
Abstract
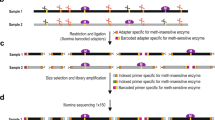
Digital Restriction Enzyme Analysis of Methylation (DREAM) is a method for quantitative map** of DNA methylation across genomes using next-generation sequencing (NGS) technology. The method is based on sequential cuts of genomic DNA with a pair of restriction enzymes (SmaI and XmaI) at CCCGGG target sites. Unmethylated sites are first digested with SmaI. This enzyme cuts the sites in the middle at CCC^GGG, leaving behind blunt ended fragments. CpG methylation completely blocks SmaI; therefore, only unmethylated sites are cleaved. The remaining methylated sites are digested with XmaI in the next step. This enzyme is not blocked by CpG methylation. It cuts the recognition site sideways at C^CCGGG forming 5′-CCGG overhangs. The sequential cuts thus create distinct methylation-specific signatures at the ends of restriction fragments: 5′-GGG for unmethylated CpG sites and 5′-CCGGG for methylated sites. The DNA fragments resulting from the digestions are ligated to NGS adapters. Sequencing libraries are prepared using hexanucleotide barcodes for sample identification. Individual libraries with distinct barcodes are pooled and sequenced using a paired ends protocol. The sequencing reads are aligned to the genome and mapped to unique CCCGGG target sites. Methylation at individual CpG sites is calculated as the ratio of sequencing reads with the methylated signature to the total number of reads map** to the site. Sequencing of 25 million reads per sample typically yields 50,000 unique CpG sites covered with hundreds of reads enabling accurate determination of DNA methylation levels. DREAM does not require bisulfite conversion, has a very low background, and has high sensitivity to low levels of methylation. The method is simple, cost-effective, quantitative, highly reproducible, and can be applied to any species.
Similar content being viewed by others


References
Laird PW (2010) Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 11:191–203
Toyota M, Ahuja N, Ohe-Toyota M et al (1999) CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 96:8681–8686
Estécio MR, Yan PS, Ibrahim AE et al (2007) High-throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res 17:1529–1536
Shen L, Kondo Y, Guo Y et al (2007) Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet 3:2023–2036
Jelinek J, Liang S, Lu Y et al (2012) Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique. Epigenetics 7:1368–1378
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Acknowledgments
We thank Amy B. Hart for editorial help. Research in the authors’ laboratories is supported by grants CA100632, CA158112, and CA049639 from the NIH.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media, LLC
About this protocol

Cite this protocol
Jelinek, J. et al. (2018). Digital Restriction Enzyme Analysis of Methylation (DREAM). In: Tost, J. (eds) DNA Methylation Protocols. Methods in Molecular Biology, vol 1708. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7481-8_13
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7481-8_13
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-7479-5
Online ISBN: 978-1-4939-7481-8
eBook Packages: Springer Protocols