
Abstract
Large-scale association analyses using whole-genome sequence data have become feasible, but understanding the functional impacts of these associations remains challenging. Although many tools are available to predict the functional impacts of genetic variants, it is unclear which tool should be used in practice. This work provides a practical guide to assist in selecting appropriate tools for variant annotation. We conducted a MEDLINE search up to November 10, 2023, and included tools that are applicable to a broad range of phenotypes, can be used locally, and have been recently updated. Tools were categorized based on the types of variants they accept and the functional impacts they predict. Sequence Ontology terms were used for standardization. We identified 118 databases and software packages, encompassing 36 variant types and 161 functional impacts. Combining only three tools, namely SnpEff, FAVOR, and SparkINFERNO, allows predicting 99 (61%) distinct functional impacts. Thirty-seven tools predict 89 functional impacts that are not supported by any other tool, while 75 tools predict pathogenicity and can be used within the ACMG/AMP guidelines in a clinical context. We launched a website allowing researchers to select tools based on desired variants and impacts. In summary, more than 100 tools are already available to predict approximately 160 functional impacts. About 60% of the functional impacts can be predicted by the combination of three tools. Unexpectedly, recent tools do not predict more impacts than older ones. Future research should allow predicting the functionality of so far unsupported variant types, such as gene fusions.
URL: https://cardio-care.shinyapps.io/VEP_Finder/.
Registration: OSF Registries on November 10, 2023, https://osf.io/s2gct.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Whole-genome sequencing (WGS) has become precise and affordable on a large scale, and several cohorts now involve hundreds of thousands of subjects (Halldorsson et al. 2022; Taub et al. 2022; The All of Us Research Program Investigators 2019). Statistical analyses have associated diseases with common and rare variants (Povysil et al. 2019), and the GWAS Catalog currently contains more than half a million associations (Sollis et al. 2023). However, the causal mechanism behind most genetic associations is unclear and can take a long time to understand. For example, even for the best-replicated locus in cardiovascular disease, it took four years to unravel its function (Harismendy et al. 2011; Wellcome Trust Case Control Consortium 2007). To accelerate the understanding of biological function, a series of computational tools have been proposed in recent years.
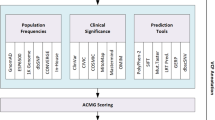
In this work, we consider Variant Effect Predictors (VEPs) to be databases or software packages that predict the functional impacts of genetic variants. Each VEP is usually specialized in annotating one or a few categories of variants, such as single nucleotide variations (SNVs), indels, missense variants, or structural variants (SVs) (Geoffroy et al. 2018; Pagel et al. 2019; Rentzsch et al. 2019; Vaser et al. 2016). The variety of VEPs and their functionalities poses the challenge of choosing the appropriate tool for a specific task, a topic that has been addressed in non-systematic reviews (Katsonis et al. 2022; Tabarini et al. 2022). Some reviews summarize VEPs for one type of variant only (Abramowicz and Gos 2018; Glusman et al. 2017). Other articles focus on variation relevant to the American College of Medical Genetics and Genomics/Association of Molecular Pathology (ACMG/AMP) guidelines (Ghosh et al. 2017; Kassahn et al. 2014). All reviews have in common that their summary tables group functional information into a few categories, usually SNVs, indels, and SVs only. This categorization limits the search for VEPs suitable for other categories of variants, such as missense mutations or copy number variation.
This work aims to provide a systematic overview of the broad range of variant types and their functional impacts across VEPs. To this end, we systematically searched MEDLINE and investigated the possible input and output of each tool. The efficient selection of the most appropriate tool for a specific task can easily be accomplished using an interactive website.
Methods
A systematic review was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (Page et al. 2021). The protocol was registered in OSF Registries on November 10, 2023 (https://doi.org/10.17605/OSF.IO/S2GCT).
Literature search
The literature search was conducted in the MEDLINE database. The search was restricted to articles published in English after January 1, 2014. This date was chosen to coincide with two milestones in genomics: the launch of the GRCh38 reference genome in December 2013 and the release of higher-throughput sequencing machines (Guo et al. 2017; Sheridan 2014). The search was performed on November 10, 2023, and results up to that date were included. The search query combined groups of terms related to variant, effect, prediction, and tools. Within each group, the terms were combined using the logical operator OR. The complete query is provided in Supplementary Table S1.
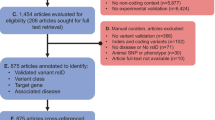
Articles containing the term “cancer” in the title were excluded to reduce the number of irrelevant hits and to find VEPs applicable across several diseases. We scanned the reference lists of review and benchmarking articles to retrieve additional eligible articles.
Study selection
Included articles described a VEP, i.e., a tool accepting human genetic variants and predicting functional impacts. The list of exclusion criteria was made to ensure that tools were reliable, broadly applicable, accessible, scalable, and reproducible (Table 1). In cases where a tool appeared to be discontinued, generally indicated by a non-functional URL in the publication, we contacted the corresponding author for confirmation. Some authors supplied a working URL, which allowed us to reassess the publication against the other exclusion criteria. We removed tools not applicable to humans or without any documentation.
Review and benchmarking articles were used to find additional eligible articles. However, only original work describing a VEP was included in this review. Web-only and GUI-only tools were deemed insufficiently reproducible and scalable and were thus excluded. In line with our accessibility requirement, tools requiring a fee were also excluded. Additionally, given the fast pace of progress in the field, we included only tools that support the GRCh38 genome build and were updated at least once since January 1, 2020. Tools that were specific to a small number of genes or a specific disease were excluded, as we were interested in the application of VEPs to a broad range of studies. If several versions of the tool existed, we only included the latest version, regardless of whether the latest version had an associated publication. Nevertheless, significant updates often coincided with a publication, such as dbNSFP v4 (Liu et al. 2020).
One author (CR) selected the studies based on the exclusion criteria (Table 1). First, titles were screened for eligibility. Second, articles were filtered based on the abstract. Third, the full text of the remaining articles was examined. Reasons for exclusion were recorded for each round.
Data extraction
First, one author (CR) extracted the tool name, variant types, functional impacts, and operating system requirements from the included publications and their latest documentation. The URLs of tools with online capabilities were retrieved. Tools that required a high-performance computer were identified. Second, another author (LG) reviewed the extracted data to confirm the accuracy of the information from the publications and documentation. Divergences were resolved through discussion. For each article, the following characteristics were automatically retrieved: PubMed ID, title, authors, citation, first author, journal, year of publication, date of PubMed entry creation, PMCID, NIHMS ID, and digital object identifier.
Sequence Ontology terms were used to describe the variant types and functional impacts wherever applicable (Eilbeck et al. 2005). In case a Sequence Ontology term was unavailable to describe a particular variant type or functional impact, a new term was coined. For terms consistent with the structure of the Sequence Ontology, a request to create the new term was made on the Sequence Ontology GitHub page (https://github.com/The-Sequence-Ontology/SO-Ontologies/issues). Eighteen new terms were requested and are awaiting approval. Examples include “enhancer variant” and “promoter variant”. The full list of Sequence Ontology terms is provided in Supplementary Table S2.
Data synthesis
Descriptive statistics were calculated for each tool, including the number of variant and functional impact categories. Linear regression was used to study the relationship between the number of functional impacts predicted by each tool and the date it was uploaded to the MEDLINE database.
Software
All analyses used R version 4.2.2; all R scripts are attached as supplementary files and were uploaded to Zenodo (see section Code availability). A website was created with the shiny package (Chang et al. 2012). Furthermore, to facilitate interoperability between different tools, we also advocate the use of controlled vocabularies to describe phenotypes (Kohler et al. 2021; Malone et al. 2010). We aim to perform a bigger update of the VEP Finder once per year and to do regular update after user input and evidence.
VEPs predicting many functional impacts, such as SnpEff, FAVOR and WGSA, represent a potential solution to the problem of tool choice. Nevertheless, the rapid evolution of the field necessitates continuous updates to keep them up to date. Furthermore, we expect specialized tools to be continuously released (Fig. 4). Consequently, systematic reviews on VEPs will be needed regularly.
Conclusion
A staggering 118 tools were available to predict approximately 160 functional impacts that ranged from molecular to phenotypic effects. About 60% of these impacts could be predicted by combining just three tools. Unexpectedly, recent tools did not necessarily predict more impacts than older ones. Despite the vast diversity of VEPs, some genetic variants were not yet supported and should be the object of future research.
The abundance of available options can complicate the tool selection process. However, this challenge is mitigated by the Shiny app developed in this review. The app enables users to filter tools based on their specific needs, narrowing down the list of suitable options.
Availability of data and material
The VEP Finder website is freely available at https://cardio-care.shinyapps.io/VEP_Finder/.
Code availability
The scripts for data wrangling, generating plots, and running linear models are stored on Zenodo and can be accessed at https://doi.org/10.5281/zenodo.10255446. They are also available in the Supplementary zip file. The code for the Shiny app is accessible at https://github.com/CristianRiccio/VEP_Finder.
References
Abramowicz A, Gos M (2018) Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet 59:253–268. https://doi.org/10.1007/s13353-018-0444-7
Bragin E, Chatzimichali EA, Wright CF, Hurles ME, Firth HV, Bevan AP, Swaminathan GJ (2014) DECIPHER: database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res 42:D993–D1000. https://doi.org/10.1093/nar/gkt937
Brookes AJ, Robinson PN (2015) Human genotype–phenotype databases: aims, challenges and opportunities. Nat Rev Genet 16:702–715. https://doi.org/10.1038/nrg3932
Chang W, Cheng J, Allaire J, Sievert C, Schloerke B, **e Y, Allen J, McPherson J, Dipert A, Borges B (2023) shiny: Web application framework for R. https://CRAN.R-project.org/package=shiny. Accessed 3 Apr 2024
Cheng N, Li M, Zhao L, Zhang B, Yang Y, Zheng CH, **a J (2020) Comparison and integration of computational methods for deleterious synonymous mutation prediction. Brief Bioinform 21:970–981. https://doi.org/10.1093/bib/bbz047
Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (austin) 6:80–92. https://doi.org/10.4161/fly.19695
Ding Q, Somerville C, Manshaei R, Trost B, Reuter MS, Kalbfleisch K, Stanley K, Okello JBA, Hosseini SM, Liston E, Curtis M, Zarrei M, Higginbotham EJ, Chan AJS, Engchuan W, Thiruvahindrapuram B, Scherer SW, Kim RH, Jobling RK (2023) SCIP: software for efficient clinical interpretation of copy number variants detected by whole-genome sequencing. Hum Genet 142:201–216. https://doi.org/10.1007/s00439-022-02494-1
Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, Ashburner M (2005) The Sequence ontology: a tool for the unification of genome annotations. Genome Biol 6:R44. https://doi.org/10.1186/gb-2005-6-5-r44
Frazer J, Notin P, Dias M, Gomez A, Min JK, Brock K, Gal Y, Marks DS (2021) Disease variant prediction with deep generative models of evolutionary data. Nature 599:91–95. https://doi.org/10.1038/s41586-021-04043-8
Geoffroy V, Herenger Y, Kress A, Stoetzel C, Piton A, Dollfus H, Muller J (2018) AnnotSV: an integrated tool for structural variations annotation. Bioinform 34:3572–3574. https://doi.org/10.1093/bioinformatics/bty304
Ghosh R, Oak N, Plon SE (2017) Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol 18:225. https://doi.org/10.1186/s13059-017-1353-5
Glusman G, Rose PW, Prlic A, Dougherty J, Duarte JM, Hoffman AS, Barton GJ, Bendixen E, Bergquist T, Bock C, Brunk E, Buljan M, Burley SK, Cai B, Carter H, Gao J, Godzik A, Heuer M, Hicks M, Hrabe T, Karchin R, Leman JK, Lane L, Masica DL, Mooney SD, Moult J, Omenn GS, Pearl F, Pejaver V, Reynolds SM, Rokem A, Schwede T, Song S, Tilgner H, Valasatava Y, Zhang Y, Deutsch EW (2017) Map** genetic variations to three-dimensional protein structures to enhance variant interpretation: a proposed framework. Genome Med 9:113. https://doi.org/10.1186/s13073-017-0509-y
Guo Y, Dai Y, Yu H, Zhao S, Samuels DC, Shyr Y (2017) Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics 109:83–90. https://doi.org/10.1016/j.ygeno.2017.01.005
Halldorsson BV, Eggertsson HP, Moore KHS, Hauswedell H, Eiriksson O, Ulfarsson MO, Palsson G, Hardarson MT, Oddsson A, Jensson BO, Kristmundsdottir S, Sigurpalsdottir BD, Stefansson OA, Beyter D, Holley G, Tragante V, Gylfason A, Olason PI, Zink F, Asgeirsdottir M, Sverrisson ST, Sigurdsson B, Gudjonsson SA, Sigurdsson GT, Halldorsson GH, Sveinbjornsson G, Norland K, Styrkarsdottir U, Magnusdottir DN, Snorradottir S, Kristinsson K, Sobech E, Jonsson H, Geirsson AJ, Olafsson I, Jonsson P, Pedersen OB, Erikstrup C, Brunak S, Ostrowski SR, Consortium DG, Thorleifsson G, Jonsson F, Melsted P, Jonsdottir I, Rafnar T, Holm H, Stefansson H, Saemundsdottir J, Gudbjartsson DF, Magnusson OT, Masson G, Thorsteinsdottir U, Helgason A, Jonsson H, Sulem P, Stefansson K (2022) The sequences of 150,119 genomes in the UK Biobank. Nature 607:732–740. https://doi.org/10.1038/s41586-022-04965-x
Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu XD, Topol EJ, Rosenfeld MG, Frazer KA (2011) 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature 470:264–268. https://doi.org/10.1038/nature09753
Imker HJ (2018) 25 years of molecular biology databases: a study of proliferation, impact, and maintenance. Front Res Metr Anal 3:18. https://doi.org/10.3389/frma.2018.00018
Kassahn KS, Scott HS, Caramins MC (2014) Integrating massively parallel sequencing into diagnostic workflows and managing the annotation and clinical interpretation challenge. Hum Mutat 35:413–423. https://doi.org/10.1002/humu.22525
Katsonis P, Wilhelm K, Williams A, Lichtarge O (2022) Genome interpretation using in silico predictors of variant impact. Hum Genet 141:1549–1577. https://doi.org/10.1007/s00439-022-02457-6
Kohler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, Danis D, Balagura G, Baynam G, Brower AM, Callahan TJ, Chute CG, Est JL, Galer PD, Ganesan S, Griese M, Haimel M, Pazmandi J, Hanauer M, Harris NL, Hartnett MJ, Hastreiter M, Hauck F, He Y, Jeske T, Kearney H, Kindle G, Klein C, Knoflach K, Krause R, Lagorce D, McMurry JA, Miller JA, Munoz-Torres MC, Peters RL, Rapp CK, Rath AM, Rind SA, Rosenberg AZ, Segal MM, Seidel MG, Smedley D, Talmy T, Thomas Y, Wiafe SA, **an J, Yuksel Z, Helbig I, Mungall CJ, Haendel MA, Robinson PN (2021) The human phenotype ontology in 2021. Nucleic Acids Res 49:D1207–D1217. https://doi.org/10.1093/nar/gkaa1043
Kuksa PP, Lee CY, Amlie-Wolf A, Gangadharan P, Mlynarski EE, Chou YF, Lin HJ, Issen H, Greenfest-Allen E, Valladares O, Leung YY, Wang LS (2020) SparkINFERNO: a scalable high-throughput pipeline for inferring molecular mechanisms of non-coding genetic variants. Bioinformatics 36:3879–3881. https://doi.org/10.1093/bioinformatics/btaa246
Kuksa PP, Greenfest-Allen E, Cifello J, Ionita M, Wang H, Nicaretta H, Cheng PL, Lee WP, Wang LS, Leung YY (2022) Scalable approaches for functional analyses of whole-genome sequencing non-coding variants. Hum Mol Genet 31:R62–R72. https://doi.org/10.1093/hmg/ddac191
Lee K, Krempely K, Roberts ME, Anderson MJ, Carneiro F, Chao E, Dixon K, Figueiredo J, Ghosh R, Huntsman D, Kaurah P, Kesserwan C, Landrith T, Li S, Mensenkamp AR, Oliveira C, Pardo C, Pesaran T, Richardson M, Slavin TP, Spurdle AB, Trapp M, Witkowski L, Yi CS, Zhang L, Plon SE, Schrader KA, Karam R (2018) Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum Mutat 39:1553–1568. https://doi.org/10.1002/humu.23650
Liu X, White S, Peng B, Johnson AD, Brody JA, Li AH, Huang Z, Carroll A, Wei P, Gibbs R, Klein RJ, Boerwinkle E (2016) WGSA: an annotation pipeline for human genome sequencing studies. J Med Genet 53:111–112. https://doi.org/10.1136/jmedgenet-2015-103423
Liu X, Li C, Mou C, Dong Y, Tu Y (2020) dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med 12:103. https://doi.org/10.1186/s13073-020-00803-9
Livesey BJ, Marsh JA (2023) Updated benchmarking of variant effect predictors using deep mutational scanning. Mol Syst Biol 19:e11474. https://doi.org/10.15252/msb.202211474
Malone J, Holloway E, Adamusiak T, Kapushesky M, Zheng J, Kolesnikov N, Zhukova A, Brazma A, Parkinson H (2010) Modeling sample variables with an experimental factor ontology. Bioinformatics 26:1112–1118. https://doi.org/10.1093/bioinformatics/btq099
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F (2016) The Ensembl variant effect predictor. Genome Biol 17:122. https://doi.org/10.1186/s13059-016-0974-4
Meier J, Rao R, Verkuil R, Liu J, Sercu T, Rives A (2021) Language models enable zero-shot prediction of the effects of mutations on protein function. Advances Neural Inf Process Syst. https://doi.org/10.1101/2021.07.09.450648
Menzies A, Teague JW, Butler AP, Davies H, Tarpey P, Nik-Zainal S, Campbell PJ (2015) VAGrENT: variation annotation generator. Curr Protoc Bioinform 52:1. https://doi.org/10.1002/0471250953.bi1508s52
Naslavsky MS, Yamamoto GL, de Almeida TF, Ezquina SAM, Sunaga DY, Pho N, Bozoklian D, Sandberg TOM, Brito LA, Lazar M, Bernardo DV, Amaro E Jr, Duarte YAO, Lebrao ML, Passos-Bueno MR, Zatz M (2017) Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat 38:751–763. https://doi.org/10.1002/humu.23220
Nelson KN, Peiris MN, Meyer AN, Siari A, Donoghue DJ (2017) Receptor tyrosine kinases: translocation partners in hematopoietic disorders. Trends Mol Med 23:59–79. https://doi.org/10.1016/j.molmed.2016.11.002
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, Shamseer L, Tetzlaff JM, Akl EA, Brennan SE, Chou R, Glanville J, Grimshaw JM, Hrobjartsson A, Lalu MM, Li T, Loder EW, Mayo-Wilson E, McDonald S, McGuinness LA, Stewart LA, Thomas J, Tricco AC, Welch VA, Whiting P, Moher D (2021) The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 372:n71. https://doi.org/10.1136/bmj.n71
Pagel KA, Antaki D, Lian A, Mort M, Cooper DN, Sebat J, Iakoucheva LM, Mooney SD, Radivojac P (2019) Pathogenicity and functional impact of non-frameshifting insertion/deletion variation in the human genome. PLoS Comput Biol 15:e1007112. https://doi.org/10.1371/journal.pcbi.1007112
Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, O’Donnell-Luria A, Harrison SM, Tavtigian SV, Greenblatt MS, Biesecker LG, Radivojac P, Brenner SE, ClinGen Sequence Variant Interpretation Working G (2022) Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet 109:2163–2177. https://doi.org/10.1016/j.ajhg.2022.10.013
Povysil G, Petrovski S, Hostyk J, Aggarwal V, Allen AS, Goldstein DB (2019) Rare-variant collapsing analyses for complex traits: guidelines and applications. Nat Rev Genet 20:747–759. https://doi.org/10.1038/s41576-019-0177-4
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acid Res 47:D886–D894. https://doi.org/10.1093/nar/gky1016
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Riesselman AJ, Ingraham JB, Marks DS (2018) Deep generative models of genetic variation capture the effects of mutations. Nat Methods 15:816–822. https://doi.org/10.1038/s41592-018-0138-4
Sheridan C (2014) Milestone approval lifts Illumina’s NGS from research into clinic. Nat Biotechnol 32:111–112. https://doi.org/10.1038/nbt0214-111
Sollis E, Mosaku A, Abid A, Buniello A, Cerezo M, Gil L, Groza T, Gunes O, Hall P, Hayhurst J, Ibrahim A, Ji Y, John S, Lewis E, MacArthur JAL, McMahon A, Osumi-Sutherland D, Panoutsopoulou K, Pendlington Z, Ramachandran S, Stefancsik R, Stewart J, Whetzel P, Wilson R, Hindorff L, Cunningham F, Lambert SA, Inouye M, Parkinson H, Harris LW (2023) The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acid Res 51:D977–D985. https://doi.org/10.1093/nar/gkac1010
Tabarini N, Biagi E, Uva P, Iovino E, Pippucci T, Seri M, Cavalli A, Ceccherini I, Rusmini M, Viti F (2022) Exploration of tools for the interpretation of human non-coding variants. Int J Mol Sci. https://doi.org/10.3390/ijms232112977
Taub MA, Conomos MP, Keener R, Iyer KR, Weinstock JS, Yanek LR, Lane J, Miller-Fleming TW, Brody JA, Raffield LM, McHugh CP, Jain D, Gogarten SM, Laurie CA, Keramati A, Arvanitis M, Smith AV, Heavner B, Barwick L, Becker LC, Bis JC, Blangero J, Bleecker ER, Burchard EG, Celedon JC, Chang YPC, Custer B, Darbar D, de Las FL, DeMeo DL, Freedman BI, Garrett ME, Gladwin MT, Heckbert SR, Hidalgo BA, Irvin MR, Islam T, Johnson WC, Kaab S, Launer L, Lee J, Liu S, Moscati A, North KE, Peyser PA, Rafaels N, Seidman C, Weeks DE, Wen F, Wheeler MM, Williams LK, Yang IV, Zhao W, Aslibekyan S, Auer PL, Bowden DW, Cade BE, Chen Z, Cho MH, Cupples LA, Curran JE, Daya M, Deka R, Eng C, Fingerlin TE, Guo X, Hou L, Hwang SJ, Johnsen JM, Kenny EE, Levin AM, Liu C, Minster RL, Naseri T, Nouraie M, Reupena MS, Sabino EC, Smith JA, Smith NL, Su JL, Taylor JG, Telen MJ, Tiwari HK, Tracy RP, White MJ, Zhang Y, Wiggins KL, Weiss ST, Vasan RS, Taylor KD, Sinner MF, Silverman EK, Shoemaker MB, Sheu WH, Sciurba F, Schwartz DA, Rotter JI, Roden D, Redline S, Raby BA et al (2022) Genetic determinants of telomere length from 109,122 ancestrally diverse whole-genome sequences in TOPMed. Cell Genom. https://doi.org/10.1016/j.xgen.2021.100084
Taylor SM, Parobek CM, Fairhurst RM (2012) Haemoglobinopathies and the clinical epidemiology of malaria: a systematic review and meta-analysis. Lancet Infect Dis 12:457–468. https://doi.org/10.1016/S1473-3099(12)70055-5
The All of Us Research Program Investigators (2019) The “All of Us” research program. N Engl J Med 381:668–676. https://doi.org/10.1056/NEJMsr1809937
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC (2016) SIFT missense predictions for genomes. Nat Protoc 11:1–9. https://doi.org/10.1038/nprot.2015.123
Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678. https://doi.org/10.1038/nature05911
Zhang X, Wakeling M, Ware J, Whiffin N (2021) Annotating high-impact 5’untranslated region variants with the UTRannotator. Bioinformatics 37 8:1171–1173. https://doi.org/10.1093/bioinformatics/btaa783
Zhou H, Arapoglou T, Li X, Li Z, Zheng X, Moore J, Asok A, Kumar S, Blue EE, Buyske S, Cox N, Felsenfeld A, Gerstein M, Kenny E, Li B, Matise T, Philippakis A, Rehm HL, Sofia HJ, Snyder G, NHGRI Genome Sequencing Program Variant Functional Annotation Working Group, Weng Z, Neale B, Sunyaev SR, Lin X (2023) FAVOR: functional annotation of variants online resource and annotator for variation across the human genome. Nucleic Acids Res 51:D1300–D1311. https://doi.org/10.1093/nar/gkac966
Acknowledgements
We thank Dr. Hugo Solleder for his feedback on the research questions and the VEP Finder website.
Author information
Authors and Affiliations
Contributions
Design, conception of the study and development of search strategy: Cristian Riccio, Max L. Jansen, Andreas Ziegler. Literature search: Cristian Riccio. Data collection: Cristian Riccio and Linlin Guo. Data analysis and interpretation: Cristian Riccio, Andreas Ziegler. Drafted manuscript: Cristian Riccio, Andreas Ziegler. Critical review and approval of final manuscript: all authors.
Corresponding author
Ethics declarations
Conflict of interest
CR, MLJ, and AZ are employees of Cardio-CARE AG, a not-for-profit company financed by the Kühne Foundation. AZ is a member of the editorial board of Human Genetics.
Ethics approval
Not Applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
439_2024_2670_MOESM1_ESM.pdf
Supplementary file1: Fig. S1. Bar plot of the number of variant types that receive support by a VEP for the first time each year (p linear regression = 0.212) (PDF 5 KB)
439_2024_2670_MOESM3_ESM.xlsx
Supplementary file3: Table S2. Terms used in this review to describe variant types and functional impacts, including Sequence Ontology terms (XLSX 14 KB)
439_2024_2670_MOESM4_ESM.xlsx
Supplementary file4: Table S3. List of publications included in this systematic review. The table lists the PubMed ID, tool name, list of variant types, list of functional impacts, a note, publication title, authors, citation, journal, year of publication, creation date of the PubMed entry, PubMed Central ID, NIHMS ID, and DOI. SO terms related to variation types are listed alphabetically and separated by semicolons under the column “Variant types”. Similarly, SO terms related to predicted functional impacts are listed under the column “Functional impacts” (XLSX 39 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Riccio, C., Jansen, M.L., Guo, L. et al. Variant effect predictors: a systematic review and practical guide. Hum. Genet. 143, 625–634 (2024). https://doi.org/10.1007/s00439-024-02670-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-024-02670-5