
Abstract
An aberrant late sodium current (INa,Late) caused by a mutation in the cardiac sodium channel (Nav1.5) has emerged as a contributor to electrical remodeling that causes susceptibility to atrial fibrillation (AF). Although downregulation of phosphoinositide 3-kinase (PI3K)/Akt signaling is associated with AF, the molecular mechanisms underlying the negative regulation of INa,Late in AF remain unclear, and potential therapeutic approaches are needed. In this work, we constructed a tachypacing-induced cellular model of AF by exposing HL-1 myocytes to rapid electrical stimulation (1.5 V/cm, 4 ms, 10 Hz) for 6 h. Then, we gathered data using confocal Ca2+ imaging, immunofluorescence, patch-clamp recordings, and immunoblots. The tachypacing cells displayed irregular Ca2+ release, delayed afterdepolarization, prolonged action potential duration, and reduced PI3K/Akt signaling compared with controls. Those detrimental effects were related to increased INa,Late and were significantly mediated by treatment with the INa,Late blocker ranolazine. Furthermore, decreased PI3K/Akt signaling via PI3K inhibition increased INa,Late and subsequent aberrant myocyte excitability, which were abolished by INa,Late inhibition, suggesting that PI3K/Akt signaling is responsible for regulating pathogenic INa,Late. These results indicate that PI3K/Akt signaling is critical for regulating INa,Late and electrical remodeling, supporting the use of PI3K/Akt-mediated INa,Late as a therapeutic target for AF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atrial fibrillation (AF) is a common heart arrhythmia characterized by structural and electrical remodeling that leads to cardiac diseases such as heart failure [2, 7, 18]. Increasing evidence suggests that ion channel regulation is an effective therapeutic target for AF [8, 14]. Although ion channel-targeting drugs are available, the mechanism underlying those therapies remains to be determined.
The cardiac sodium channel (Nav1.5) plays a pivotal role in the initiation and propagation of action potential (AP) via membrane depolarization [22]. Mutations in Nav1.5 are mainly caused by aberrant sodium currents (INa) that lead to arrhythmogenesis and produce conditions such as long QT syndrome, Brugada syndrome, and AF [30]. Those arrhythmic events are mainly caused by abnormal inactivation of Nav1.5, called “late INa” (INa,Late) [25]. Importantly, an increase in INa,Late can affect myocyte excitability through abnormal Nav1.5 inactivation, thereby inducing proarrhythmic events such as AF [32]. Thus, INa,Late regulation could be a potential target for AF treatment.
Recent studies revealed that phosphoinositide-3-kinase (PI3K) can be an effective treatment for arrhythmic events [33]. PI3K is a heterodimeric protein with catalytic subunits, such as p110α, that play crucial roles in cell survival, growth, and proliferation. Its downstream molecule, Akt, is involved in the regulation of Nav1.5 [3, 29]. A previous report revealed that PI3K inhibition caused the prolongation of AP duration (APD) and increased INa,Late in diabetic mouse hearts [20]. Moreover, mouse hearts lacking PI3K p110ɑ showed longer APD caused by increasing INa,Late [21]. However, the underlying mechanisms of PI3K/Akt signaling that regulate INa,Late following the development of AF remain unclear. In this work, we investigated the effects of PI3K/Akt signaling and its potential roles in INa,Late and AF using tachypacing-induced HL-1 myocytes.
Materials and methods
HL-1 atrial cell line culture and rapid electrical pacing
The HL-1 atrial myocyte cell line used (Merck Millipore) was derived from the AT-1 mouse atrial carcinoma cell line [9]. Cells were cultured on gelatin-fibronectin-coated culture dishes with Claycomb medium (Sigma) supplemented with 10% fetal bovine serum (Merck), 10 μM norepinephrine (Sigma), 2 mM l-glutamine (Sigma), and 1% penicillin/streptomycin (Gendepot) in a 37 °C, 5% CO2 atmosphere. To induce tachypacing, HL-1 atrial myocytes were subjected to rapid electrical pacing for 6 h at 1.5 V/cm, 4 ms, and 10 Hz in a 37 °C, 5% CO2 incubator. Cells not treated with pacing for 6 h were used as baseline controls.
Drug application
Ranolazine (Abcam, CAS #. 142,387–99-3) was dissolved in dimethyl sulfoxide (DMSO, Biosesang) and applied to cells at a final concentration of 50 μM for 2 h before pacing was terminated. PI-103, a PI3K inhibitor, was dissolved in DMSO (Biosesang) and applied to cells at a final concentration of 100 nM for 2 h in the presence or absence of ranolazine. MK-2206, an Akt inhibitor, was dissolved in DMSO and applied to cells at a final concentration of 1 μM for 2 h. The maximum concentration of DMSO in the culture medium was 0.2%.
Cell viability assay
Cell viability was analyzed via trypan blue exclusion. Cells were trypsinized and resuspended in culture medium. The cell suspension was mixed with the same volume of 0.4% trypan blue solution (Gendepot) and incubated for 1 min. Cell viability was determined using a C-Chip hemocytometer (Incyto) by counting the number of live and dead cells under a light microscope (Olympus).
Electrophysiology
APs and Na+ currents (INa) were recorded from HL-1 cells placed onto the recording chamber of a microscope (Nikon Eclipse Ti2), and an Axopatch 200B amplifier (Axon Instrument) was used for voltage and current clam** at room temperature (23 ± 1 °C). HL-1 cells were placed in a chamber mounted on an inverted microscope. Patch pipettes were pulled from thin-walled borosilicate capillaries (Clark Electromedical Instruments) using a P-97 Flaming/Brown Micropipette Puller (Sutter Instrument Company), and the resistance was 3–5 MΩ when pipettes were filled with pipette solution. Seals were created using negative pressure to form a whole-cell configuration with a giga seal. All recordings were made within 5 min after the formation of the whole-cell configuration. The voltage and current signals were filtered using a 4-pole Bessel-type low-pass filter at 10 kHz and sampled at a rate of 25 kHz. Cell capacitance (pF) and access resistance (MΩ) were automatically calculated and used to compensate for capacitive current and normalize ion current (pA/pF). Data acquisition and analysis were performed using digitizers (DigiData 1550B) and analysis software pClamp 10.7 (Molecular Devices). Tetrodotoxin (TTX, 10 μM) was used to block voltage-gated Na+ channels.
HL-1 myocytes were placed in a chamber mounted on an inverted microscope, and AP measurements were made in the whole-cell configuration just described. We used K+-rich pipette filling solution containing (composition in mM) KCl 140, EGTA 5, glucose 5, HEPES 5, Mg-ATP 5, and MgCl2 1 (pH 7.2 adjusted with KOH). Cells were continuously superfused with Normal Tyrode (NT) solution containing (composition in mM): NaCl 143, KCl 5.4, HEPES 5, NaH2PO4 0.33, MgCl2 0.5, CaCl2 1.8, and glucose 10 (pH 7.4 adjusted with NaOH). Briefly, cells were stimulated with a 0.7 nA current pulse for 2 ms, and pulse trains were elicited at 0.5 Hz. Delayed afterdepolarizations (DADs) were counted more than two times from 10 consecutive APs and averaged by dividing by 20 s. AP amplitude, maximal upstroke velocity (dv/dtmax), resting membrane potential, and repolarization of APD at 50% and 90% (APD50 and APD90) were analyzed from 10 consecutive APs using pClamp 10.7 software (Molecular Devices).
To record peak and late INa (INa,Peak and INa,Late), cells were incubated in NT solution and switched to a tetraethylammonium (TEA) solution after being placed in the whole-cell configuration. The TEA solution contained (composition in mM) NaCl 120, TEA-Cl 20, KCl 5.4, CaCl2 1.8, MgCl2 1, HEPES 10, and glucose 10 (pH 7.4 adjusted with NaOH). Pipettes were filled with pipette solution containing (composition in mM) NaCl 10, CsCl 50, CsF 30, l-aspartic acid 50, EGTA 5, and HEPES 1 (pH 7.3 adjusted with CsOH). INa,Peak was elicited by 50-ms voltage steps at 5-mV increments to potentials ranging from − 100 to + 60 mV, with a holding potential of − 140 mV. INa,Late was elicited by 200-ms voltage steps in 10-mV increments to potentials ranging from − 120 to − 10 mV. The average was determined as between 50 and 150 ms and normalized to the peak current after subtracting the difference between that found in the presence and absence of TTX (10 μM) [12]. In some experiments, we added 1 μM phospholipids (all di-C8, Echelon Bioscences) in pipette solution. Voltage-dependent activation of INa was assessed by measuring the peak conductance (G), which was calculated as G = INa (V − Vrev), where INa is the current for each voltage step (V), and Vrev is the reversal potential. Data were normalized by maximum peak conductance (Gmax) and fitted with the Boltzmann function, giving values for V1/2 and slope k where G/Gmax = 1/[1 + exp ((V − V1/2)/k)]. The voltage dependence of steady-state inactivation curves was measured using standard two-pulse protocols from 120 mV of holding potential, and cells were held ranging from − 140 to + 20 mV for 500 ms (pre-pulse) and then subjected to a − 40-mV test pulse for 20 ms. Currents (I) were normalized to maximum I (Imax), and curves were fitted by the Boltzmann function, giving values for the V1/2 and slope k where I/Imax = 1/[1 + exp ((V − V1/2)/k)].
Confocal Ca2+ imaging
To measure intracellular Ca2+ signals, HL-1 myocytes were loaded with 3 μM fluo-4 AM for 30 min. The cells were continuously superfused with 1.8 mM Ca2+ containing NT solution composed of (in mM) 137 NaCl, 5.4 KCl, 10 HEPES, 1 MgCl2, and 10 glucose (pH of 7.4) at 36.5 °C. HL-1 cells grown to > 80% confluence normally show regular occurrences of Ca2+ transients in the absence of electrical stimulation [16]. Ca2+ fluorescence was imaged at 60 or 120 Hz in two dimensions using a laser scanning confocal imaging system (A1, Nikon) attached to an inverted microscope (Eclipse Ti, Nikon) fitted with a × 60 oil immersion objective lens (Plan Apo, Numerical Aperture 1.4, Nikon) [17]. Dyes were excited at 488 nm using an Ar laser (Ommichrome), and fluorescence emissions at > 510 nm were detected. Images were recorded and analyzed with workstation software, NIS Elements AR (v3.2, Nikon). To estimate Ca2+ increases, the average resting fluorescence intensity (F0) was calculated from several frames immediately before the Ca2+ upstroke. Tracings of local Ca2+ signals are shown as the average fluorescence of each region-of-interest normalized to the F0 (F/F0). The average F0, measured in normal HL-1 cells, was used to calculate the F/F0 and obtain Ca2+ traces in rapidly paced cells.
Immunofluorescence
Cells grown on gelatin/fibronectin-coated coverslips were washed with phosphate-buffered saline (PBS) 2 times and fixed with 4% paraformaldehyde (Biosesang) for 20 min. The fixed cells were then blocked with PBS containing 1% bovine serum albumin (Gendepot) for 1 h. For intracellular proteins, 0.1% Triton X-100 was added in blocking buffer, and coverslips were incubated with primary antibodies (phosphorylated Akt from Cell signaling, PI3K p110ɑ (1:100, Abcam), and phosphorylated Akt (1:100, Cell Signaling) mixed in each blocking buffer using titers recommended in the antibody datasheets. The coverslips were then washed with 1 × PBS buffer 2 times and incubated with secondary antibodies labeled with Alexa Fluor 488 (Abcam). The coverslips were PBS-washed 2 times, counterstained with diamidino-2-phenylindole (Abcam), and mounted on slide glasses. Visualization of the slides was performed under a fluorescence microscope (Olympus). Total fluorescence intensity was measured and analyzed with ImageJ.
Immunoblotting
The cells were washed with 1 × PBS 2 times and lysed in a Nonidet P-40 buffer (GenDepot) containing protease inhibitors (GenDepot) and a phosphatase inhibitor cocktail (GenDepot). The lysate was sonicated and incubated on ice for 5–15 min with intermittent vortexing, and then, the lysate was centrifuged at 14,000 rpm at 4 °C for 25 min. Supernatants were collected in clean microtubes. Total proteins were quantified via Bradford protein assay, and then, the proteins were size separated with acrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Atto). The membranes were blocked for 1 h with 5% skim milk (Biosesang) in TBS-T (Biosesang) and incubated with primary antibodies overnight at 4 °C. The primary antibodies were as follows: Nav1.5 (1:200, Alomone), anti-PI3K p110ɑ (1:1000, Abcam), phosphorylated Akt (1:1000, cell signaling), Akt (1:1000, Abcam), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (1:5000, Enogene). We used horseradish peroxidase–conjugated secondary anti-rabbit goat antibody (Enzo) to detect blots with enhanced chemiluminescence solution (Thermo). The blots were detected and analyzed with Chemidoc (Bio-Rad) and Image Studio™ analysis software (LI-COR Biosciences).
Experimental animals
Adult (10 weeks old) male C57BL/6 mice were used for this study. The mice were maintained in a specific pathogen-free animal facility at Korea Medical University under standardized conditions with 12-h light and dark cycles (light on from 8:00 am–8:00 pm) at 18–25 °C and free access to chow and water. All experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the Republic of Korea. Experimental procedures were also approved by the Committee on Animal Research at Korea University College of Medicine.
Electrocardiogram (ECG)
For in vivo ECGs, the mice were anesthetized by an intraperitoneal injection of Avertin (12.5 mg/ml; Sigma-Aldrich). The mice were kept on a heating pad (37 ± 0.5 °C) and baseline ECGs were recorded using four subcutaneous electrodes for lead II. For injections, PI-103 and ranolazine were dissolved in DMSO (Biosesang) and injected at a dose of 10 mg/kg and 20 mg/kg for 2 weeks, respectively. Control (CTRL) mice were injected with the same volume of DMSO. Surface ECGs were recorded at baseline, 1, and 2 weeks. ECG data recorded the RR interval, PR interval, QRS interval, and QT interval according to Bazett’s formula using the Powerlab acquisition system, and were analyzed using LabChart 8 Pro software (AD Instruments, Sydney, Australia).
Statistical analysis
All experimental data are presented as the mean ± standard error of mean (SEM). For comparisons between groups, statistical differences were evaluated by one-way ANOVA with Tukey’s multiple comparison test using Prism 8 (GraphPad Software, La Jolla, CA, USA). P-values < 0.05 were considered to indicate significant differences between groups.
Results
I Na,Late inhibition prevents myocyte remodeling in tachypacing-induced HL-1 myocytes
Rapid electrical stimulation is an established method for constructing AF models that has been used in many in vitro and in vivo studies [6]. In this study, HL-1 myocytes were exposed to rapid electrical stimulation to construct a cellular AF model. We found that 6-h tachypacing significantly lowered cell viability (Fig. 1a). Moreover, 6-h tachypacing lowered the expression of PI3K p110α and pAkt compared with 0-h tachypacing (Fig. 1b–d) and caused tachypacing-induced myocyte remodeling.

Effects of PI3K/Akt signaling on INa,Late inhibition in tachypacing-induced HL-1 myocytes. a Cell viability in tachypacing-induced HL-1 myocytes was analyzed in a time-dependent manner (n = 4 per group). b Representative immunoblot images of PI3K p110α and pAkt (n = 6 per group). c, d Quantified expression of PI3K p110ɑ and phosphorylated Akt (n = 6 per group). e Representative image of immunoblots: PI3K p110ɑ, phosphorylated Akt, total Akt, and GAPDH expression from HL-1 myocytes in the non-pacing (NP), rapid pacing (TP), and TP + ranolazine (TP + Ran) groups. f, g Quantitative protein levels of PI3K p110ɑ and phosphorylated Akt (n = 6 per group). h Representative immunofluorescence and quantified intensity of PI3K p110ɑ (n = 20 per group). i Immunofluorescence and quantified intensity of pAkt (n = 30 per group). Values are the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Comparison using one-way ANOVA followed by Tukey’s post hoc test
To assess whether decreased PI3K/Akt signaling could be reversed by INa,Late inhibition, we applied ranolazine (50 μM), an INa,Late blocker, for 2 h before the termination of tachypacing. We found that the reduction in PI3K p110α and pAkt expression induced by tachypacing was significantly recovered by INa,Late inhibition (Fig. 1e–g). Moreover, tachypacing lowered PI3K p110α and pAkt fluorescence, and INa,Late inhibition significantly reversed that trend in HL-1 myocytes (Fig. 1h, i). These results revealed that the potential effectiveness of INa,Late inhibition occurs via the upregulation of PI3K/Akt signaling in tachypacing-induced HL-1 myocytes.
I Na,Late inhibition recovers abnormal myocyte excitability in tachypacing-induced HL-1 myocytes
The enhancement of INa,Late prolongs APD and induces triggered activity (early and delayed after depolarization) that can lead to severe arrhythmic disorders [5, 31]. To explore the effects of INa,Late inhibition on atrial excitability, we made AP recordings using the patch-clamp technique. Tachypacing caused severe DADs, and INa,Late inhibition markedly decreased DAD-induced arrhythmogenesis (Fig. 2a, b). Moreover, tachypacing significantly prolonged APD at 50% and 90% compared with non-paced HL-1 myocytes and that effect was markedly decreased by INa,Late inhibition (Fig. 2c, d). This observation supports the idea that INa,Late plays a major role in the atrial excitability of tachypacing-induced HL-1 myocytes.

Effects of myocyte excitability on INa,Late inhibition in tachypacing-induced HL-1 myocytes. a Representative spontaneous AP trace (0.5 Hz) recorded from NP, TP, and TP + R HL-1 myocytes. b The occurrence of delayed afterdepolarizations (DADs) was analyzed from 10 consecutive APs in HL-1 myocytes. c, d Representative single AP traces and the repolarization of AP duration (APD) at 50% and 90% at 0.5 Hz. Values are the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Comparison using one-way ANOVA followed by Tukey’s post hoc test
I Na,Late inhibition suppresses abnormal I Na properties in tachypacing-induced HL-1 myocytes
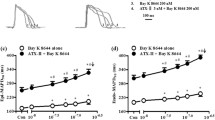
To investigate the effects of INa,Late inhibition on INa properties, we first recorded INa,Peak using the patch-clamp technique. As shown in Fig. 3a, whole-cell measurements of INa,Peak showed that tachypacing reduced INa,Peak density compared with non-paced HL-1 myocytes (Fig. 3b, c). INa,Late inhibition also decreased INa,Peak density compared with that in non-paced HL-1 myocytes, while INa,Late-inhibited and tachypacing-induced HL-1 myocytes did not differ significantly from each other (Fig. 3b, c). However, tachypacing-induced cells displayed a more positive shift in INa activation than non-paced HL-1 myocytes and that change was significantly recovered by INa,Late inhibition (Fig. 3b, c), possibly by preventing the conduction defect [10].

INa,Late inhibition affects the kinetics of INa activation in tachypacing-induced HL-1 myocytes. a, b Representative images of whole-cell INa,Peak and the current–voltage relationship (I-V curve) from NP, TP, and TP + Ran HL-1 atrial myocytes. c INa,Peak density was calculated at − 25 mV (n = 12–14 per group). d, e The voltage-dependent activation curve was determined by the Boltzmann function, and V1/2 was analyzed (n = 9–14 per group). Values are the mean ± SEM. **p < 0.01. Comparison using one-way ANOVA followed by Tukey’s post hoc test
The regulation of INa,Late is crucial in the management of AF, although it is observed to ~ 1% of INa,Late [13]. To test its effects on pathological atrial remodeling, we next recorded INa,Late using a depolarizing voltage of − 10 mV (with − 140 mV of holding potential) and calculated normalized INa,peak between 50 and 150 ms, after subtracting the difference between that measured in the presence and absence of 10 μM TTX (Fig. 4a).

Dysregulated INa,Late is recovered by ranolazine treatment in tachypacing-induced HL-1 myocytes. a INa,Late was recorded after subtracting the difference measured in the presence and absence of TTX (10 μM) and elicited by a holding potential of − 120 mV from a 200-ms pulse at − 10 mV. b, c Representative traces of INa,Late are calculated as percentage of INa,Peak at − 10 and 0 mV (n = 10 per group). d, e The voltage-dependent steady-state inactivation curve was determined by the Boltzmann function, and V1/2 was analyzed (n = 9–11 per group). f, g Representative immunoblot image and analysis of Nav1.5 (n = 8 per group). Values are the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Comparison using one-way ANOVA followed by Tukey’s post hoc test
Those direct recordings showed that INa,Late was significantly higher in tachypacing-induced than non-paced HL-1 myocytes, and INa,Late inhibition markedly abolished that increase (Fig. 4b, c). Interestingly, tachypacing produced a positive shift in INa inactivation, which was markedly recovered by INa,Late inhibition (Fig. 4d, e), suggesting that INa,Late inhibition effectively reversed the aberrant kinetics of Nav1.5. Although we also detected Nav1.5 expression, we found no differences between groups (Fig. 4f, g). Our results revealed the potential role of INa,Late inhibition in INa open probability.
Suppression of tachypacing-induced arrhythmic Ca2+ release by I Na,Late inhibition
INa,Late increases intracellular Na+, which can enhance the Ca2+ influx, ultimately inducing Ca2+ overload and electrical abnormality [4]. Thus, we next examined whether increases in INa,Late affected Ca2+ handling in tachypacing-induced HL-1 myocytes. Figure 5a (i) and b (i) (left panel) show representative Ca2+ traces measured from HL-1 myocytes under normal conditions. Those myocytes showed autorhythmic regular Ca2+ transients at a confluence of > 80% [16]. Caffeine (10 mM) was applied to those cells after we measured rhythmic Ca2+ releases to estimate the sarcoplasmic reticulum (SR) Ca2+ loading status. In HL-1 myocytes treated with 6 h of tachypacing, we observed irregular Ca2+ releases of varying magnitudes and higher basal Ca2+ levels (Fig. 5b (ii), c). In addition, we found more Ca2+ spikes than under control conditions. The average magnitude of Ca2+ transients in HL-1 myocytes with tachypacing was significantly smaller than that in HL-1 myocytes under control conditions (~ 60% of control levels; Fig. 5c). However, it should be noted that tachypacing-induced HL-1 myocytes had larger and more prolonged Ca2+ releases than control cells. Percentages of arrhythmic Ca2+ spikes relative to the number of regular Ca2+ peaks were estimated from Ca2+ traces recorded for 10 s. We found that the percentage of irregular Ca2+ transients was dramatically increased in tachypacing-induced HL-1 myocytes (Fig. 5b (ii), c). The amplitudes of caffeine-induced Ca2+ release increased by approximately twofold after 6 h of tachypacing (Fig. 5b (ii) and c), suggesting that the increase in SR Ca2+ load was caused by tachypacing.

Disturbance of rhythmic Ca2+ signaling by tachypacing and its recovery by ranolazine in HL-1 myocytes. a Confocal image of representative HL-1 myocytes (labeled with the numbers 1–9) showing Ca2+ signals (left) and the regions-of-interest used to measure the Ca2+ signals (right). The confocal Ca2+ images were measured from normal (non-paced) HL-1 myocytes with a confluence of > 90% (i) and similarly confluent HL-1 myocytes subjected to tachypacing at 10 Hz for 6 h (ii) and tachypacing (10 Hz, 6 h) with a 2-h incubation in 50 μM ranolazine (iii). b Ca2+ traces measured from normal cells (i; 1–3) and tachypacing-induced HL-1 myocytes without (ii; 4–6) and with ranolazine (iii; 7–9), showing regular Ca2+ transients followed by caffeine (10 mM)-induced Ca2+ transients. c Comparisons of the basal Ca2+ level, Ca2+ transient magnitudes, the magnitude of caffeine-induced Ca2+ transients (SR Ca2+ load), the rate of Ca2+ transient occurrence (Ca2+ spikes/s), and the % of irregular Ca2+ spikes between normal cells (3 batches, 144 cells) and tachypacing-induced cells (10 Hz, 6 h) without (3 batches, 143 cells) and with (2 h, 10 μM; 3 batches, 128 cells) ranolazine. Values are the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Comparison using one-way ANOVA followed by Tukey’s post hoc test
We examined the effects of ranolazine (50 μM), which suppresses INa,Late, on abnormal and arrhythmic Ca2+ signals in tachypacing-induced HL-1 myocytes. Ranolazine was added to the external solution for the last 2 h of the 6-h tachypacing period. Interestingly, most of the cells treated with ranolazine showed regular Ca2+ transients (Fig. 5b (iii), c). In addition, basal Ca2+ level was completely reversed to the control level in those cells. Nevertheless, the magnitude of Ca2+ transients, SR Ca2+ loading, and the rate of Ca2+ spiking remained higher than the control levels after the application of ranolazine, although SR Ca2+ loading was slightly reduced (Fig. 5b (iii), c). These results suggest that INa,Late could play an important role in the disruption of beating rhythm and basal Ca2+ increases in atrial myocytes under prolonged high-frequency electrical excitations that mimic AF. These data also suggest that INa,Late inhibition can reverse tachypacing-induced abnormalities in Ca2+ transients, maybe by suppressing Na+ influx-mediated Ca2+ increases. However, it should also be noted that the increased SR Ca2+ loading and higher beating rate induced by tachypacing might be only slightly attenuated by INa,Late inhibition.
Dysregulation of myocyte excitability by PI3K inhibition is reversed by I Na,Late inhibition
To determine whether PI3K inhibition impairs atrial excitability and further contributes to pathogenic INa,Late, we measured the APs in HL-1 myocytes treated with PI-103 (100 nM) as a PI3K inhibitor or with co-application of PI-103 and ranolazine (50 μM) for 2 h and then divided the cells into three groups: control (CTRL), PI-103 treatment (PI), and co-application of PI-103 and ranolazine (PI + Ran). After PI3K inhibition, as shown in Fig. 6a, immunoblot analyses showed that PI3K p110ɑ and pAkt were significantly decreased compared with the CTRL group, and INa,Late inhibition completely reversed those changes to the CTRL level, demonstrating that INa,Late inhibition upregulated PI3K/Akt signaling (Fig. 6b, c).

INa,Late is regulated by the PI3K/Akt pathway. a, b Representative traces of INa,Late, calculated as a percentage of INa,Peak at − 10 or 0 mV (n = 7–9 per group). c Representative immunoblot images of PI3K p110α and pAkt from CTRL, PI, and PI + Ran groups of HL-1 myocytes (n = 6 per group). d, e Representative traces of APs and DADs were analyzed from CTRL, PI, and PI + Ran groups of HL-1 myocytes (n = 9–15 per group). f, g Representative single traces of APs and the repolarization of APD at 50% and 90% (n = 9–15 per group). h, i Representative traces of INa,Late, calculated as a percentage of INa,Peak at − 10 or 0 mV (n = 7–9 per group). j, k Sample trace of INa,Late in non-pacing (NP), tachypacing (TP), and TP with or without infusion of PIP3 (n = 5–6 per group). l, m Representative images of INa,Late in control (CTRL), Akt inhibition (Akti), and Akti with or without infusion of PIP3 (n = 6 per group). n, o Representative immunoblot image and analysis for Nav1.5 (n = 8 per group). Values are the mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001. Comparison using one-way ANOVA followed by Tukey’s post hoc test
To investigate the effects of atrial excitability on PI3K inhibition, we recorded APs using the patch-clamp technique. As shown in Fig. 6d, PI3K inhibition caused severe DADs, which was significantly abolished by INa,Late inhibition (Fig. 6e). Moreover, PI3K inhibition displayed prolonged APD50 and APD90 compared with control HL-1 myocytes (Fig. 6f, g), implying that PI3K inhibition had detrimental effects on atrial excitability. In contrast, INa,Late inhibition significantly reduced APD50 and APD90 compared with the PI-103-induced HL-1 myocytes (Fig. 6f, g). These results support the idea that PI3K/Akt signaling plays a pivotal role in INa,Late-regulated myocyte excitability.
PI3K/Akt is required for I Na,Late regulation
Our findings open the possibility that PI3K/Akt signaling has an anti-arrhythmic effect on INa,Late regulation. Thus, we formulated our central hypothesis that PI3K/Akt signaling regulates pathogenic INa,Late. To elucidate the direct implications of PI3K/Akt signaling for INa,Late regulation, we made direct recordings of INa,Late. PI3K inhibition significantly increased INa,Late compared with control HL-1 myocytes and that increase was recovered by ranolazine (Fig. 6h, i). To test whether increasing INa,Late under pathological condition is associated with PI3K regulation, we recorded INa,Late in HL-1 myocytes with tachypacing or Akt inhibition by applying phosphatidylinositol 3,4,5-trisphosphate (PIP3), the second messenger produced by PI3K. Increasing INa,Late in tachypacing-induced HL-1 myocytes was recovered by infusion of PIP3 to non-pacing levels (Fig. 6j, k). Moreover, Akt inhibition also caused an increase in INa,Late but was significantly decreased by PIP3 infusion (Fig. 6l, m). Therefore, pathogenic INa,Late might be due to alterations in PI3K/Akt signaling. Although we performed immunoblotting of Nav1.5 expression, we found no significant difference between groups (Fig. 6n, o).
To elucidate the in vivo electrophysiological properties, we next recorded electrocardiograms (ECGs) from mice, which were divided into 3 groups: control (CTRL, DMSO injection), PI (PI-103 injection), and PI + Ran (PI-103 with ranolazine injection). We intraperitoneally injected PI-103 (10 mg/kg) alone or PI-103 with ranolazine (20 mg/kg) every day for 2 weeks. We found that PI3K inhibition displayed prolongation of QT interval, which was significantly shortened by INa,Late inhibition (Fig. 7a, b), leading to an anti-arrhythmic effect. However, there were no significant differences in the RR, PR, and QRS interval between groups (Fig. 7c–e). Our results imply that INa,Late inhibition is sufficient to alter electrophysiological properties and enhances PI3K/Akt signaling.

Prolongation of QT in PI3K inhibition is reversed by INa,Late inhibition in mouse heart. a Representative ECGs from CTRL (n = 6), PI (n = 6), and PI + Ran (n = 6) injected mice at pre-injection, 1, and 2 weeks. b–e Comparison of ECG parameters including QT, RR, PR, and QRS interval in CTRL, PI, and PI + Ran mice at baseline, 1, and 2 weeks. Values are the mean ± SEM. *p < 0.05 vs. CTRL, #p < 0.05 vs. PI + Ran. Comparison using one-way ANOVA followed by Tukey’s post hoc test
Taken together, our findings support the anti-arrhythmic effect of PI3K/Akt signaling on pathogenic INa,Late regulation, which prevents electrophysiological dysfunction and arrhythmic effects under AF conditions.
Discussion
In this study, we demonstrated that inhibiting INa,Late reversed abnormal Ca2+ handling and AP (prolonged APD and DADs) in tachypacing-induced HL-1 myocytes and that this recovery effect is related to the enhancement of PI3K/Akt signaling. Intriguingly, PI3K inhibition produced marked increases in INa,Late and subsequent contributions to aberrant AP morphology that were recovered by exposure to the INa,Late blocker ranolazine, suggesting that PI3K/Akt signaling plays a beneficial role in regulating pathogenic INa,Late and alterations in electrical remodeling. Together, these findings provide new insight into the potential role of the PI3K/Akt pathway in INa,Late and indicate that such regulation is critical to the treatment of AF (Fig. 8).

Scheme to demonstrate the mechanism for AF after INa,Late inhibition in tachypacing-induced HL-1 myocytes. Tachypacing or PI3K inhibition reduces PI3K p110α and pAkt expression, which further increases INa,Late, and those detrimental effects are recovered by ranolazine treatment, which also reduces the accumulation of Na+ and Ca2+ and thereby prevents AF
Nav1.5 mutations appear to play a pivotal role in the management of myocyte remodeling in AF. In particular, dysregulation of INa amplitude and duration contributes to incidence-triggered activity and prolonged AP and is related to severe AF [23, 24, 30]. Reductions in INa,Peak have been observed in arrhythmic disorders such as Brugada syndrome, long QT syndrome, and AF [10, 11, 28]. This effect further increases INa,Late by altering biophysical properties [19]. Thus, the combined gain and loss of INa function contributes to arrhythmic activity [4]. Previous studies reported reduced INa,Peak and enhanced INa,Late with biophysical defects in activation and inactivation in SCAN5A 1472del mutations of tsA201 cells [10]. Moreover, Lebek et al. [19] demonstrated that a negative shift in steady-state inactivation decreased INa,Peak and increased INa,Late while causing no difference in the activation curves of atrial myocytes in patients with sleep-disordered breathing. Although previous findings in clinical and experimental studies reported the dysregulation of INa in arrhythmia, the underlying mechanism and potential therapeutic options remain unclear. To clarify the pathological role of INa regulation in AF and the effect of INa,Late inhibition on INa, we examined both INa,Peak and INa,Late amplitudes in tachypacing-induced HL-1 myocytes. In those cells, we observed decreased INa,Peak and increased INa,Late, along with decreased kinetics of activation and inactivation and a depolarized shift rightward compared with non-paced HL-1 myocytes. Importantly, the consequences of pathological remodeling were all significantly reversed by INa,Late inhibition. Moreover, INa,Late inhibition recovered irregular Ca2+ handling and AP morphology. These results identify INa,Late as an efficient modulator of INa function and changes in electrical remodeling that could be used to prevent arrhythmic effects [15].
Importantly, a sustained increase in INa,Late is also a contributor to rising intracellular Na+, which induces intracellular Ca2+ accumulation and ultimately induces DADs and severe AF [4, 32]. We have demonstrated that increasing INa,Late contributes to Ca2+ dysregulation and the incidence of DADs, and those detrimental effects were abolished by INa,Late inhibition, suggesting that INa,Late could also be crucial in the regulation of Ca2+ handling. Of note, excessive intracellular Na+ overload caused by increasing INa,Late could be a result of reduced PI3K signaling, which leads to further intracellular Ca2+ overload [34]. Although we did not observe that Ca2+ signaling had any effect on PI3K signaling, it is possible that PI3K-mediated INa,Late regulation is responsible, at least in part, for alternating irregular Ca2+ release with basal Ca2+ levels. Further studies will be needed to examine the role of PI3K signaling in Ca2+ signaling under AF conditions.
Reducing or inhibiting PI3K/Akt signaling induces an increase in INa,Late through alterations of the INa gating property and exacerbates the cardiac remodeling related to arrhythmias [34]. Therefore, modulation of PI3K/Akt signaling could be informative in regulating pathogenic INa,Late and treating AF. Our data support the hypothesis that PI3K/Akt signaling plays an important role in INa,Late regulation for the management of AF in tachypacing-induced HL-1 myocytes. That loss of PI3K signaling significantly enhanced INa,Late due to decreased kinetics of activation and inactivation, which further contributed to prolonged APD and DADs, which are related to AF. Of note, PI3K inhibition is implicated as a consequence of INa,Late dysfunction and subsequent arrhythmia [21]. In fact, PI3K inhibition for 2 h by means of PI-103 or nilotinib resulted in increases in INa,Late in canine ventricular myocytes. In addition, Yang et al. [33] demonstrated that inhibiting the PI3Kɑ-specific subunit caused increases in INa,Late and further contributed to prolonged APD and triggered activity. Our results demonstrate that PI3K inhibition leads to increased INa,Late, aberrant APD, and the occurrence of DADs, which are all abolished by INa,Late inhibition and the consequent upregulation of PI3K 110α and pAkt. Moreover, we discovered that a reduction in INa,Late occurred after application of PIP3 in tachypacing- or Akt-inhibited HL-1 myocytes. This suggests that a reduction in PI3K/Akt signaling contributes to the enhancement of INa,Late and the dysregulation of INa open probability, which can lead to further accumulation of intracellular Na+ under AF conditions.
Our study has several limitations. First, the constructed cellular AF model induced myocyte remodeling and thus might not fully represent the environments and circumstances of AF in vivo. Because upregulation of the PI3K/Akt pathway is involved in oncogenesis, appropriate dosages and the potential side effects of using ranolazine to treat AF need to be further investigated [27]. Second, our animal experiment revealed prolongation of the QT interval with a slight increase in RR interval (decreased heart rate) on PI3K inhibition. QT interval prolongation is a major determinant of the risk of arrhythmia and is caused by electrical repolarization abnormalities [1, 26]. Although we did not examine the inducibility of atrial arrhythmia, it is possible that prolongation of QT interval by PI3K inhibition is due to pathogenic INa,Late regulation as well as an increase in APD.
In conclusion, these findings highlight the role of PI3K/Akt signaling in pathogenic INa,Late and suggest ways that it might be used against AF. This study supports the identification of PI3K/Akt as an effective factor for regulating pathogenic INa,Late and preventing AF caused by aberrant electrical remodeling.
References
Amin AS, Asghari-Roodsari A, Tan HL (2010) Cardiac sodium channelopathies. Pflugers Arch 460:223–237. https://doi.org/10.1007/s00424-009-0761-0
Baek YS, Choi JI, Kim YG, Lee KN, Roh SY, Ahn J, Kim DH, Lee DI, Hwang SH, Shim J, Kim JS, Kim DH, Park SW, Kim YH (2020) Atrial substrate underlies the recurrence after catheter ablation in patients with atrial fibrillation. J Clin Med 9. https://doi.org/10.3390/jcm9103164
Ballou LM, Lin RZ, Cohen IS (2015) Control of cardiac repolarization by phosphoinositide 3-kinase signaling to ion channels. Circ Res 116:127–137. https://doi.org/10.1161/CIRCRESAHA.116.303975
Belardinelli L, Shryock JC, Fraser H (2006) Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92(Suppl 4):iv6–iv14. https://doi.org/10.1136/hrt.2005.078790
Biet M, Morin N, Lessard-Beaudoin M, Graham RK, Duss S, Gagne J, Sanon NT, Carmant L, Dumaine R (2015) Prolongation of action potential duration and QT interval during epilepsy linked to increased contribution of neuronal sodium channels to cardiac late Na+ current: potential mechanism for sudden death in epilepsy. Circ Arrhythm Electrophysiol 8:912–920. https://doi.org/10.1161/CIRCEP.114.002693
Brundel B (2004) Calpain inhibition prevents pacing-induced cellular remodeling in a HL-1 myocyte model for atrial fibrillation. Cardiovascular Research 62(3):521–528. https://doi.org/10.1016/j.cardiores.2004.02.007
Choi JI, Baek YS, Roh SY, Piccini JP, Kim YH (2019) Chromosome 4q25 variants and biomarkers of myocardial fibrosis in patients with atrial fibrillation. J Cardiovasc Electrophysiol 30:1904–1913. https://doi.org/10.1111/jce.14104
Choi JI, Jung JS, Kim MK, Sim J, Kim JS, Lim HE, Park SW, Kim YH (2016) Effects of angiotensin-II receptor blocker on inhibition of thrombogenicity in a canine atrial fibrillation model. Korean Circ J 46:335–342. https://doi.org/10.4070/kcj.2016.46.3.335
Claycomb WC, Lanson NA Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ Jr (1998) HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 95:2979–2984. https://doi.org/10.1073/pnas.95.6.2979
Detta N, Frisso G, Zullo A, Sarubbi B, Cozzolino C, Romeo E, Wang DW, Calabro R, Salvatore F, George AL Jr (2013) Novel deletion mutation in the cardiac sodium channel inactivation gate causes long QT syndrome. Int J Cardiol 165:362–365. https://doi.org/10.1016/j.ijcard.2012.08.032
Dolz-Gaiton P, Nunez M, Nunez L, Barana A, Amoros I, Matamoros M, Perez-Hernandez M, Gonzalez de la Fuente M, Alvarez-Lopez M, Macias-Ruiz R, Tercedor-Sanchez L, Jimenez-Jaimez J, Delpon E, Caballero R, Tamargo J (2013) Functional characterization of a novel frameshift mutation in the C-terminus of the Nav1.5 channel underlying a Brugada syndrome with variable expression in a Spanish family. PLoS One 8:e81493. https://doi.org/10.1371/journal.pone.0081493
Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ (2015) Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation 132:567–577. https://doi.org/10.1161/CIRCULATIONAHA.114.015218
Han D, Tan H, Sun C, Li G (2018) Dysfunctional Nav1.5 channels due to SCN5A mutations. Exp Biol Med (Maywood) 243:852–863. https://doi.org/10.1177/1535370218777972
Hancox JC, James AF, Marrion NV, Zhang H, Thomas D (2016) Novel ion channel targets in atrial fibrillation. Expert Opin Ther Targets 20:947–958. https://doi.org/10.1517/14728222.2016.1159300
Horvath B, Hezso T, Kiss D, Kistamas K, Magyar J, Nanasi PP, Banyasz T (2020) Late sodium current inhibitors as potential antiarrhythmic agents. Front Pharmacol 11:413. https://doi.org/10.3389/fphar.2020.00413
Kim JC, Son MJ, Subedi KP, Li Y, Ahn JR, Woo SH (2010) Atrial local Ca2+ signaling and inositol 1,4,5-trisphosphate receptors. Prog Biophys Mol Biol 103:59–70. https://doi.org/10.1016/j.pbiomolbio.2010.02.002
Kim JC, Woo SH (2015) Shear stress induces a longitudinal Ca(2+) wave via autocrine activation of P2Y1 purinergic signalling in rat atrial myocytes. J Physiol 593:5091–5109. https://doi.org/10.1113/JP271016
Kim YG, Han KD, Choi JI, Choi YY, Choi HY, Boo KY, Kim DY, Lee KN, Shim J, Kim JS, Park YG, Kim YH (2021) Non-genetic risk factors for atrial fibrillation are equally important in both young and old age: a nationwide population-based study. Eur J Prev Cardiol 28:666–676. https://doi.org/10.1177/2047487320915664
Lebek S, Pichler K, Reuthner K, Trum M, Tafelmeier M, Mustroph J, Camboni D, Rupprecht L, Schmid C, Maier LS, Arzt M, Wagner S (2020) Enhanced CaMKII-dependent late INa induces atrial proarrhythmic activity in patients with sleep-disordered breathing. Circ Res 126:603–615. https://doi.org/10.1161/CIRCRESAHA.119.315755
Lu Z, Jiang YP, Wu CY, Ballou LM, Liu S, Carpenter ES, Rosen MR, Cohen IS, Lin RZ (2013) Increased persistent sodium current due to decreased PI3K signaling contributes to QT prolongation in the diabetic heart. Diabetes 62:4257–4265. https://doi.org/10.2337/db13-0420
Lu Z, Wu CY, Jiang YP, Ballou LM, Clausen C, Cohen IS, Lin RZ (2012) Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Transl Med 4:131ra150. https://doi.org/10.1126/scitranslmed.3003623
Maier LS, Sossalla S (2013) The late Na current as a therapeutic target: where are we? J Mol Cell Cardiol 61:44–50. https://doi.org/10.1016/j.yjmcc.2013.03.001
Makiyama T, Akao M, Shizuta S, Doi T, Nishiyama K, Oka Y, Ohno S, Nishio Y, Tsuji K, Itoh H, Kimura T, Kita T, Horie M (2008) A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol 52:1326–1334. https://doi.org/10.1016/j.jacc.2008.07.013
Musa H, Kline CF, Sturm AC, Murphy N, Adelman S, Wang C, Yan H, Johnson BL, Csepe TA, Kilic A, Higgins RS, Janssen PM, Fedorov VV, Weiss R, Salazar C, Hund TJ, Pitt GS, Mohler PJ (2015) SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc Natl Acad Sci U S A 112:12528–12533. https://doi.org/10.1073/pnas.1516430112
Potet F, Beckermann TM, Kunic JD, George AL Jr (2015) Intracellular calcium attenuates late current conducted by mutant human cardiac sodium channels. Circ Arrhythm Electrophysiol 8:933–941. https://doi.org/10.1161/CIRCEP.115.002760
Sabir IN, Li LM, Jones VJ, Goddard CA, Grace AA, Huang CL (2008) Criteria for arrhythmogenicity in genetically-modified Langendorff-perfused murine hearts modelling the congenital long QT syndrome type 3 and the Brugada syndrome. Pflugers Arch 455:637–651. https://doi.org/10.1007/s00424-007-0326-z
Samuels Y, Ericson K (2006) Oncogenic PI3K and its role in cancer. Curr Opin Oncol 18:77–82. https://doi.org/10.1097/01.cco.0000198021.99347.b9
Selga E, Sendfeld F, Martinez-Moreno R, Medine CN, Tura-Ceide O, Wilmut SI, Perez GJ, Scornik FS, Brugada R, Mills NL (2018) Sodium channel current loss of function in induced pluripotent stem cell-derived cardiomyocytes from a Brugada syndrome patient. J Mol Cell Cardiol 114:10–19. https://doi.org/10.1016/j.yjmcc.2017.10.002
Song J-Q, Teng X, Cai Y, Tang C-S, Qi Y-F (2009) Activation of Akt/GSK-3β signaling pathway is involved in intermedin1–53 protection against myocardial apoptosis induced by ischemia/reperfusion. Apoptosis 14:1299. https://doi.org/10.1007/s10495-009-0398-7
Song W, Shou W (2012) Cardiac sodium channel Nav1.5 mutations and cardiac arrhythmia. Pediatr Cardiol 33:943–949. https://doi.org/10.1007/s00246-012-0303-y
Song Y, Shryock JC, Belardinelli L (2008) An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol 294:H2031-2039. https://doi.org/10.1152/ajpheart.01357.2007
Wu L, Ma J, Li H, Wang C, Grandi E, Zhang P, Luo A, Bers DM, Shryock JC, Belardinelli L (2011) Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation 123:1713–1720. https://doi.org/10.1161/CIRCULATIONAHA.110.000661
Yang T, Meoli DF, Moslehi J, Roden DM (2018) Inhibition of the alpha-subunit of phosphoinositide 3-kinase in heart increases late sodium current and is arrhythmogenic. J Pharmacol Exp Ther 365:460–466. https://doi.org/10.1124/jpet.117.246157
Zhabyeyev P, Chen X, Vanhaesebroeck B, Oudit GY (2019) PI3Kalpha in cardioprotection: cytoskeleton, late Na(+) current, and mechanism of arrhythmias. Channels (Austin) 13:520–532. https://doi.org/10.1080/19336950.2019.1697127
Funding
This work was supported by grants from Korea University, Korea University Anam Hospital, Seoul, Republic of Korea, the Korean Heart Rhythm Society (No. KHRS2015-3 to J-I.C), and by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT, Ministry of Science and ICT) (No. 2021R1A2C2011325 to J-I.C).
Author information
Authors and Affiliations
Contributions
J. I. Choi had full access to all data in this study and is responsible for data integrity and analytical accuracy. The study concept and design were formulated by J. I. Choi. Electrophysiology and molecular experiments were performed by T. H. Ko, D. Jeong, B. Yu, and J.E. Song. Ca2+ transients were measured and interpreted by Q. Ahn L and S. H. Woo. Data analysis and interpretation were performed by T.H. Ko, D. Jeong, and J. I. Choi. T.H. Ko and D. Jeong performed the statistical analysis. T.H. Ko and D. Jeong drafted the article. S. H. Woo and J. I. Choi reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ko, T.H., Jeong, D., Yu, B. et al. Inhibition of late sodium current via PI3K/Akt signaling prevents cellular remodeling in tachypacing-induced HL-1 atrial myocytes. Pflugers Arch - Eur J Physiol 475, 217–231 (2023). https://doi.org/10.1007/s00424-022-02754-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-022-02754-z