chem 295(50): 17365–17373. https://www.sciencedirect.com/science/article/pii/S0021925817506225
Mengist HM, Dilnessa T, ** T et al (2021) Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front Chem 12(9):622898. https://www.frontiersin.org/article/10.3389/fchem.2021.622898
Chen YW, Yiu CPB (2020) Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 9:129. https://doi.org/10.12688/f1000research.22457.2
He J, Hu L, Huang X et al (2020) Potential of coronavirus 3C-like protease inhibitors for the development of new anti-SARS-CoV-2 drugs: insights from structures of protease and inhibitors. Int J Antimicrob Agents 56(2):106055. https://doi.org/10.1016/j.ijantimicag.2020.106055
Chuck C-P, Chong L-T (2010) Profiling of substrate specificity of SARS-CoV 3CLpro. PLoS One 5(10):e13197. https://doi.org/10.1371/journal.pone.0013197
Kneller DW, Phillips G, O’Neil HM et al (2020) Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat Commun 11(1):3202. https://doi.org/10.1038/s41467-020-16954-7
Moustaqil M, Ollivier E, Chiu H-P et al (2021) SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerging Microbes & Infections 10(1):178–195. https://doi.org/10.1080/22221751.2020.1870414
** Z, Du X, Xu Y et al (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811):289–293. https://www.ncbi.nlm.nih.gov/pubmed/32272481
Marzi M, Vakil MK, Bahmanyar M et al (2022) Paxlovid: mechanism of action, synthesis, and in silico study. Biomed Res Int 2022: 7341493. https://www.ncbi.nlm.nih.gov/pubmed/35845944
Unoh Y, Uehara S, Nakahara K et al (2022) Discovery of S-217622, a noncovalent oral SARS-CoV-2 3cl protease inhibitor clinical candidate for treating COVID-19. J Med Chem 65(9): 6499–6512. https://www.ncbi.nlm.nih.gov/pubmed/35352927
Gao S, Sylvester K, Song L et al (2022) Discovery and crystallographic studies of trisubstituted piperazine derivatives as non-covalent SARS-CoV-2 main protease inhibitors with high target specificity and low toxicity. J Med Chem 65(19):13343–13364. https://doi.org/10.1021/acs.jmedchem.2c01146
Lockbaum GJ, Reyes AC, Lee JM et al (2021) Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses-Basel 13(2):174. https://doi.org/10.3390/v13020174
Rossetti GG, Ossorio MA, Rempel S et al (2022) Non-covalent SARS-CoV-2 M-pro inhibitors developed from in silico screen hits. Sci Rep 12(1):2505. https://doi.org/10.1038/s41598-022-06306-4
Mesecar AD, St John S et al (2020) A taxonomically-driven approach to development of potent, broad-spectrum inhibitors of coronavirus main protease including SARS-CoV-2 (COVID-19). PDB. https://doi.org/10.2210/pdb6W79/pdb
Dai Q et al (2021) AncPhore: a versatile tool for anchor pharmacophore steered drug discovery with applications in discovery of new inhibitors targeting metallo-beta-lactamases and indoleamine/tryptophan 2,3-dioxygenases. Acta Pharm Sin B 11(7):1931–1946. https://doi.org/10.1016/j.apsb.2021.01.018
SARS-CoV-2 Data(NCBI). Available online: https://www.ncbi.nlm.nih.gov/sars-cov-2/
Greasley SE, Noell S, Plotnikova O et al (2022) Structural basis for the in vitro efficacy of nirmatrelvir against SARS-CoV-2 variants. J Biol Chem 298(6):101972. https://doi.org/10.1074/jbc.M109089200
Tan K, Maltseva NI, Endres MJ et al The crystal structure of SARS-CoV-2 Omicron Mpro (P132H) in complex with masitinib. PDB. https://doi.org/10.2210/pdb7TVX/pdb.
Kneller DW, Li H, Galanie S et al (2021) Structural, electronic, and electrostatic determinants for inhibitor binding to subsites S1 and S2 in SARS-CoV-2 main protease. J Med Chem 64(23):17366–17383. https://doi.org/10.1021/acs.jmedchem.1c01475
Pairwise Structure Alignment(RCSB PDB). Available online: https://www.rcsb.org/alignment.
**ong G, Wu ZX, Yi JC et al (2021) ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res 49(W1): W5–W14. https://doi.org/10.1093/nar/gkab255
Alhossary A, Handoko SD, Mu YG et al (2015) Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics 31(13):2214–2216. https://doi.org/10.1093/bioinformatics/btv082
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Dai W, Zhang B et al (2020) Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368(6497):1331–1335. https://doi.org/10.1126/science.abb4489
Vuong W, Khan MB et al (2020) Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun 11(1):4282. https://doi.org/10.1038/s41467-020-18096-2
Zhang L, Lin D et al (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368(6489):409–412. https://doi.org/10.3390/v13020174
Andi B et al (2022) Hepatitis C virus NS3/4A inhibitors and other drug-like compounds as covalent binders of SARS-CoV-2 main protease. Sci Rep 12(1):12197. https://doi.org/10.1038/s41598-022-15930-z
Dampalla CS, Zheng J et al (2021) Postinfection treatment with a protease inhibitor increases survival of mice with a fatal SARS-CoV-2 infection. Proc Natl Acad Sci U S A 118(29):e2101555118. https://doi.org/10.1073/pnas.2101555118
Narayanan A, Narwal M et al (2022) Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun Biol 5(1):169. https://doi.org/10.1038/s42003-022-03090-9
Clyde A, Galanie S et al (2022) High-throughput virtual screening and validation of a SARS-CoV-2 main protease noncovalent inhibitor. J Chem Inf Model 62(1):116–128. https://doi.org/10.1021/acs.jcim.1c00851
Dampalla CS et al (2021) Structure-guided design of potent inhibitors of SARS-CoV-2 3CL protease: structural, biochemical, and cell-based studies. J Med Chem 64(24):17846–17865. https://doi.org/10.1021/acs.jmedchem.1c01037
Kneller DW, Li H et al (2022) Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat Commun 13(1):2268. https://doi.org/10.1038/s41467-022-29915-z
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592. https://doi.org/10.1002/jcc.22885
Neese F (2018) Software update: the ORCA program system, version 4.0. WIREs Comput Mol Sci 8:e1327. https://doi.org/10.1002/wcms.1327
Lu T, Sobtop, Version 1.0(dev3.0), http://sobereva.com/soft/Sobtop (accessed on 26 Mar 2022 )
Weng YL et al (2021) Molecular dynamics and in silico mutagenesis on the reversible inhibitor-bound SARS-CoV-2 main protease complexes reveal the role of lateral pocket in enhancing the ligand affinity. Sci Rep 11(1):7429. https://doi.org/10.1038/s41598-021-86471-0
Mahmud S et al (2021) Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Brief Bioinform 22(2):1402–1414. https://doi.org/10.1093/bib/bbaa428
Valdés-Tresanco MS et al (2021) gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput 17(10):6281–6291. https://doi.org/10.1021/acs.jctc.1c00645
Funding
This research was funded by the Branch Office of Science and Technology in Sichuan Province (grant number 2020JDS0039).
Author information
Authors and Affiliations
Contributions
Conceptualization, Yunfan Shi and Qiufu Li; methodology, Yunfan Shi; software, Qiufu Li and Liting Dong; validation, Yunfan Shi; formal analysis, Yunfan Shi; investigation, Zhuang Ju and Liting Dong; resources, Zhuang Ju and Yanru Cui; data curation, Yunfan Shi; writing original draft preparation, Yunfan Shi; writing review and editing, Yunfan Shi; visualization, Yunfan Shi and Liting Dong; supervision, Yiran Liu and Jiaoyu He; project administration, ** Ding; funding acquisition, ** Ding. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• The study demonstrates the high conserved sequence and structure for Mpro of SARS-CoV-2.
• The study used the complete process of virtual screening.
• The study identified 5 potential inhibitors for Mpro of SARS-CoV-2.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shi, Y., Dong, L., Ju, Z. et al. Exploring potential SARS-CoV-2 Mpro non-covalent inhibitors through docking, pharmacophore profile matching, molecular dynamic simulation, and MM-GBSA. J Mol Model 29, 138 (2023). https://doi.org/10.1007/s00894-023-05534-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05534-3