
Abstract
Purpose
Poor maternal nutrition sensitises to the development of metabolic diseases and obesity in adulthood over several generations. The prevalence increases when offspring is fed with a high-fat (HF) diet after weaning. This study aims to determine whether such metabolic profiles can be transmitted to the second generation and even aggravated when the mothers were exposed to overnutrition, with attention to potential sex differences.
Methods
Pregnant Wistar rats were subjected to ad libitum (control) or 70% food-restricted diet (FR) during gestation (F0). At weaning, F1 females were allocated to three food protocols: (1) standard diet prior to and throughout gestation and lactation, (2) HF diet prior to and standard diet throughout gestation and lactation, and (3) HF diet prior to and throughout gestation and lactation. F2 offspring was studied between 16 and 32 weeks of age.
Results
FR-F2 offspring on standard diet showed normal adiposity and had no significant metabolic alterations in adulthood. Maternal HF diet resulted in sex-specific effects with metabolic disturbances more apparent in control offspring exposed to HF diet during gestation and lactation. Control offspring displayed glucose intolerance associated with insulin resistance in females. Female livers overexpressed lipogenesis genes and those of males the genes involved in lipid oxidation. Gene expression was significantly attenuated in the FR livers. Increased physical activity associated with elevated corticosterone levels was observed in FR females on standard diet and in all females from overnourished mothers.
Conclusions
Maternal undernutrition during gestation (F0) improves the metabolic health of second-generation offspring with more beneficial effects in females.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The developmental origins of health and disease (DOHaD) hypothesis states that malnutrition, especially when occurring during fetal and/or neonatal periods, increases the risks of develo** metabolic diseases in adulthood [1,2,3]. Offspring have to be nutritionally thrifty to adapt to tide over poor nutrition in anticipation of future nutritional conditions identical to the maternal environment [4, 5]. Any environmental mismatch with the predictions then results in maladaptation and increases disease risk in individuals [6,7,8]. Experimental and epidemiological evidence suggests that risk is not only limited to exposed individuals, but also to subsequent generations, even though nutritional conditions are favorable [9,10,11,12,13]. Intergenerational transmission of disease thus contributes to the pandemic of obesity and diabetes in the world. Offspring (grandchildren) of prenatally undernourished people have an increased adiposity and are prone to chronic diseases later on [14]. In agreement, animal models have shown that malnutrition in utero and/or during lactation increased also the risk of obesity, glucose intolerance, insulin resistance, and type 2 diabetes in the following generation [15,16,17,18,19,20,21].
Sex differences in the inheritance of programmed diseases have been observed, with male often more affected than females [13, 17, 22, 23]. Sexual dimorphism is partly explained by the fact that the growth trajectories of male and female fetuses are different in both normal and pathological pregnancies. Growth velocity is accelerated in male fetuses and maintained when confronted with an adverse environment at the expense of their future health. In contrast, female fetuses have slower growth, allowing them to better adapt to intrauterine disturbances with a prognosis of favorable long- term survival [13, 14]. Placenta contributes to sex-specific adaptations to pregnancy perturbations [24, 25].
Using a rat model, we have previously reported that male offspring from prenatally food-restricted mothers (70% caloric restriction, FR30 animals) exhibit intrauterine growth restriction, low birth weight and develop adiposity, glucose intolerance and hypertension with aging [26, 27], particularly when fed a high-fat (HF) diet post-weaning [28,29,30]. Although FR-F1 females are less susceptible to develo** metabolic diseases compared with males [31, 32], under the physiological demands of pregnancy, they can exhibit during gestation metabolic disorders such as glucose intolerance and gestational diabetes [13]. Thus, gestation may unmask the metabolic dysfunctions in predisposed females, especially if pregnant mothers are overnourished. This in turn induces a suboptimal intrauterine environment for the develo** F2 fetus and compromises his future health in adulthood.
Maternal nutrition during lactation plays a critical role in determining the offspring’s metabolic phenotype [33,34,35], and maternal obesity and/or maternal HF diet affects the lipid composition of breast milk [36]. This suggests that exposure to hypercaloric diet in susceptible females during gestation and/or lactation can increase vulnerability to metabolic diseases in the subsequent F2 generation.
In the present study, we assessed the intergenerational transmission of programmed disease outcomes through the maternal lineage. Pregnant mothers, F0, were exposed to undernutrition throughout gestation. F1 females from food-restricted (FR) mothers were allocated to either standard or HF diets at weaning and bred to yield the F2 generation. Here, we tested the hypothesis that FR-F2 generation, born to F1 females on HF diet, prior and/or during gestation and lactation was more affected. We therefore determined whether maternal undernutrition during gestation (1) increased the risk of develo** metabolic disease in the F2 offspring, (2) accentuated the effects induced by the HF diet of F1 females and (3) induced sexual differences.
Materials and methods
Animals
The rat model of fetal growth restriction has been previously described [27]. Pregnant Wistar rats (Charles Rivers Laboratories, L’Arbresle, France) were randomly maintained with unrestricted (Control, C) access to standard (S) chow diet (Safe, Augy, France) or restricted to 70% (food-restricted, FR30 animals) during gestation to yield the first generation (F1). At birth, pups (4 males and 4 females) remained with their respective mothers throughout lactation. At weaning, F1 females were fed either standard diet (8.4% fat) or high-fat (HF) diet (45.8% fat, purified diet U8954, safe) (Supplemental Table 1). At 13 weeks of age, F1 females were bred with control males to generate the second generation (F2). We performed three paradigms of maternal diet (Fig. 1): (1) S diet prior to mating and throughout gestation and lactation (SSS); (2) HF diet prior to mating and S diet throughout gestation and lactation (HFSS) and (3) HF diet prior to mating and throughout gestation and lactation (HFHFHF). F2 offspring were fed a standard diet from weaning to killing. Food intake and spontaneous physical activity were measured at 16 weeks of age using the Physiocage System and Metabolism v2.1 software (Panlab-bioseb, Chaville, France). A glucose tolerance test was performed at 30 weeks of age. At 31 weeks of age, a tail blood sample was collected to determine the basal (nonfasted) corticosterone levels. At 32 weeks of age, animals were fasted overnight and euthanized with isoflurane. Livers were rapidly dissected and directly snap frozen in liquid nitrogen. Animals were maintained at 22 °C on a 12:12 h light/dark cycle with light off at 19:00 and on at 07:00.The accreditation for animal experimentation has been granted to our laboratory by the French Ministry of Agriculture (no. 04860). All experiments were conducted according to European Communication Council Directive of November 24, 1986 (86/609/EEC), and approved by the French Department Direction of Veterinary Services Committee (DDSV/59-009228).

Experimental design presenting the breeding scheme and nutritional strategies for the second generation (F2). Diet in F0 mothers during pregnancy is denoted as control (C, ad libitum) or food restricted (FR, 30% of daily intake) (first letter). Diet in F1 female’s offspring is denoted as standard diet (S) or high-fat diet (HF) and is allocated prior to gestation (second letter) or during gestation (third letter) and lactation (fourth letter)
Oral glucose tolerance test
After an overnight fasting, d-glucose (2 mg/g body weight) was injected per os in conscious rats. Blood glucose levels were measured using ACCU-CHEK Performa (Roche Diagnostics, Meylan, France) at 0, 30, 60, 90 and 120 min via tail puncture. The total area under the curve (AUC) was calculated using the trapezoidal method (GraphPad Prismv6.01, La Jolla, CA, USA). Homeostasis model assessment for insulin resistance (HOMA-IR) was calculated as previously described [37].
Plasma analysis
Insulin and corticosterone plasma levels were determined using rat-specific commercial ELISA kits (DRG International, Inc, USA; Bertin Pharma, Montigny-le Bretonneux, France; Immunodiagnostic Systems Ltd, Boldon, UK). Plasma non-esterified fatty acids (NEFAs) and triglyceride (TGs) concentrations were analyzed using enzymatic assay kits (Wako Chemicals, Neus, Germany; Sobioda, Montbonnot, France).
Hepatic triglyceride content
Liver triglycerides were extracted according to a previously described method with some modifications [38]. Lipids were extracted by isopropanol–hexane–water/hexane–diethyl ether technique. Triglycerides were measured using the Infinity Triglyceride Reagent (Thermo Fisher Scientific, MA, USA). Triolein (Sigma-Aldrich, St. Louis, MO, USA) was used as standard.
Real-time quantitative polymerase chain reaction
Total RNA was extracted from snap-frozen liver samples (20 mg), using the RNeasy minikit (Qiagen, Crawley, UK). RNA (1 µg), was reverse-transcribed by the use of the ThermoScript cDNA Reverse Transcription Kit (Invitrogen Life Technologies, Carlsbad, CA, USA). Real-time PCR experiments were performed via a Light-Cycler 2700 System (Roche Biochemicals, France). All cDNA samples were analyzed in duplicate and the reaction was carried out with QuantiTect SYBR Green PCR Master Mix (Qiagen). Relative quantification of each gene was calculated after normalization to β-actin (housekee** gene) using the comparative threshold cycle method. The primers used are listed in Supplemental Table 2.
Statistical analysis
Statistical analyses were performed using GraphPad Prism v6.01.Multiple comparisons were performed using Kruskal–Wallis analysis of variance followed by Mann–Whitney post hoc test. Body weight and oral glucose tolerance test (OGTT) values were analyzed by a two-way repeated measures analysis of variance (RM-ANOVA) with Bonferroni post hoc test. Differences between male and female values were analyzed with unpaired t test with Welch’ correction. Data are expressed as mean ± SD or median with interquartile range as appropriate. Significance was determined at P value < 0.05.
Results
Maternal undernutrition effects on growth and metabolism of F2 offspring
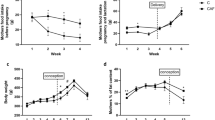
F1 females born to FR dams with unrestricted access to standard diet (FRSS) had the same weight as F1 control females (CSS) prior to breeding (Table 1). There were no significant differences between FR-F1 females and C-F1 females with respect to mother weight gain during gestation (Table 1). Litter size was similar in the two groups (Table 1). Despite this, weight at 4 days of age was significantly reduced in the FR-F2 offspring (FRSSS) compared with the C-F2 offspring (CSSS) (Fig. 2). Growth restriction remained until 8 weeks and 16 weeks of age in males and females, respectively (Fig. 2). Food intake and spontaneous activity did not differ between controls and FRSSS males at 16 weeks of age (Fig. 3, Supplemental Figs. 1A and 2A). However, FRSSS females despite normal food intake (Fig. 3a) demonstrated increased spontaneous activity compared with controls (Fig. 3b, Supplemental Fig. 2B). FRSSS offspring had similar weight and visceral adiposity to CSSS offspring at 32 weeks of age (Table 2). Thus, FR-F1 females led to F2 offspring that was lighter in early postnatal life and failed to show catch-up growth and increased adiposity in adulthood. To define glucose homeostasis, we performed OGTT at 30 weeks of age. Glucose tolerance was unimpaired in FRSSS offspring (Fig. 4) and HOMA-IR indicated normal insulin sensitivity (Table 3). Plasma levels of NEFAs were increased in FRSSS males compared to controls, but not in females (Table 3). In contrast, TGs levels were similar in FRSSS and control offspring (Table 3). Furthermore, nonfasting (baseline) corticosterone levels were increased in FRSSS compared to CSSS only in females (Table 3). We next explored the influence of maternal undernutrition on liver function in F2 offspring. We were not able to detect significant difference in liver mass in FRSSS offspring when compared with controls (Table 2). Despite that FRSSS offspring were not exposed to nutritional stress in utero and during breastfeeding, TGs content and the expression of genes involved in lipid metabolism were modulated in female livers but not male livers. Expression of genes from lipid biosynthesis such as Dgat2 and Fasn was reduced in FRSSS livers (Fig. 5). Likewise, the gene expression of Cpt1α that regulates lipid oxidation was also decreased (Fig. 6). Changes in gene expression in female livers were associated with higher levels of TGs compared to controls (Table 3).

Growth curves for the F2 offspring. Mean body weight of male and female offspring from 1 to 21 days of age (left panel) and from 1 to 32 weeks of age (right panel). Data are presented as mean ± SD, n = 7–10 males and females (from 5 to 7 litters). *P < 0.05, ***P < 0.001 HF vs S diet; †P < 0.05 FR vs C (two way repeated-measures ANOVA)

Food intake and spontaneous locomotor activity of F2 offspring at 16 weeks of age. Food intake (a) and locomotor activity (b) in males and females were measured at 2-h intervals for 24 h using metabolic cages. Data are presented as median with interquartile range, n = 5 males and females (from 5 to 7 litters). **P < 0.01, ***P < 0.001 HF vs S diet; †††P < 0.001 FR vs C (ANOVA)

Glucose tolerance in F2 offspring at 30 weeks of age. Oral glucose tolerance test (2 mg/g body weight) was performed after an overnight fast. Mean glucose values of male and female offspring (left panel). Data are presented as mean ± SD (two-way repeated-measures ANOVA). Glucose tolerance test area under the curve (AUC) of male and female offspring (right panel). Data are presented as median with interquartile range. *P < 0.05, **P < 0.01 HF vs S diet; †P < 0.05 FR vs C (ANOVA). n = 7–10 males and females (from 5 to 7 litters)

Expression of lipogenesis genes in the liver of F2 adult males and females. The expression of DGAT2, Plin2 and FASN from male and female livers was determined by real-time polymerase chain reaction at 32 weeks of age. Data are presented as median with interquartile range, n = 5–6 males and females (from 5 to 7 litters). *P < 0.05, **P < 0.01 HF vs S diet; †P < 0.05, ††P < 0.01 FR vs C (ANOVA)

Expression of lipid oxidation genes in the liver of F2 adult males and females. The expression of CPT1α and Acadvl from male and female livers was determined by real-time polymerase chain reaction at 32 weeks of age. Data are presented as median with interquartile range, n = 5–6 males and females (from 5 to 7 litters). *P < 0.05, **P < 0.01 HF vs S diet; †P < 0.05, ††P < 0.01 FR vs C (ANOVA)
Maternal high-fat diet effects on growth and metabolism in F2 offspring
Maternal body weight prior to breeding was similar in HF dams (CHFS and FRHFS) compared to controls, indicating that none of the F1 females fed the HF diet became obese after 10–12 weeks of feeding (Table 1). In pregnancies of F1 females fed HF diet before breeding, both maternal weight gain and litter size were similar between CHFS and FRHFS groups (Table 1). The consequences on F2 offspring born to mothers fed HF diet prior to breeding were dependent on the study groups. FR-F2 offspring (FRHFSS) were heavier from 1 to 7 days of age compared to those born to mothers on the control diet (Fig. 2). Post-weaning and through 32 weeks of age, FRHFSS offspring demonstrated weight gain similar to controls (Fig. 2), whereas FRHFSS females presented visceral adiposity by 32 weeks of age (Table 2). Food intake was similar to controls, whereas spontaneous activity increased in CHFSS females (Fig. 3b, Supplemental Fig. 2B). Glucose and insulin levels and HOMA-IR were similar in FRHFSS and CHFSS offspring compared to controls (Table 3), whereas glucose tolerance was impaired in CHFSS males (Fig. 4). Likewise, plasma NEFAs levels were increased in CHFSS males and females, but not in FRHFSS animals (Table 3). However, NEFAs levels were significantly reduced in FRHFSS compared to CHFSS when considering only females. In contrast, TGs levels were unaltered in both CHFSS and FRHFSS offspring. We observed that liver mass was increased in CHFSS males (Table 2) and TGs content was increased in CHFSS females (Table 3) but not in FRHFSS animals. Liver gene expression was mostly modulated in FRHFSS females. Strikingly, expression of Dgat2, Plin2, Fasn and Cpt1α genes was significantly increased in FRHFSS livers (Figs. 5, 6), whereas TGs content remained unchanged (Table 3). These data support that FR-F1 females fed HF diet during the pregestational period led to F2 offspring with no significant metabolic disorders compared to C-F2 offspring. However, maintaining FR-F1 females on an HF diet induced significant changes in the expression of lipid genes in livers from FR-F2 females.
As maternal stress experienced during gestation may unmask programmed disease in FR females, an HF diet was continued during gestation and throughout lactation. We observed that C-F1 females (CHFHF) showed a tendency to reduced weight gain during gestation compared to controls (Table 1). In contrast, FR-F1 females (FRHFHF) significantly gained the least weight in gestation (Table 1). There were no significant differences with respect to sex ratio (data not shown) and litter size compared with controls (Table 1). Unfortunately, the weight of lactating mothers had not been evaluated in the study, and thus we were not able to determine if the HF diet affected their body weight during breastfeeding. The impact on the growth in F2 offspring was similar between C and FR lineages. F2 newborns at day 1 had low weight before experiencing rapid weight gain until 21 days of age during sucking (Fig. 2). Post-weaning and through 32 weeks of age, both CHFHFHF and FRHFHFHF offspring had body weight (Fig. 2) and visceral adiposity (Table 2) similar to controls. Food intake was not different from controls, whereas spontaneous activity was increased in CHFHFHF and FRHFHFHF females (Fig. 3b, Supplemental Fig. 2B). Glucose levels in F2 offspring did not differ from controls. However, CHFHFHF females had reduced insulin sensitivity, as indicated by higher insulin levels and HOMA-IR values compared to controls (Table 3). Glucose tolerance was impaired in CHFHFHF males and females relative to controls (Fig. 4). In contrast, FRHFHFHF males and females exhibited normal insulin sensitivity and glucose tolerance. Likewise, plasma NEFAs levels were increased in CHFHFHF males and females, but not in FRHFHFHF animals when compared to control (Table 3). However, NEFAs levels were significantly reduced in FRHFHFHF females compared to CHFHFHF females. A similar pattern was not observed in plasma TGs levels which increased in FRHFHFHF males, but not in FRHFHFHF females and CHFHFHF animals (Table 3). TGs levels were significantly increased in FRHFHFHF males compared with CHFHFHF males. Furthermore, corticosterone concentrations were increased in CHFHFHF females with levels significantly higher than that in FRHFHFHF females (Table 3). Plasma corticosterone was unaltered in both CHFHFHF and FRHFHFHF males. With regard to liver function, we found that liver mass (Table 2) and TGs content (Table 3) remained increased in CHFHFHF males and CHFHFHF females, respectively, but not in FRHFSS animals. However, exposure to maternal HF diet in utero and during the sucking period induced differential expression of lipid genes between FR-F2 and C-F2 offspring. Expression of Plin2, Fasn (Fig. 5) and Cpt1α (Fig. 6) was increased in livers of CHFHFHF males, whereas Cpt1α and Acadvl (Fig. 6) were overexpressed in livers of FRHFHFHF males. The expression of Plin2, Fasn and Cpt1α was reduced in livers of FRHFHFHF males compared with CHFHFHF males. Dgat2, Plin2 and Fasn gene expression was also increased in CHFHFHF female livers (Fig. 5), whereas Dgat2, Fasn and Cpt1α (Fig. 6) were overexpressed in livers of FRHFHFHF females. The expression of Dgat2, Plin2, Fasn and Cpt1α was reduced in livers of FRHFHFHF females compared with CHFHFHF females. Collectively, these data indicate that F1 females maintained on the HF diet throughout gestation and lactation led to more metabolic disorders in the F2 offspring, with significant differences according to lineage and sex. Maternal HF diet impaired glucose tolerance and insulin sensitivity and increased NEFAs circulating levels in C-F2 animals. Conversely, FR-F2 offspring were glucose tolerant with intact insulin sensitivity and the lipid profile was improved in females, but not in males that have developed dyslipidemia. The livers from C-F2 offspring displayed some functional alterations that were associated with greater lipid gene expression compared to FR-F2 animals. There were also sex-specific differences because genes that regulate TGs biosynthesis were more modulated in female livers, whereas those involved in fatty acid oxidation were more modulated in male livers.
Biological sexual dimorphism
In general, 32-week-old male offspring had higher plasma insulin and TGs levels, and HOMA-IR values as compared to female offspring, irrespective of lineage and F1 dam’s diet. In contrast, plasma NEFAs levels were higher in females than males irrespective of F1 dam’s diet in C lineage not in FR lineage.
Discussion
Maternal malnutrition during gestation increases the risk of develo** metabolic diseases and obesity in the F1 generation with a sex-specific sensitivity, as female offspring are generally less susceptible compared with males. However, when these predisposed F1 females become pregnant, they often display some physiological alterations that may, in turn, create an adverse intrauterine environment for the growth of F2 fetus that can contribute to disease transmission through subsequent generations. Such transmission of environment-induced traits to the F2 generation through the maternal lineage must be defined as a multigenerational effect, rather than transgenerational transmission. We have previously shown that 70% caloric restriction during gestation in pregnant females (F0) predisposes F1 males to develop metabolic disorders when they are exposed to consumption of HF diet later in life [28, 29]. Hence, we assessed intergenerational effects of maternal undernutrition (F0) on the growth and metabolic profile in the F2 generation through the maternal lineage. To date, multigenerational effects of maternal malnutrition were investigated on the F1 generation that was fed a standard diet [16,17,18,19,20,21]. In this context, our study was conducted (1) to determine whether F2 offspring born to FR-programmed F1 females on standard diet or hypercaloric diet were destined to develop metabolic diseases in adulthood, (2) to define the most vulnerable period for the effects of HF diet (prior to mating vs prior to mating until lactation) and (3) to evaluate whether transmission of phenotypes was sex-specific.
We report that the FR-F2 offspring, born to mothers on standard diet, show reduced body weight from early life until 8–16 weeks of age. Thus, the growth-restricted phenotype is transmitted to the second generation through the maternal lineage, supporting the previously published data on other rodent models [19, 39, 40]. Heritable growth restriction is associated with reduced H19 expression, the imprinted gene integral to fetal growth and development via epigenetic modifications [41]. Strikingly, we found that growth-restricted F2 generation was not prone to develop obesity and associated metabolic disorders, contrary to previous studies [19, 40]. Here, we assessed the transmission of programmed diseases to the second generation through the maternal lineage, since F1 females exposed to undernutrition in utero were mated with healthy males. In contrast, when undernourished males were used for breeding and mated with undernourished or healthy females, their offspring had increased risk for obesity-associated metabolic diseases. This indicates that susceptibility toward obesity and/or diabetes requires either the paternal lineage only or a combination of the two parental lineages. However, we show that in utero undernutrition in female influences the expression of genes involved in lipid metabolism in livers of F2 females. Expression of genes that regulate TGs biosynthesis and fatty acid oxidation is decreased in female livers. Reduced turnover of lipid indicates that the liver of FR females is programmed to optimize cellular energy efficiency according to the thrifty phenotype hypothesis. Interestingly, lipid gene expression is unaltered in livers of F2 males. Sexual dimorphism can be explained, in part, by distinct sex hormone signaling between males and females in the liver [42]. Alternatively, deregulated lipid gene expression in the liver can be attributed to heritable epigenetic modifications that occur in a sex-specific way. It is likely that the sex steroid hormones synthesized by the fetus and the placenta may modulate the effects of maternal undernutrition during gestation on epigenetic modifications in the offspring in a sex-dependent manner. Epigenetic mechanisms have been proposed to explain how the early life environment can affect genome function and gene expression with long-term consequences on health [43]. Furthermore, some epigenetic changes induced by the environment in the parent may be inherited through the germline and involve maternal or paternal lineages [44]. Several studies have reported that suboptimal maternal nutrition, such as low-protein diet, and high-fat feeding, leads to epigenetic modifications, notably DNA methylation [45], histone modifications [46, 47] and non-coding microRNAs (miRNA) expression [48] in the offspring livers. Furthermore, it has been shown that undernutrition in utero influenced the expression of lipogenic genes in livers from F2 males through the paternal lineage [49]. Importantly, the authors identified epigenetic marks in the male livers that are already present in germ cells from their progenitor F1 males. This demonstrates that under-nourishment in utero leads to epigenome changes in the gametes of the male progeny that are inherited in the subsequent generations. Whether epigenetic alterations could be propagated into the second-generation offspring through the maternal lineage remains a future topic of research.
To test the hypothesis that a nutritional challenge imposed on the FR females can further increase the risk of develo** diseases in their offspring, we nourished F1 females with high-fat diet prior to and throughout gestation and lactation. We examined the effects of each of these exposure periods on offspring phenotype. We found that F1 females fed the HF diet are not obese prior to breeding and then gain the least weight during gestation. This indicates that dietary components, such as protein, fat and carbohydrate contents as well as sucrose and saturated fatty acid levels are essential for maternal diet-induced obesity. Consequently, the effects of HF diet on the offspring can be different from those induced by maternal obesity. Consumption of an HF diet prior to mating in FR-F1 females was associated with overgrowth in FR-F2 offspring during early postnatal life without adiposity and metabolic alterations in adulthood. The HF feeding in C-F1 females did not impact on body weight of C-F2 offspring, but resulted in an impaired glucose tolerance and elevated NEFAs levels. Examination of gene expression in the liver demonstrated programming effects of maternal undernutrition in F2 females, because lipogenic and oxidative genes exhibited increase in FR livers. This suggests that maternal undernutrition programs a reduced expression of genes involved in lipid metabolism in the livers of F2 females born to mothers on the control diet, whereas it augments expression in livers of F2 females from mothers on HF diet. Thereby, the liver of FR females is endowed with remarkable metabolic plasticity allowing a better adaptation to maternal nutritional challenges. Consumption of an HF diet prior to and throughout gestation and lactation impacts the growth of F2 offspring irrespective of the maternal lineage. This results in low weight at P1 and accelerated growth until weaning in both C and FR offspring, showing the importance of HF feeding rather than programming on postnatal growth. Growth restriction in the newborns may reflect fetal malnutrition as HF-fed dams gain less weight during pregnancy and HF diet has been reported to cause placental insufficiency [50, 51]. Catch-up growth displayed during the suckling period emphasizes the impact of maternal milk on newborns’ development. Nevertheless, maternal HF diet has not impacted the body weight and adiposity of F2 offspring in adulthood, unlike published models of maternal obesity [35, 52]. This emphasizes that maternal obesity and weight gain during gestation are predictive of adiposity/obesity in the offspring. On the other hand, maternal HF diet influenced differentially the metabolic profile of the F2 offspring according to the lineage and sex. Impaired glucose tolerance and insulin sensitivity were evident in C-F2 males and females, whereas glucose homeostasis was preserved in the FR-F2 offspring. The lipid plasma profile was different between sexes, with FR-F2 males demonstrating elevated TGs levels, whereas FR-F2 females displayed lower NEFAs concentrations compared with the C-F2 offspring. Despite their normal glucose tolerance and insulin sensitivity, FR-F2 males had mild dyslipidemia. Nonetheless, we reported that consumption of an HF diet by F1 females perturbed lipid metabolism in livers of C-F2 and FR-F2 offspring with sex-dependent effect. Indeed, expression of genes involved in TGs synthesis and/or storage within lipid droplets increases in F2 female livers, whereas F2 male livers present an increase in gene expression related to lipid oxidation. This reveals the programming effects of maternal HF diet during both gestation and lactation on lipid gene expression in the F2 offspring livers. Thus, we think that the female-specific gene profile in the liver would promote nonalcoholic steatosis without significant cellular damage, while that of males could be associated with mitochondrial oxidative stress that is known to be involved in a more severe form of liver disease named nonalcoholic steatohepatitis (NASH). These data are in accordance with the sexual dimorphism of the nonalcoholic fatty liver disease (NAFDL) programming [53]. In addition, our study shows that exposure to maternal HF diet leads to further functional changes in C-F2 livers that are associated with greater gene expression. So, the attenuated expression of genes in FR-F2 livers would prevent lipid accumulation and avoid the emergence and/or progression of NAFLD. Taken together, our results suggest that FR-F2 offspring exposed to maternal HF diet through gestation and lactation remain healthier compared to the C-F2 offspring.
We previously demonstrated that maternal undernutrition programs the HPA axis in F1 males [54]. Here, we show that maternal programming effect continues in the second-generation offspring in females only. The persistent alteration in the set point of the HPA axis may be considered as an imprinting of food-restricted exposure during the early postnatal period. The effects of the maternal environment on the HPA activity often involve epigenetic modifications of the glucocorticoid receptor genes that are potentially reversible [55]. In this context, we could speculate that some epigenetic marks are conserved in FR-F2 females. Intriguingly, we found that consumption of an HF diet by the F1 females also increased HPA axis activity in F2 females irrespective of the lineage. Maternal HF diet acts as a stressor increasing corticosterone levels in pregnant dams and disturbing the fetal HPA axis development [56]. However, it remains to be determined if the mechanisms of HPA axis programming by maternal food restriction or overnutrition overlap or not. Sex differences in the programming of the HPA axis agree with more vulnerability in females in terms of reactivity [57]. Finally, we observed that both maternal undernutrition programming and HF diet result in increased spontaneous physical activity (SPA) in F2 females, but not in males. Conversely, studies have reported physical inactivity and reduced energy expenditure in female offspring exposed in utero and during the postnatal life to overnutrition [58, 59]. The phenotype was associated with obesity, unlike in our study where adiposity of females was not affected. The reasons for this discrepancy as well as underlying mechanisms regulating individual propensity for physical activity in response to maternal nutrition remain largely unknown.
We conclude that FR-F2 offspring born to F1 mothers on the control diet or fed an HF diet have low risk of metabolic diseases, indicating that in utero undernutrition (F0) seems to have a favorable impact on the metabolic health of second-generation offspring. This finding challenges the hypothesis that the thrifty phenotype makes an individual more sensitive to chronic diseases. At least, these results suggest reduced susceptibility to diseases in offspring when associated with the maternal lineage. We report evidence that the metabolic profile is improved in FR-F2 females, and that the pattern of gene expression in the liver is predictive of fatty liver disease, supporting the sex differences in the pathophysiology of metabolic diseases [60]. Sexual dimorphism reveals that offspring females may be less vulnerable to the influence of nutritional environment during early developmental periods. Sexual differences can impact the occurrence and gravity of pathologies and thus compromise the life expectancy of the individual.
In this context, animal studies should be systematically conducted in both males and females to design therapeutic sex-specific interventions that aim at preventing metabolic diseases and improving the health of future generations.
References
Barker DJ (2007) The origins of the developmental origins theory. J Intern Med 261:412–417
Gluckman PD, Hanson MA, Cooper C, Thornburg KL (2008) Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359:61–73
Warner MJ, Ozanne SE (2010) Mechanisms involved in the developmental programming of adulthood disease. Biochem J 427:333–347
Hales CN, Barker DJ (2001) The thrifty phenotype hypothesis. Br Med Bull 60:5–20
Wells JC (2011) The thrifty phenotype: an adaptation in growth or metabolism? Am J Hum Biol 23:65–75
Ross MG, Desai M (2005) Gestational programming: population survival effects of drought and famine during pregnancy. Am J Physiol Regul Integr Comp Physiol 288:R25–R33
Vaag AA, Grunnet LG, Arora GP, Brøns C (2012) The thrifty phenotype hypothesis revisited. Diabetologia 55:2085–2088
Hanson MA, Gluckman PD (2014) Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev 94:1027–1076
Patti ME (2013) Intergenerational programming of metabolic disease: evidence from human populations and experimental animal models. Cell Mol Life Sci 70:1597–1608
Aiken CE, Ozanne SE (2014) Transgenerational developmental programming. Hum Reprod Update 20:63–75
Vickers MH (2014) Developmental programming and transgenerational transmission of obesity. Ann Nutr Metab 64:26–34
Jimenez-Chillaron JC, Ramon-Krauel M, Ribo S, Diaz R (2016) Transgenerational epigenetic inheritance of diabetes risk as a consequence of early nutritional imbalances. Proc Nutr Soc 75:78–89
Cheong JN, Wlodek ME, Moritz KM, Cuffe JS (2016) Programming of maternal and offspring disease: impact of growth restriction, fetal sex and transmission across generations. J Physiol 594:4727–4740
Veenendaal MVE, Painter RC, de Rooij SR et al (2013) Transgenerational effects of prenatal exposure to the 1944–45 Dutch famine. BJOG 120:548–553
Benyshek DC, Johnston CS, Martin JF (2004) Post-natal diet determines insulin resistance in fetally malnourished, low birthweight rats (F1) but diet does not modify the insulin resistance of their offspring (F2). Life Sci 74:3033–3041
Benyshek DC, Johnston CS, Martin JF (2006) Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. Diabetologia 49:1117–1119
Zambrano E, Martínez-Samayoa PM, Bautista CJ et al (2005) Sex differences in transgenerational alterations of growth and metabolism in progeny (F2) of female offspring (F1) of rats fed a low protein diet during pregnancy and lactation. J Physiol 566:225–236
Pinheiro AR, Salvucci ID, Aguila MB, Mandarim-de-Lacerda CA (2008) Protein restriction during gestation and/or lactation causes adverse transgenerational effects on biometry and glucose metabolism in F1 and F2 progenies of rats. Clin Sci (Lond) 114:381–392
Jimenez-Chillaron JC, Isganaitis E, Charalambous M et al (2009) Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes 58:460–468
Peixoto-Silva N, Frantz ED, Mandarim-de-Lacerda CA, Pinheiro-Mulder A (2011) Maternal protein restriction in mice causes adverse metabolic and hypothalamic effects in the F1 and F2 generations. Br J Nutr 106:1364–1373
Hanafi MY, Saleh MM, Saad MI, Abdelkhalek TM, Kamel MA (2016) Transgenerational effects of obesity and malnourishment on diabetes risk in F2 generation. Mol Cell Biochem 412:269–280
Dunn GA, Morgan CP, Bale TL (2011) Sex-specificity in transgenerational epigenetic programming. Horm Behav 59:290–295
Aiken CE, Ozanne SE (2013) Sex differences in developmental programming models. Reproduction 14:R1–R13
Rosenfeld CS (2015) Sex-specific placental responses in fetal development. Endocrinology 156:3422–3434
Tarrade A, Panchenko P, Junien C, Gabory A (2015) Placental contribution to nutritional programming of health and diseases: epigenetics and sexual dimorphism. J Exp Biol 218:50–58
Rivière G, Michaud A, Breton C et al (2005) Angiotensin-converting enzyme 2 (ACE2) and ACE activities display tissue-specific sensitivity to undernutrition-programmed hypertension in the adult rat. Hypertension 46:1169–1174
Breton C, Lukaszewski MA, Risold PY et al (2009) Maternal prenatal undernutrition alters the response of POMC neurons to energy status variation in adult male rat offspring. Am J Physiol Endocrinol Metab 296:E462–E472
Lukaszewski MA, Mayeur S, Fajardy I et al (2011) Maternal prenatal undernutrition programs adipose tissue gene expression in adult male rat offspring under high-fat diet. Am J Physiol Endocrinol Metab 301:E548–E559
Lukaszewski MA, Butruille L, Moitrot E et al (2013) The hypothalamic POMC mRNA expression is upregulated in prenatally undernourished male rat offspring under high-fat diet. Peptides 43:146–154
Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD (2000) Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol Endocrinol Metab 279:E83–E87
Vickers MH, Ikenasio BA, Breier BH (2001) IGF-I treatment reduces hyperphagia, obesity, and hypertension in metabolic disorders induced by fetal programming. Endocrinology 142:3964–3973
Howie GJ, Sloboda DM, Vickers MH (2012) Maternal undernutrition during critical windows of development results in differential and sex-specific effects on postnatal adiposity and related metabolic profiles in adult rat offspring. Br J Nutr 108:298–307
Desai M, Jellyman JK, Han G, Beall M, Lane RH, Ross MG (2014) Maternal obesity and high-fat diet program offspring metabolic syndrome. Am J Obstet Gynecol 211:237.e1–237.e13
Alfaradhi MZ, Kusinski LC, Fernandez-Twinn DS et al (2016) Maternal obesity in pregnancy developmentally programs adipose tissue inflammation in young, lean male mice offspring. Endocrinology 157:4246–4256
Wankhade UD, Thakali KM, Shankar K (2016) Persistent influence of maternal obesity on offspring health: mechanisms from animal models and clinical studies. Mol Cell Endocrinol 435:7–19
Bautista CJ, Montaño S, Ramirez V et al (2016) Changes in milk composition in obese rats consuming a high-fat diet. Br J Nutr 115:538–546
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Schwartz DM, Wolins NE (2007) A simple and rapid method to assay triacylglycerol in cells and tissues. J Lipid Res 48:2514–2520
Goodspeed D, Seferovic MD, Holland W et al (2015) Essential nutrient supplementation prevents heritable metabolic disease in multigenerational intrauterine growth-restricted rats. FASEB J 29:807–819
Hardikar AA, Satoor SN, Karandikar MS et al (2015) Multigenerational undernutrition increases susceptibility to obesity and diabetes that is not reversed after dietary recuperation. Cell Metab 22:312–319
Gonzalez-Rodriguez P, Cantu J, O’Neil D et al (2016) Alterations in expression of imprinted genes from the H19/IGF2 loci in a multigenerational model of intrauterine growth restriction (IUGR). Am J Obstet Gynecol 214:625.e1–625.e11
Zheng D, Wang X, Antonson P, Gustafsson JA, Li Z (2018) Genomics of sex hormone receptor signaling in hepatic sexual dimorphism. Mol Cell Endocrinol 471:33–41
Gluckman PD, Hanson MA, Beedle AS (2007) Non-genomic transgenerational inheritance of disease risk. Bioessays 29:145–154
Guerrero-Bosagna C, Skinner MK (2012) Environmentally induced epigenetic transgenerational inheritance of phenotype and disease. Mol Cell Endocrinol 354:3–8
Burdge GC, Slater-Jefferies J, Torrens C et al (2007) Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br J Nutr 97:435–439
Suter MA, Ma J, Vuguin PM et al (2014) In utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. Am J Obstet Gynecol 210:463.e1–463.e11
Zhou D, Wang H, Cui H, Chen H, Pan YX (2015) Early-life exposure to high-fat diet may predispose rats to gender-specific hepatic fat accumulation by programming Pepck expression. J Nutr Biochem 26:433–440
Puppala S, Li C, Glenn JP et al (2018) Primate fetal hepatic responses to maternal obesity: epigenetic signalling pathways and lipid accumulation. J Physiol Mar. https://doi.org/10.1113/JP275422
Martínez D, Pentinat T, Ribó S et al (2014) In utero undernutrition in male mice programs liver lipid metabolism in the second-generation offspring involving altered Lxra DNA methylation. Cell Metab 19:941–951
Hayes EK, Tessier DR, Percival ME et al (2014) Trophoblast invasion and blood vessel remodeling are altered in a rat model of lifelong maternal obesity. Reprod Sci 21:648–657
Kim DW, Young SL, Grattan DR, Jasoni CL (2014) Obesity during pregnancy disrupts placental morphology, cell proliferation, and inflammation in a sex-specific manner across gestation in the mouse. Biol Reprod 90:130,1–11
Lecoutre S, Deracinois B, Laborie C et al (2016) Depot- and sex-specific effects of maternal obesity in offspring’s adipose tissue. J Endocrinol 230:39–53
Norheim F, Hui ST, Kulahcioglu E et al (2017) Genetic and hormonal control of hepatic steatosis in female and male mice. J Lipid Res 58:178–187
Vieau D, Sebaai N, Léonhardt M et al (2007) HPA axis programming by maternal undernutrition in the male rat offspring. Psychoneuroendocrinology 32:S16–S20
Buschdorf JP, Meaney MJ (2015) Epigenetics/programming in the HPA axis. Compr Physiol 6:87–110
Bellisario V, Panetta P, Balsevich G, Baumann V et al (2015) Maternal high-fat diet acts as a stressor increasing maternal glucocorticoids’ signaling to the fetus and disrupting maternal behavior and brain activation in C57BL/6J mice. Psychoneuroendocrinology 60:138–150
Gifford RM, Reynolds RM (2017) Sex differences in early-life programming of the hypothalamic-pituitary-adrenal axis in human. Early Hum Dev 114:7–10
Li G, Kohorst JJ, Zhang W et al (2013) Early postnatal nutrition determines adult physical activity and energy expenditure in female mice. Diabetes 62:2773–2783
Baker MS, Li G, Kohorst JJ, Waterland RA (2015) Fetal growth restriction promotes physical inactivity and obesity in female mice. Int J Obes (Lond) 39:98–104
Della Torre S, Maggi A (2017) Sex differences: a resultant of an evolutionary pressure? Cell Metab 25:499–505
Acknowledgements
The authors thank Dr. Marie-Amelie Lukaszewski for technical assistance during blood and tissue sampling. Funding was provided by Agence Nationale de la Recherche.
Author information
Authors and Affiliations
Contributions
CL and IF designed the research; OC, FD, ADC, VM and EM performed the experiments; OC, IF and CL analyzed the data; CL and DV wrote the manuscript; OC and FD contributed to the discussion. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Fig. 1
Food intake of F2 offspring during light and dark phases. Food intake was measured in 2-hours intervals for 24h using metabolic cages in males (A) and females (B) at 16 weeks of age. Data are presented as median with interquartile range, n = 5 males and females (from 5 to 7 litters). *P < .05 HF vs S diet (ANOVA) (PDF 22 KB)
Supplemental Fig. 2
Locomotor activity of F2 offspring during light and dark phases. Locomotor activity was measured in 2-hours intervals for 24h using metabolic cages in males (A) and females (B) at 16 weeks of age. Data are presented as median with interquartile range, n = 5 males and females (from 5 to 7 litters). **P < .01, ***P < .001 HF vs S diet; ††P < .01, †††P < .001 FR vs C (ANOVA) (PDF 23 KB)
Rights and permissions
About this article

Cite this article
Cissé, O., Fajardy, I., Delahaye, F. et al. Effect of diet in females (F1) from prenatally undernourished mothers on metabolism and liver function in the F2 progeny is sex-specific. Eur J Nutr 58, 2411–2423 (2019). https://doi.org/10.1007/s00394-018-1794-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00394-018-1794-y