
Abstract
Objective
The COVID-19 epidemic is raging around the world, with the emergence of viral mutant strains such as Delta and Omicron, posing severe challenges to people’s health and quality of life. A full understanding life cycle of the virus in host cells helps to reveal inactivation mechanism of antibody and provide inspiration for the development of a new-generation vaccines.
Methods
In this work, molecular recognitions and conformational changes of SARS-CoV-2 spike protein mutants (i.e., Delta, Mu, and Omicron) and three essential partners (i.e., membrane receptor hACE2, protease TMPRSS2, and antibody C121) both were compared and analyzed using molecular simulations.
Results
Water basin and binding free energy calculations both show that the three mutants possess higher affinity for hACE2 than WT, exhibiting stronger virus transmission. The descending order of cleavage ability by TMPRSS2 is Mu, Delta, Omicron, and WT, which is related to the new S1/S2 cutting site induced by transposition effect. The inefficient utilization of TMPRSS2 by Omicron is consistent with its primary entry into cells via the endosomal pathway. In addition, RBD-directed antibody C121 showed obvious resistance to Omicron, which may have originated from high fluctuation of approaching angles, high flexibility of I472-F490 loop, and reduced binding ability.
Conclusions
According to the overall characteristics of the three mutants, high infectivity, high immune escape, and low virulence may be the future evolutionary selection of SARS-CoV-2. In a word, this work not only proposes the possible resistance mechanism of SARS-CoV-2 mutants, but also provides theoretical guidance for the subsequent drug design against COVID-19 based on S protein structure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As the largest RNA virus, coronaviruses contain about 30 kb of a non-segmental positive-sense genome, which can cause different diseases in poultry and mammals. The current three coronavirus causing human disease are all β-CoVs, including the severe acute respiratory syndrome coronavirus (SARS-CoV), the Middle East respiratory syndrome coronavirus (MERS-CoV), and the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1]. SARS-CoV was initially reported in Guangdong China in 2002 [2], and infected over 8000 people worldwide with fatality rate of 10% [3, 4]. MERS-CoV emerged in Saudi Arabia in June 2012 [5], showing low infection rate but higher fatality rate of 34.4% [1]. On February 11, 2020, the acute pneumonia caused by SARS-CoV-2 has been named COVID-19. It has led to over 500 million infections and 6 million deaths worldwide due to its rapid global spread, long incubation period, and proclivity to evolve new variants [6]. Rabaan et al. revealed that SARS-CoV-2 is structurally similar to SARS-CoV in general, but slightly different in spike protein [7]—the most important structural protein [8]. Compared with SARS and MERS, SARS-CoV-2 is less pathogenic, while possessing higher transmission rate in humans due to the presence of a furin-like cleavage site in spike and high affinity with human angiotensin-converting enzyme 2 (hACE2) receptor [8, 9].
For searching an effective therapy, it is necessary to understand the transmission of SARS-CoV-2 between host populations, especially multibody interactions of key viral proteins including spike. The SARS-CoV-2 spike consists of S1 and S2 subunits: the former comprises a receptor binding domain (RBD) to initiate viral entry into the target cell, as well as an N-terminal domain (NTD); [10] the latter contains six domains (i.e., fusion peptide, HR1 (heptapeptide repeat 1), central helix, HR2 (heptapeptide repeat 2) junction domain, transmembrane domain, and cytoplasmic tail), which mediate the fusion of viral to host cellular membranes after significant conformational rearrangements [1, 11,12,13,14]. The S protein binds to hACE2 via RBD, which is a key step of membrane fusion and is closely related to the infection, tropism, and pathogenicity of SARS-CoV-2 [8, 15, 16]. The up- and down-conformations of spike were observed prior to membrane fusion, in which the up-state was more conducive to molecular recognition by hACE2 receptor [17,18,19]. S protein is then cleaved and activated by furin-like protease and transmembrane protease serine 2 (TMPRSS2) in turn, undergoing significant structural rearrangements [20,21,22,23,24]. After membrane fusion and endocytosis mediated by transmembrane proteases, the genomic RNA is eventually expressed and replicated to assemble new coronaviruses particles. In sum, RBD conformational state of spike, the interactions respectively with hACE2 and TMPRSS2 are all key scientific questions to reveal the life cycle of SARS-CoV-2 [25].
In the face of the COVID-19 pandemic, the structure and transmission of the pathogen SARS-CoV-2 have received the greatest attention, and numerous effective intervention measures have emerged successively. RNA vaccines, live attenuated vaccines, and inactivated vaccines are all extensively used around the world as the primary means of prevention and treatment against COVID-19. In addition to them, there are several types of vaccines, all of which are in phases 3 or 4 clinical trials (see Fig. 1) [26]. By April 11, 2022, total 344 SARS-CoV-2 vaccine candidates have been developed and tested in various stages of clinical trials, and 31 of them in use [27]. Universal vaccination appears to prevent the severe course of illness and deaths caused by all occurring variants of concern (VOCs) [28, 29]. By April 13, 2022, a total of 11,294,502,059 vaccine doses have been administered all over the world, with 65.5% of the population having received at least one dose of a COVID-19 vaccine [30]. Given the increasing number of infections, booster jab of COVID-19 vaccine had been widely adopted to help enhance the action of antibodies and prolong protection [31, 32]. In addition, some antiviral drugs and therapeutic antibodies are also recommended, such as bamlanivimab, etesevimab [33], sotrovimab, casirivimab, imdevimab [34, 35], Evusheld [36] (acting on RBD to affects the binding with hACE2), and Actemra (inhibiting IL-6 induction) [37]. The previously authorized use of sotrovimab will be discontinued until April 5, 2022, as it may not be effective against the Omicron variant. At present, there are few antiviral drugs, only baricitinib (selective inhibitor against Janus-activated kinases 1 and 2), Paxlovid, and molnupiravir (targeting autophagy and unfolded protein) were reported [38].

Mutants, infection mechanisms, and treatment strategies against SARS-CoV-2. A Variants emerging from March 2020 to January 2022, new weekly cases and deaths as well as treatment strategies at the corresponding time; B the life cycle of SARS-CoV-2 and the action course of vaccines
After more than 2 years of fighting against the epidemic, the situation remained dire, and many types of variants of concern (VOCs) have emerged, including Delta (B.1.617.2), Mu (B.1.621), and Omicron (B.1.1.529) [39]. The epidemic storm dominated by Delta was popular for a time, showing high infectivity and hospitalization rate [40, 41]. Despite the many existing treatment strategies that have been available, Delta still remained more prevalent than other subtypes in quite a long period of time, with the second highest weekly new cases and moderate weekly deaths (see Fig. 1). The Mu variant was first identified in Colombia in January 2021 [42] and has become a new member of variant of interest (VOI). Subsequently, Mu was shown to be remarkably resistant not only to the sera from COVID-19 convalescents and BNT162b2-vaccinated individuals, but also to the inactivated vaccine-elicited antibodies [43]. On November 26, 2021, the WHO named the new variant B.1.1.529 to be Omicron with a large number of mutations in S protein RBD [44, 45]. Studies on spike VSV pseudovirus have shown that the neutralizing activity against Omicron is substantially reduced after 2 or 3 doses (booster) of inactivated vaccine, falling below the protective threshold in several cases [46, 47]. So far, Omicron has proven to be a highly contagious variant and may evolve new mechanisms infecting humans [47,48,49].
There are a variety of new drugs and treatments for COVID-19 that are still under continuous development. Nevertheless, inadequate vaccination rates and a declining ability to neutralize viral variants both made the situation difficult [50, 51]. In this work, based on molecular simulations of four strains (WT, Delta, Mu, and Omicron) spike. we demonstrated how mutations respectively affect the recognition of S protein with hACE2, TMPRSS2, and C121 (a neutralizing antibody that blocks the association with hACE2) during infection, and then revealed the possible mechanism of vaccine inactivation. It provides theoretical guidance for the development, evaluation, and design of the next generation of vaccines against COVID-19.
Materials and methods
Preparation of simulation systems
The initial S protein structure was taken from the RCSB protein data bank (PDB), with all missing residues completed by the SWISS-MODEL server [52]. Considering that the up-state of spike is necessary for our study the crystal structure of wild-type spike with one RBD up (PDB ID: 7KJ2, bond with hACE2) was selected as the template for subsequent system construction [53]. After deleting the amino acid sequence of deletion mutation and hACE2, the model with deletion mutation was established by SWISS-MODEL. The whole mutants are obtained by completing the residual mutation in the Pymol mutagenesis module [54]. Then, HADDOCK 2.4 sever [55] was used to build the following twelve systems: (1) four spike trimer complexes with hACE2 receptor; (2) four spike trimer docked complex models with TMPRSS2; (3) four spike trimer docked complex models with antibody C121. The structures of TMPRSS2 and C121 were available from PDBs 7MEQ [56] and 7K8X [57], respectively. For convenience of analysis, the twelve SP complex systems are denoted by WT_hACE2/_TMPRSS2/_C121, Delta_hACE2/_TMPRSS2/_C121, Mu_hACE2/_TMPRSS2/_C121, and Omicron_hACE2/_TMPRSS2/_C121, respectively.
Molecular dynamics simulation
Molecular dynamics (MD) simulation has been widely used for exploring the thermodynamic characteristics of biomolecules at the atomic level. In order to analyze how the three drug resistance mutations (i.e., Delta, Mu, and Omicron) affect molecular recognition of S protein and its essential partners, comparative MD simulations were conducted for the twelve S protein complex systems using ff14SB force field and Amber 19 package [58, 59]. All solutes were solvated in octahedral box with the boundary of 15.0 Å by TIP3P water model [60].
To reduce the space collision in simulated systems, two energy optimizations (EM) were carried out. The first EM stage is composed of 5000 steps of steepest descent and 5000 steps of conjugate gradient optimizations with constraint force constant of 500 kcal·mol−1·Å−2; in the second EM stage, the solute constraint is completely removed, also containing the same optimization steps and convergence criterion with energy gradient less than 0.01 kcal·mol−1·Å−2.
After EM procedure, MD simulations were also divided into two stages: (1) 5 ns solute-constrained MD simulations with a force constant of 10 kcal·mol−1 Å−2, where temperature gradually increased from 0 to 300 K; (2) 95 ns solute-unconstrained productive MD simulations, adopting SHAKE algorithm [61] to constrain the hydrogen-containing atoms. During the simulation, non-bonded interaction radius and integration step were set to 10 Å and 2 fs, respectively, and the conformational fluctuation was monitored by VMD 1.9.3 package [62]. The atomic coordinates were stored every 10 ps, and a total of 10,000 conformations were collected for the subsequent analyses of molecular recognition and conformational change.
Binding free energy calculation
As an important physical and chemical parameter, binding free energy can be applied to effectively judge molecular recognition between receptors and ligands, and is also a key criterion for evaluating drug activity. Molecular mechanics/Poisson Boltzmann (MM/PBSA) method was used to calculate the binding free energies between four spike trimers (i.e., the WT, Delta, Mu, and Omicron variants) with hACE2, TMPRSS2, and C121. Total 25 snapshots were collected from each MD trajectory every 4 ns intervals from 1 to 100 ns, and used for the calculation of average binding free energy. The calculation formula is as follows:
where ∆H represents the total enthalpy change, T is the absolute temperature in Kelvin, and ∆S refers to the total entropy change calculated using the normal mode method [63]. ∆EVDW indicates intramolecular van der Waals energy under vacuum, while ∆EELE refers to the electrostatic fraction. ∆GPBELE and ∆GPBSUR represent the hydrophilic and hydrophobic parts of solvation binding free energy, respectively.
Identification of key residues
Energy decomposition was carried out by molecular mechanics/generalized Born area (MM/GBSA) method [64]. According to MM/GBSA, the binding energy values between receptors and ligands can first be assigned to each residue, then be subdivided into internal energy in vacuum (including polar electrostatic interactions and non-polar van der Waals interactions), polar solvation energy and non-polar solvation energy, and eventually be decomposed into the main chain and side chain of each residue. Here, the polar and non-polar solvation energy can be calculated with generalized Born (GB) model and LCPO algorithm [65], respectively.
The identification of hot spots between the PPIs plays a key part in the detection of active sites of receptors. By now, hierarchical clustering (HC) analysis has been an efficient technology for determining the hot spots and common binding characteristics [66]. In this work, key residues were identified by HC analysis based on energy contributions per amino acid using R statistical package. Manhattan distance is used to describe the similarity levels among vectors. It is defined as:
where i indicates the dimensional index of individual binding energies a and b at residual level. To implement cluster discrimination, the Ward’s minimum variance method was adopted here [67]. Finally, the calculation result files were processed using the online tree generator iTOL to formulate the hierarchical tree graph shown in color-coded modes [68]. Through energy decomposition and HC analyses, the key residues in the PPIs of S protein with three essential partners can be obtained.
Solvent accessible surface area (SASA) calculation
SASA is one of the key parameters to be considered in the study of biomolecular recognition [69, 70]. SASA is generally defined as the surface area formed by a water probe ball rolling around the surface of biomolecules. The larger the SASA, the weaker the hydrophobic effect. In the actual calculation, SASA can be assigned into protein residual level. It is worth mentioning that some amino acids are buried in the proteins deeply and the corresponding SASA is zero. In addition, the buried SASA (Sburied) is an important criterion for evaluating the strength and effectiveness of molecular recognition between proteins. The computational formula is as follows:
Here, SA and SC are used to describe the SASAs of spike protein and complexes, respectively. In this work, the SASA of four trimers (i.e., WT, Delta, Mu, Omicron) and their complexes with hACE2/TMPRSS2/C121 can be computed from MD trajectories with gmx sasa module using the algorithms of Eisenhaber et al. [71].
Self-defined parameters
In order to more accurately describe the mutual recognition between S protein and three essential partners (i.e., hACE2, TMPRSS2, and C121), the following three self-defined parameters are provided. (1) Water basin: formed by the part near mass center in the up-state RBD (chain A, E516-G566) and the adjacent HR1 (chain C, G910-D985) and NTD (chain B, N30-F55), in which the amount of water varies with the open and closed state of RBD [72]. In water basin, the less the amount of water is, the easier it is for RBD to maintain the up-state and bind with hACE2, and not vice versa. (2) Tetrahedron height: the catalytic triplet of TMPRSS2 (i.e., H296, S441, S460) were found to interact with the cleavage sites (i.e., R815, S816) of SARS-CoV-2 spike [73]. In the spike complexes with TMPRSS2, tetrahedron height representing vertical distance from the center points of R815/S816 to the plane formed by Cα atoms of the catalytic triplet. The size and stability of tetrahedron height can reflect the affinity of S protein and TMPRSS2 to a certain extent: the farther the distance, the harder it is to bind. (3) Approaching angle: the normal vectors of the three planes (i.e., S2 region horizontal plane, antibody plane interacting with RBD, and RBD plane interacting with antibodies) were defined, and approaching angle 1 is formed by the first two vectors, with approaching angle 2 by the latter vectors. The Cα of three amino acids is connected to form a plane, and we define three such planes. The horizontal plane of S2 region is defined by T998 of the three chains (located in the stable spiral region). The other two are represented respectively by three points of the interface between RBD and C121 (RBD: G446, E484, V503; C121: T28, V54, S88). Different types of neutralizing antibodies have special approaching angle to S protein interface, which is related to its neutralization effect [74].
Results
Introduction to simulation systems
The interactions of WT S protein and three mutants respectively with three partners (i.e., hACE2, TMPRSS2, and C121) was explored in this work. Figure 2 shows their molecular structure and the list of mutation sites. Spike protein is composed of three chains with residue range from A27 to S1147. According to the order of residues, the noteworthy domains in spike are as follows: (1) RBD (R319-N540), its transition from down- to up-conformation is a key step before binding with hACE2, being a hot spot targeted by mainstream antibodies; (2) NTD (S13-F318), the target region of another potent antibody, although its specific biological function is unclear; (3) 630 loop (V620-S640), the transition between ordered and disordered conformation affects the opening and closing efficiency of RBD; (4) S2’ (R815/S816), as the cleavage site of TMPRSS2, it is closely related to virus replication; (5) FPPR (F823-P862), it not only affects the recognition by TMPRSS2, but also acts synergistically with 630 loop to affect the transition of down- to up-conformation of RBD; (6) HR1 (G910-D985), as the most important part of the transmembrane helices, they insert the fusion peptides into the target cell membrane, by undergoing significant conformational rearrangement. In terms of mutation sites, compared with WT, Delta possesses 7 mutations and 2 deletions, with Mu having 9 mutations, as well as Omicron having 30 mutations, 5 deletions, and 1 insertion. Excessive mutations suggest that Omicron spike may exhibit novel characteristics including substrate recognition, conformational change, and immunogenicity.

Molecular structure of S protein trimer and its essential partners (A), as well as the list of mutation sites (B). The circled part is the binding site for the three essential partners, where the red parts of TMPRSS2, C121, and hACE2 represent the recognition interface by spike. S protein trimer is shown using surface model, and solid-ribbon model is added to the outermost chain A. The binding regions for the three partners with spike are Q24-L45/L79-Y83/T324-D355 of hACE2, H296-A486 of TMPRSS2, and H: G26-T30/W50-S75/Y95-Y112 of C121, which are represented in red in the upper right a/b/c panels
Overall structural convergence
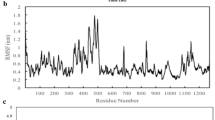
Figure 3A, S1A, and S2A show the potential energy of all 12 systems over simulation time, as well as the mean and standard deviation. The systems all reach potential energy equilibrium after 5 ns, indicating that the simulation process is smooth and the trajectory is reliable. By measuring the overall difference between the conformational snapshot and the first frame, RMSD values can be used to effectively evaluate whether the trajectories of biomolecules have reached the equilibrium state in the MD simulation. As shown from Fig. 3B, S1B, and S2B, RMSD values keep relatively stable after 50 ns, and the equilibrium trajectory in this period can be used for subsequent analyses of conformational change and molecular recognition. Overall, the fluctuation of RMSD values is similar for the 12 investigated systems, ranging from 3.47 Å (Mu_TMPRSS2) to 7.41 Å (Mu_hACE2). The relatively high RMSD value is due to the large simulation system with more than 3500 residues and excessive number of mutations.

Conformation convergence parameters for MD simulations of the WT_hACE2, Delta_hACE2, Mu_hACE2, and Omicron_hACE2 systems. The variation of potential energy (A) and RMSD (B) with time; RMSF distribution at residue level of S protein chain A in the four systems (C); RMSF correlation of S protein chain A between Delta_hACE2 and WT_hACE2 (D)
RMSF can provide flexibility difference of the simulation system at residual level. As shown in Fig. 3C, the RMSF distribution in four Spike_hACE2 systems are highly similar. Specially, HR1(G910-D985) has the lowest flexibility, which is closely related to the high rigidity of transmembrane helical [ The relative position of TMPRSS2 catalytic triplet and spike protein S2’ cleavage site, as well as the variation of tetrahedron height over time in WT_/Delta_/Mu_/Omicron_TMPRSS2 systems
A comprehensive consideration of the binding energy and cleavage efficiency of TMPRSS2 and S protein is crucial for the correct evaluation of the infectivity and virulence from different SARS-CoV-2 virus strains. Specially, WT is easily cut by TMPRSS2, but its low binding ability greatly reduces infectivity and toxicity. The Delta mutants showed strong viral replication and pathogenicity, suggesting that TMPRSS2 inhibitors may have high antiviral potential against this type of strain [88]. Although Mu spike has the strongest binding capacity to TMPRSS2, while accompanied by a weak cutting ability, so the transmembrane efficiency is low, which may be one of the reasons for the narrow epidemic area of Mu virus strain. Omicron obviously abandoned the transmembrane of TMPRSS2 pathway with much lower binding ability and cutting ability than that of Delta, which was consistent with the significant decrease of syncytial formation [100]. In other words, the endosomal pathway has emerged as a novel option for transmembrane of Omicron strain [101]. Mutations of more than 30 residues resulted in reduced S1/S2 cleavage efficiency, accompanied by an increase in the positive charge of the S protein, which all contribute to Omicron mutants favoring endosomal pathway [102].
Molecular recognition of S protein and C121
The purpose of vaccination for COVID-19 patients is to make the body produce corresponding antibodies, which is the main strategy against SARS-CoV-2. Study on escape the mechanism of viral mutants to antibodies can provide a basis for the development of vaccine and the selection of clinical diagnosis and treatment. C121, encoded by VH3-53, is a strongly neutralizing antibody that can be repeatedly found in the plasma of most convalescent or vaccinated patients [103]. Current neutralization mechanisms of antibodies targeting SARS-CoV-2 are diverse, such as blocking receptor binding, steric hindrance, locking S protein in down-conformation, and so on. Antibody C121 binds to RBD’s receptor-binding ridge (i.e., the main epitopes) and overlaps with the polar and hydrophobic residues in the plane responsible for the interaction with hACE2, exerting inhibitory activity through the mechanism of competitive binding [57]. Currently, the most mainstream antibodies used to treat COVID-19 are RBD-directed type. In addition to antibody C121, there are some antibodies with the same epitope, such as C002, C009, 5A6, and COVA2-39 [63].
Antibodies targeting different epitopes bind to S protein at different approaching angles, whereas those from the same epitope having similar approaching angles [67]. Given the right approaching angle is the key factor for antibody C121 to capture spike antigen epitope, Fig. 8 comparatively shows their variation over simulation time in the WT_C121, Delta_C121, Mu_C121, and Omicron_C121 systems. In terms of approaching angle 1 used to characterize the recognition of spike by C121 at the macro level, the values from high to low are Omicron_C121 > Mu_C121 > Delta_C121 ≈ WT_C121; while for the approaching angle 2 more suitable for representing the C121-RBD recognition at the micro level, the systems are Omicron_C121 > WT_C121 ≈ Delta_C121 ≈ Mu in descending order. By comparing the deviation of approaching angles in three mutants to those in the WT, Omicron mutants shows obvious immune escape from this kind of RBD-directed antibodies.

Two approaching angles for RBD-directed neutralizing antibody C121 (A), as well as their variation with simulation time in the WT_C121, Delta_C121, Mu_C121, and Omicron_C121 systems (B)
Fig. S10 shows the top 10 residues with the largest contribution of Sburied on the recognition interface in the four Spike_C121 systems. The mean Sburied values of WT_C121, Delta_C121, Mu_C121, and Omicron_C121 are 6.51, 6.12, 5.3, and 5.22 Å2, initially showing the binding potency of descending order. Key residues with high frequency including Y449, L455, F456, E484, F486, Y489, F490, Q493, and Q498 have all been reported previously [63]. The key amino acids in WT_C121 and Delta_C121 are almost the same, and all of them appear at high frequencies except T500, which also guarantees their similar Sburied values. The key residues in the Omicron_C121 system change significantly. some high frequency residues (e.g., L455 and F456) that appear in the other three systems are no longer present, while new contribution sites (e.g., G485, S494, and C488) and mutation residues (e.g., E484A and Q498R) both lead to the minimal Sburied. The variation degree of key residues in Mu is between Delta and Omicron, with P479 only appearing in this system.
Several studies have confirmed that residue mutations (e.g., K417N, E484K, L452R, N501Y) located at the interface of RBD (R319-N540) can reduce the neutralizing activity of antibodies to varying degrees [63, 104, 105]. By superimposing the RBMs (N437-P507) of the four Spike_C121 complexes, it is found that there is significant conformational change in the I472-F490 loop, which also covers some key epitope residues (e.g., E484, F486, Y489, and F490). In fact, a series of previous studies have also shown that the I472-F490 loop exhibits great flexibility in the Alpha, Beta, Delta, Mu, and Omicron mutants. In order to quantitatively compare the conformational changes of the loop regions, the changes in the distance between Ca atoms of residues P479, E484, and Q493 were measured for the four Spike_C121 systems (see Fig. 9). These residues were selected for the following reasons: P479 is a unique contact residue in the Mu system; Q493, not far from the loop, is a good reference point with a constant position; the high-frequency contact residue E/K/A484 appears in all spike systems except Mu. The mean E484-Q493 distances of Mu_C121 and WT_121 are 22.41 and 20.60 Å, while they decrease to 14.22 and 14.32 Å for Delta_C121 and Omicron_C121. In addition, Mu_C121 has a shorter P479-Q493 distance than the other three systems with a decrease of about 3 Å, which is consistent with the fact that P479 is only the key contact residue of Mu_C121. In summary, the I472-F490 loop in the four Spike_C121 systems underwent obvious conformational change, which greatly affected the Sburied value of the interface, intermolecular binding force, and antibody efficacy [106].

Molecular recognition of antibody C121 with RBM in the four Spike_C121 complexes (A), as well as the distance of key residues E/K/A484-Q493 and P479-Q493 as a function of simulation time (B)
Fig. S11 shows binding free energies of spike trimer to antibody C121. Under the same calculation parameters, the descending order of the predicted binding capacity with C121 was WT, Delta, Mu, and Omicron. Based on energy decomposition data of key residues, three different clusters (C1–C3) especially C1 (Y489, F490, F486) and C3 (V483, E484, F456, L455, Y449) conducive to the association of S protein and C121 were provided by HC analysis. In the Omicron and Mu systems, the decrease of binding free energy is mainly due to the weakening of energy contribution of key residues in clusters C1 and C3. Mutations such as E484K in Mu and E484A/Q493R/Q498R in Omicron were not conducive to molecular recognition of SP by antibody C121. In fact, E484K not only causes spatial collisions, but also leads to a positively charged environment at the antibody binding site, while E484A weakens some of the original hydrogen bonds of E484K [107]. According to the binding modes of Spike_C121, the hydrogen bond and hydrophobic interactions of Mu_C121 and Omicron_C121 were partially damaged compared with WT_C121, which reasonably explains the significant decrease in affinity between the viral mutants and the antibody.
Comparative molecular dynamics simulations were performed for 12 complexes. Potential energy, RMSD, and RMSF analyses all show that MD simulation trajectories are stable and reliable, and the flexibility of S protein RBM and S2’ cleavage sites decreased slightly with the binding of hACE2 and TMPRSS2. In the Spike_hACE2 system, the amount of water in the basin is WT > Mu ≈ Omicron > Delta. The order of 630 loop is WT > Mu > Delta > Omicron. The Sburied of Omicron and Delta are larger than Mu and WT. The affinity of mutants to hACE2 was higher than that of WT, and Delta was the strongest. Omicron does not rely on traditional key residues to maintain its affinity with hACE2, but R493. In the Spike_TMPRSS2 system, mutants use a different binding pattern than WT to recognize TMPRSS2. Because of the shift effect, the mutant may have the ability of double-site cutting. The short distance between S2’ site and catalytic triplet corresponding to the increase of Sburied is reflected in Mu and Delta systems. On the contrary, Omicron has a catalytic triplet with an increased Sburied but far away from TMPRSS2. The calculated results of binding free energy show that Mu > Delta > Omicron > WT. In Spike_C121 system, both Mu and Omicron have strong approaching angle fluctuation, especially Omicron. The average E484-Q493 distance of Delta_C121 and Omicron_C121 is significantly shorter, and the P479-Q493 distance of Mu_C121 is shorter than that of the other three systems, reducing by about 3 Å. The affinity of the mutants to C121 became weaker, among which Omicron was the weakest. HC analysis showed that E484, Q493, and Q498 had the most significant effect on affinity.
Discussion
SARS-CoV-2 spike protein is first recognized and adsorbed by hACE2, then cleaved by proteases such as TMPRSS2, and finally enters human host cells through virus-cell membrane fusion and endocytosis. The interactions between S protein and hACE2 receptors is a pivotal step in viral replication cycle, and its transmission efficiency depends to a large extent on this process [108, 109]. The S protein mutants enhance its affinity with hACE2 receptor by increasing hydrogen bonds, buried area, electrostatic interaction, stabilizing hot spot residues, and other mechanisms, so as to make it spread faster [76]. Several studies have confirmed that the replication rate of Omicron is slower than that of Delta, because the former is mediated by endocytosis independent of TMPRSS2 cleavage modification [84, 101]. Factually, the difference in cell entry pathway is related to clinical manifestation and severity of the disease [96]. Alex et al. estimated that Omicron-induced disease severity was about half that of Delta, with less infection of human lung cells and organs [81, 101, 110, 111]. The improvement of transmembrane efficiency will contribute to syncytial formation, directly inducing stronger viral pathogenicity [112, 113]. The syncytia formed by micron spikes were significantly attenuated in the Omicron mutant, which was consistent with the results from animal experiments [100, 102].
Mu and Omicron both showed severe resistance to a class of antibodies with the same epitope as C121, because the E484K/A mutation impaired the interactions with antibodies, as reported in previous studies [114]. Moreover, Omicron accumulated 15 mutations in RBD, which provided a solid structural foundation for its obvious immune escape. A variety of RBD-directed antibodies, including REGN10933, REGN1098, COV2-2196, LY-CoV555, C135, and S309, have been shown to significantly reduce their neutralizing activity against Omicron [115]. At present, the development of NTD-directed antibodies has met some bottlenecks, such as G142D and del143-145 both leading to immune escape of Omicron strains. The former cannot stably bind to NTD-directed neutralizing monoclonal antibodies such as S2X333 due to steric hindrance; the latter Y144del has been proven to cause a significant reduction in neutralization activity of NTD-supersite antibodies [105].
Based on MD simulation of WT/Delta/Mu/Omicron S protein in complex with hACE2/TMPRSS2/C121, Table 1 comparatively lists their conformational change and molecular affinity. Generally, the large-scale epidemic of a virus is determined by transmission efficiency, pathogenicity, immune escape, and other factors. As shown from Table 1, SARS-CoV-2 variants have a potential biological balance among transmission efficiency, pathogenicity, and immune escape, rather than showing an obvious monotonic trend. As the main epidemic strain of COVID-19 in the world, Omicron shows strong immune escape and weakened pathogenicity, which undoubtedly belongs to evolutionary direction conducive to widespread spread of the virus. There is no doubt that the protective immunity on patients infected with Omicron strains is significantly reduced by vaccination [111, 116, 117]. Fortunately, booster doses of the vaccine still provide better neutralization response, and antiviral drugs remain effective against Omicron [118]. Under the severe situation of epidemic wave caused by Omicron, in addition to physical prevention such as wearing masks and reducing crowd gathering, as well as improving vaccination rate, it is necessary to explore more conserved epitopes and develop more specific vaccines and therapeutic antibodies. Epitope prediction appeared in the eyes of vaccine developers as early as the outbreak of MERS-CoV, breaking through the stagnant research and development process and providing new vaccine candidates [119]. This technology is still in continuous application and development, and has been used in a variety of research and development of specific vaccines in recent years, such as monkeypox vaccine [120] and orthohantavirus vaccine [121]. Among them, multi-epitope vaccine is an effective way to deal with the continuous mutation of the virus, which brings good news for the future development of SARS-CoV-2 vaccine.
Conclusion
To explore recognition differences and conformational changes, revealing viral evolutionary characteristics and possible vaccine inactivation mechanism is purpose of the study. In Spike_hACE2 systems, compared with WT, the mutants still maintain high affinity with hACE2 by kee** the RBD UP, increasing the entrapment area, or using new residues to provide energy (such as R493 to bring about favorable electrostatic interactions). In the Spike_TMPRSS2 systems, the increased Sburied provides enough affinity for the mutant. The inconsistency between the binding free energy of Spike_TMPRSS2 and the distance between S2’ site and catalytic triplet indicates that the binding ability of the two is separated from the cutting ability of TMPRSS2. It is clear that Delta is significantly higher than WT in terms of TMPRSS2-dominated cleavage efficiency, while Omicron may select a novel cell fusion mechanism different from other mutants. In Spike_C121 system, the high fluctuation of approaching angles and I472-F490 loop, as well as the reduced recognition area, all explain the resistance of Mu and Omicron mutants to C121 antibody. Overall, Delta strain has high infectivity and pathogenicity with weak immune escape ability; while Omicron strain possesses strong infectivity and immune escape ability with weak pathogenicity; and Mu strain is somewhere in between. This work not only proposed the possible mechanisms of infectivity, virulence, and drug resistance of the variants, but also provided theoretical guidance for the design of a new generation of therapeutic antibodies and vaccines against COVID-19.
