
Abstract
Cross-linking, in general, involves the covalent linkage of two amino acid residues of proteins or protein complexes in close proximity. Mass spectrometry and computational analysis are then applied to identify the formed linkage and deduce structural information such as distance restraints. Quantitative cross-linking coupled with mass spectrometry is well suited to study protein dynamics and conformations of protein complexes. The quantitative cross-linking workflow described here is based on the application of isotope labelled cross-linkers. Proteins or protein complexes present in different structural states are differentially cross-linked using a “light” and a “heavy” cross-linker. The intensity ratios of cross-links (i.e., light/heavy or heavy/light) indicate structural changes or interactions that are maintained in the different states. These structural insights lead to a better understanding of the function of the proteins or protein complexes investigated. The described workflow is applicable to a wide range of research questions including, for instance, protein dynamics or structural changes upon ligand binding.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Proteins and protein complexes are key players in the cell. Their function is often modulated by structural changes upon loss or formation of interactions with their ligands, such as proteins, sugars, nucleotides, ions, or lipids. Assessing these structural changes is therefore important for fully understanding their function. Quantitative cross-linking coupled with mass spectrometric analysis allows analyzing these structural changes in solution and is, therefore, well suited to study dynamic proteins and protein complexes in different structural states [1]. In this chapter we describe the workflow of quantitative cross-linking, including the cross-linking reaction, sample preparation , mass spectrometric analysis, as well as data analysis.
1.1 Cross-Linking
Cross-linking of proteins or protein complexes, in general, is the covalent linkage of two functional groups of amino acid side chains resulting in inter- or intramolecular linkage. Partial hydrolysis of the cross-linker can further lead to modification of the reactive amino acids. Two strategies are commonly followed: photo-induced or chemical cross-linking. Chemical cross-linking reagents usually contain two identical (homobifunctional) or different (heterobifunctional) reactive groups as well as a linker [2]. Two amino acids are cross-linked when the linker length correlates with the distance between these two amino acids. Zero-length cross-linkers, consequently, do not introduce a linker and only link reactive groups in close proximity [3, 4]. Chemical cross-linkers are, for instance, reactive toward primary amines [5], arginine residues [6], sulfhydryl groups [7,8,9], or in the case of bifunctional cross-linkers, carboxyl, and amine groups [10, 11]. In addition, there are photo-reactive cross-linkers such as the UV-inducible amino acids photo-methionine or photo-leucine [12]. In some cases, cross-linking reagents contain a third functional group which is used for affinity enrichment (e.g., through a biotin moiety) [13]. The variety of chemical specificity and linker length therefore provides a selection of cross-linking approaches which can be applied to answer diverse research questions [14].
Of the available reagents, N-hydroxysuccinimide (NHS) esters are most commonly used [15]. One prominent, water soluble example is bis(sulfosuccinimidyl)suberate (BS3), which targets primary amines of lysine residues or the proteins’ N-termini. Side reactions with hydroxyl groups of serine, threonine, and tyrosine residues also occur when using BS3 or analogue cross-linkers [16]. Due to their polarity, lysine residues are typically located at the solvent-accessible surface of proteins and therefore represent a suitable amino acid target making NHS-esters popular cross-linking reagents.
1.2 Sample Preparation and LC-MS/MS Analysis
Following the cross-linking reaction, proteins or protein complexes are hydrolyzed using a specific endoproteinase such as trypsin. Enzymatic hydrolysis yields linear and cross-linked peptides. The latter include intra-peptide cross-links (so-called loop links) and inter-peptide cross-links [17]. Of these, the inter-peptide cross-links provide information on distance constraints between the cross-linked amino acids of different proteins or within the same protein.
Cross-linked peptides are usually low abundant when compared with their linear counterparts. Therefore, enrichment strategies including affinity chromatography, size exclusion chromatography, or ion-exchange chromatography are employed. Cross-linking reagents containing biotin labels or click-based affinity tags have been developed for affinity chromatography [18,19,20,21] while size exclusion chromatography [22] or cation exchange chromatography [23] make use of the increased size or higher charge of cross-linked di-peptides.
Obtained cross-linked peptides are then analyzed by liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS) following standard proteomic procedures. To increase the identification rate of cross-linked peptides, doubly charged precursors, which mostly correspond to linear peptides or intra-cross-linked peptides (see above), are often excluded from selection for MS/MS while highly charged cross-linked di-peptides are preferably analyzed [24].
1.3 Data Analysis
Identification of cross-linked peptides is challenging for the following reasons: the masses of the two cross-linked peptides and the linker mass of the cross-linker add up to the precursor mass. Depending on the sample complexity and the required database, a large number of cross-linked peptide combinations are possible, and the analysis can be computationally challenging [25]. In addition, fragmentation of a precursor ideally leads to series of fragment ions of both peptides. However, during collision-induced dissociation or high-energy collision dissociation of cross-linked peptides, the fragmentation of one peptide is typically favored [26]. Moreover, tryptic peptides often yield more y-ions than b-ions [27]. This suboptimal product ion distribution further affects the correct and confident assignment of cross-linked peptides. Therefore, specialized software has been developed; Examples are: xQuest [28], XlinkX [29], Kojak [30], StavroX [31], MeroX [32], **Search [33], and pLink [34]. Although the various software tools are constantly developed and improved, the identification of cross-linked peptides often relies on manual validation [15]. Particularly, the analysis of complex samples can therefore be laborious.
Having identified a set of cross-links, these are further analyzed and often visualized in network plots, cartoon representations, or, if available, in high-resolution structures. Available visualization tools are, for instance, xiView/xiNet [35, 36], XLink-DB [37], xVis [38], ProXL [39], or CLMSVault [40]. If crystal structures are available, distance restraints of cross-linked amino acid residues can be extracted using software tools such as Xwalk [41] or Xlink Analyer [42].
1.4 Quantitative Cross-Linking
Applied in the described way, cross-linking contributed considerably to the structural and functional understanding of molecular machines [43]. Recently, the field of cross-linking was expanded, and methods for quantitative cross-links were developed. Note that label-free quantification, which is often employed in proteomic studies, is not fully applicable to cross-linking analyses, mostly due to the difficulties in reproducibility of low abundant peptides such as cross-linked peptides (see above). Therefore, targeted approaches such as selected reaction monitoring or parallel reaction monitoring have been explored [44, 45]. Very recently, a quantitative cross-linking workflow including data-independent acquisition was developed [46, 47] enabling large scale quantitative analysis with high reproducibility [46].
On the other hand, a variety of label-based quantification methods is more commonly used. These are based on metabolic labelling (e.g., SILAC; stable isotope labelling by amino acids in cell culture) [48, 49], isobaric labelling using tandem mass tags (TMT [50] or iTRAQ [51]), or isotope labelled cross-linkers. Isotope labelled cross-linking reagents were first introduced to facilitate the identification of cross-links in complex samples [52]. They differ in the number of incorporated heavy stable isotopes leading to a characteristic mass shift in the mass spectrum and resulting in peak pairs of the cross-linked peptides. Nonetheless, differentially labelled cross-linkers (light and heavy) have the same physicochemical properties such as elution time, ionization, and fragmentation. Cross-linking different states of the protein with light and heavy cross-linkers, respectively, therefore allows the quantification of protein interactions in the different states through intensities of the peak pairs [53]. More precisely, by extracting ion chromatograms and comparing the intensity ratios of the two states (light versus heavy), protein interactions that vary or are maintained in the two states are observed. Differences in peak intensities represent structural changes, while the same intensities correspond to interactions that are not affected.
Initially, several strategies have been followed to analyze quantitative cross-linking data, including manual [53] as well as automatic data analysis [54]. To facilitate data analysis, specialized software has been developed and improved for quantitative cross-linking experiments, for instance, xTract [55], **Q [54], or Spectronaut (Biognosys) [56, 46].
1.5 Quantitative Cross-Linking Workflow and Example Data
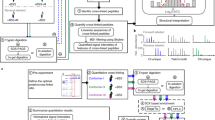
Usually, prior to quantitative cross-linking, optimal experimental conditions are determined by varying the cross-linker concentration or modifying the reactions conditions. For this, the protein or protein complex of interest in one state, for instance, in the apo-form without bound ligand, is used. Applying these conditions, the different states of the protein (or protein complex) are then cross-linked with differentially labelled cross-linkers (e.g., BS3-d0 and BS3-d4, respectively). After cross-linking, the two samples are pooled in a 1:1 ratio, the samples are prepared for LC-MS/MS analysis , and cross-link identification is processed. Finally, protein interactions of the different protein states are quantified through their abundances in the mass spectra. The complete workflow is shown in Fig. 1.

Workflow of quantitative cross-linking. First, experimental cross-linking conditions such as reaction time, protein or cross-linker concentration, etc. are optimized and followed, for instance, by gel electrophoresis (lhs). The protein or protein complex is then cross-linked in different states using light (e.g., BS4-d0) and heavy (e.g., BS3-d4) cross-linkers, respectively (rhs). After cross-linking, the samples are pooled and processed for LC-MS/MS analysis and identification of cross-linked peptides. As a control, the differentially labelled cross-linking reagents are swapped for the different states
Figures 2 and 3 show comparative cross-linking results of Calmodulin (CaM) in the Ca2+-free and Ca2+-bound states which both regulate the function of many proteins [57, 58]. The Ca2+-free form of CaM differs structurally from the Ca2+-bound form (compare high-resolution structures shown in Figs. 2 and 3). Due to structural changes upon Ca2+-binding, lysine residues 76 and 95 reach close proximity (Fig. 2a). Probing the two states, i.e., Ca2+-free and Ca2+-bound, with a light and heavy cross-linker, different intensities in the mass spectrum of the cross-link between these two residues are observed (Fig. 2b). Peak intensities corresponding to the apo-CaM form are lower than those observed for the Ca2+-bound form. In other words, the structural changes occurring due to Ca2+-binding result in a closer proximity of lysine 76 and 95. On the other hand, the abundance of the loop-link observed between lysine residues 76 and 78 does not differ for the two states (Fig. 3). Ca2+-binding has no effect on this region of the protein structure (Fig. 3a), and equal intensities are observed for the loop-linked peptide cross-linked with the light (BS3-d0) and heavy (BS3-d4) cross-linkers (Fig. 3b). The sequences of the cross-linked peptides and the localization of the cross-linked residues were confirmed by their tandem mass spectra (Figs. 2c and 3c).

Quantitative cross-linking of CaM in two states. (a) Crystal structures of apo-CaM (blue, lhs, PDB: 1DMO, state 1) and CaM with Calcium (red, rhs, PDB: 1PRW, state 2) are shown. Residues 76 and 95 are highlighted (green). (b) Mass spectra of the cross-link CaM (K76-K95) obtained from CaM cross-linked in state 1 using BS3-d0 and in state 2 using BS3-d4 (lhs). The light and heavy cross-linkers were also swapped (rhs). The isotope envelope corresponding to the cross-link of CaM in state 1 (blue) and in state 2 (red) are highlighted. (c) Tandem mass spectrum of the cross-link CaM (K76-K95). Series of y-ions of the two cross-linked peptides are assigned (green and purple)

Identification and quantification of a loop-link between residues K76 and K78 of CaM. (a) Crystal structures of apo-CaM (blue, top, PDB: 1DMO, state 1) and CaM with Calcium (red, bottom, PDB: 1PRW, state 2) are shown. Residues K76 and K78 are highlighted (green). (b) Mass spectra of the cross-link CaM (K76-K78) obtained from CaM cross-linked in state 1 using BS3-d4 and in state 2 using BS3-d0. The isotope envelope corresponding to the cross-link of CaM in state 1 (blue) and in state 2 (red) are highlighted. (c) Tandem mass spectrum of the loop-link CaM (K76-K78). Series of y-ions of the peptide are assigned (purple)
2 Materials
Prepare all solutions using ultra-pure water (HPLC grade). All other reagents should have highest available purity.
2.1 Chemical Cross-Linking
-
1.
Protein or protein complex in a suitable buffer (see Note 1).
-
2.
BS3-d0 stock solution: Dissolve 1.43 mg of BS3-d0 in 100 μL water to obtain 25 mM stock. Dilute 10-times to obtain 2.5 mM stock solution. Dilute with water to obtain lower cross-linker concentrations. Freshly prepare the solution before use (see Note 2).
-
3.
BS3-d4 stock solution: Dissolve 1.43 mg of BS3-d4 in 100 μL water to obtain 25 mM stock. Dilute 10-times to obtain 2.5 mM stock solution. Dilute with water to obtain lower cross-linker concentrations. Freshly prepare the solution before use (see Note 2).
-
4.
1 M Tris–HCl, pH 7.5: Dissolve 6.04 g Tris in 40 mL water. Adjust pH to 7.5 using HCl, and add water to a final volume of 50 mL. Store buffer in a glass bottle at room temperature for up to 3 months.
-
5.
Gel electrophoresis materials (sample buffer, running buffer, polyacrylamide gels, molecular weight marker, coomassie staining solution, gel electrophoresis chamber , power supply, etc.).
2.2 In-Solution Digestion
2.2.1 Ethanol Precipitation
-
1.
3 M sodium acetate, pH 5.3: Dissolve 24.6 g of sodium acetate in 100 mL of water and adjust the pH to 5.3 using acetic acid. Store the solution in a glass bottle for several months.
-
2.
Ethanol 100% (v/v) can be stored in a glass bottle for several months at −20 °C to provide an ice-cold solution.
-
3.
80% (v/v) ethanol: Mix 80 mL 100% ethanol and 20 mL water, and store the solution in a glass bottle at −20 °C.
2.2.2 In-Solution Digestion in the Presence of RapiGest
-
1.
25 mM ammonium bicarbonate (NH4HCO3), pH 8.0: Dissolve 19.75 mg NH4HCO3 in 10 mL water. Freshly prepare before use (see Note 3).
-
2.
1% (m/v) RapiGest : Dissolve one aliquote (1 mg) RapiGest (RapiGest SF Surfactant, Waters Corporation) in 100 μL 25 mM NH4HCO3. The solution can be stored at −20 °C for several weeks.
-
3.
50 mM dithiothreitol (DTT): Dissolve 7.7 mg in 1 mL 25 mM NH4HCO3. Freshly prepare before use.
-
4.
100 mM iodoacetamide (IAA): Dissolve 18.5 mg in 1 mL 25 mM NH4HCO3. Freshly prepare the solution before use and protect from light by wrap** with aluminum foil.
-
5.
Trypsin stock solution: 0.1 μg/μL sequencing grade modified trypsin (Promega Corporation) in 50 mM acetic acid. Dissolve 20 μg trypsin in 200 μL 50 mM acetic acid (see Note 4).
-
6.
5% Trifluoroacetic acid (TFA): Mix 95 μL water and 5 μL TFA.
2.3 Enrichment of Cross-Links
-
1.
Size-exclusion chromatography system equipped with size-exclusion chromatography column for peptides.
-
2.
Mobile phase: 30% (v/v) acetonitrile (ACN), 0.1% (v/v) TFA.
2.4 LC-MS/MS Analysis
-
1.
Sample solution: 2% (v/v) ACN, 0.1% (v/v) formic acid (FA). Mix 98 mL water with 2 mL ACN and 100 μL FA. Store the solution in a glass bottle for several weeks at −4 °C.
-
2.
Mobile phase A: 0.1% (v/v) FA. Mix 100 mL water and 100 μL FA. Degas the solution for 30 min in an ultrasonic bath.
-
3.
Mobile phase B: 80% (v/v) ACN and 0.1% (v/v) FA. Mix 20 mL water, 80 mL ACN and 100 μL FA. Degas the solution for 30 min in an ultrasonic bath.
- 4.
2.5 Identification of Cross-Linked Peptides
-
1.
Cross-link analysis software (see Note 7).
-
2.
Data processing tool (see Note 8).
-
3.
Software tool for visualization of cross-links (see Note 9).
2.6 Quantification of Identified Cross-Links
-
1.
MS software (see Note 10).
3 Methods
3.1 Chemical Cross-Linking
3.1.1 Identification of Optimal Cross-Linker Concentration
-
1.
Prepare 5–10 sample tubes containing 10 μM of the protein or protein complex and add increasing amounts of BS3-d0. The BS3-d0 concentration should be ranging from 0 to 5 mM.
-
2.
Incubate for 1 h at 25 °C and 350 rpm.
-
3.
Quench the reaction by addition of 1 M Tris–HCl to a final concentration of 10–20 mM Tris.
-
4.
Prepare samples for gel electrophoresis according to manufacturer’s protocols and perform gel electrophoresis. After gel-electrophoresis, wash and stain the gel using coomassie staining solution.
-
5.
Examine the gel and determine optimal protein to cross-linker ratio (see Note 11).
3.1.2 Quantitative Cross-Linking
-
1.
Prepare 2 reaction mixtures: One containing 10 μM of the protein in state 1 (mixture A) and the other in state 2 (mixture B). Add the optimal BS3-d0 concentration to mixture A (i.e., state 1) and BS3-d4 to mixture B (state 2). For determination of optimal cross-linker concentration, see Subheading 3.1.1.
-
2.
Incubate for 1 h at 25 °C and 350 rpm.
-
3.
Quench the reaction by addition of Tris (10–20 mM final concentration; see also Subheading 3.1.1, step 3).
-
4.
Pool equal amounts of the protein cross-linked with BS3-d0 (mixture A) and BS3-d4 (mixture B).
-
5.
Repeat steps 1–4 and swap light and heavy cross-linkers (i.e., incubate state 2 with BS3-d0 and state 1 with BS3-d4).
3.2 Sample Preparation for LC-MS/MS Analysis
3.2.1 Ethanol Precipitation and in-Solution Digestion
-
1.
Add water to the cross-linked proteins to reach a final volume of 200 μL.
-
2.
Add 20 μL (1/10 vol.) 3 M sodium acetate (pH 5.3).
-
3.
Add 600 μL (3 vol.) 100% (v/v) ice-cold ethanol and vortex the sample for several seconds.
-
4.
Incubate the sample for at least 2 h at −20 °C (see Note 12).
-
5.
Centrifuge the sample mixture for 30 min at 16,200 × g and 4 °C.
-
6.
Carefully remove the supernatant and wash the protein pellet by addition of 1 mL 80% (v/v) ice-cold ethanol (see Note 13).
-
7.
Centrifuge for 30 min at 16,200 × g and 4 °C.
-
8.
Carefully remove the supernatant (see Note 13).
-
9.
Dry the pellet in a vacuum centrifuge.
-
10.
Dissolve the protein pellet in 10 μL of 1% (m/v) RapiGest (see Note 14).
-
11.
Add 10 μL of 50 mM DTT and incubate for 30 min at 60 °C and 500 rpm.
-
12.
Add 10 μL of 100 mM IAA and incubate for 1 h at 37 °C and 500 rpm.
-
13.
Add 70 μL of trypsin solution in 25 mM NH4HCO3 at a trypsin-to-protein ratio of 1:20 (w/w).
-
14.
Incubate over night at 37 °C.
-
15.
Add 20 μL of 5% (v/v) TFA and incubate for 2 h at 37 °C.
-
16.
Centrifuge for 30 min at 16,200 × g.
-
17.
Transfer the supernatant into a new tube (see Note 15).
-
18.
Dry the peptides in a vacuum centrifuge.
3.2.2 Enrichment of Cross-Linked Di-Peptides
-
1.
Dissolve the peptides in 60 μL of 30% (v/v) ACN, 0.1% (v/v) TFA and sonicate for 1 min.
-
2.
Centrifuge for 1 min at 16,200 × g.
-
3.
Load 50 μL of the peptide solution into the sample loop of the chromatography system.
-
4.
Separate the peptides isocratically at a flow rate of 50 μL/min using a size exclusion column. Collect 50 μL fractions (see Note 16).
-
5.
Dry peptides in a vacuum centrifuge.
3.3 LC-MS/MS Analysis
-
1.
Dissolve dried peptides in 2% (v/v) ACN, 0.1% FA. Typically, each fraction is dissolved in 8 μL and 5 μL are injected onto the nano-LC system (see Note 17).
- 2.
3.4 Identification of Cross-Linked Peptides
The identification of cross-links can be performed using different software. Detailed tutorials are available (see Note 7). General aspects are listed in the following steps.
-
1.
Generate a protein database containing the proteins of interest in FASTA format.
-
2.
Configure protein database and mass of the cross-linker (for instance, deuterated and non-deuterated BS3).
-
3.
Upload your raw data (see Note 18).
-
4.
Specify search-parameters, e.g., enzyme used for hydrolysis, number of allowed missed cleavage sites, fixed and variable modifications, instrument parameters.
-
5.
The result file usually contains information on inter- and intra-molecular cross-links as well as mono- and loop-links (see Note 8).
-
6.
Inspect spectra of cross-linked peptides and manually validate peptide sequences and localization of cross-linked residues (see Notes 19 and 20). Inspect corresponding MS spectra, and identify the characteristic isotope patterns of light and heavy cross-linked peptides (see Note 21). Discard false-positive hits, i.e., cross-links without characteristic peak pair or with low-quality MS/MS spectra.
-
7.
Visualize validated cross-links manually or using available software tools (see Note 9).
3.5 Quantification of Identified Cross-Links
-
1.
Extract ion chromatograms for all validated cross-links. Extract ion chromatograms for both forms, the light and heavy cross-linked peptides (see Note 10).
-
2.
Calculate BS3-d0-to-BS3-d4 ratios for each cross-linked peptide pair.
-
3.
Analyze the calculated ratios to deduce structural information on your protein or protein complex (see Note 22).
4 Notes
-
1.
Purification and handling of the protein sample is specific to the protein or protein complex of interest. If possible, use ammonia-free buffers like PBS or HEPES to avoid quenching of the cross-linker.
-
2.
Avoid repeated freeze-thaw cycles and long storage duration in solution.
-
3.
Filter the solution using a syringe filter. The pH should be between 7.5 and 9.
-
4.
The use of modified trypsin reduces proteolytic self-digestion. Depending on the product specification of trypsin, different resuspension buffers are used.
-
5.
Every laboratory uses its own setup. Usually, peptides are first loaded onto a reversed-phase C18 pre-column to concentrate the peptides. A long HPLC column is used to achieve optimal separation of low abundant cross-linked peptides and to enable a high analytical depth during LC-MS/MS analysis.
-
6.
The settings of the mass spectrometer strongly depend on the instrument used. Typically, mass spectra are acquired in data-dependent mode, and tandem-MS spectra are acquired for triply or higher charged peptides. The application of high-resolution mass spectrometers facilitates identification of cross-linked peptides.
-
7.
Following software can be used for identification of cross-links: xQuest [28], XlinkX [29], Kojak [30], StavroX [31], MeroX [32], **Search [33], and pLink [34].
-
8.
The Croco [59] software tool can be employed to convert result files from several search engines into other formats.
-
9.
xiView/xiNet [35, 36], XLink-DB [37], xVis [38], ProXL [39], CLMSVault [40], Xwalk [41], or Xlink Analyer [42] are used for downstream analysis and visualization of cross-links.
-
10.
Extracted ion chromatograms (XIC) can be generated manually using vendor-specific software (e.g., Thermo Xcalibur Qual Browser v4.0, Thermo Fisher scientific) or computationally using xTract [55], **Q [54], or Spectronaut (Biognosys) [56, 46] software.
-
11.
With increasing amounts of BS3, protein gel bands are usually less resolved (Fig. 1). For protein complexes, intensities of monomeric bands decrease, while protein bands of covalently linked proteins are observed at higher molecular weight. Too high concentrations of the cross-linker might induce protein aggregation, and aggregates might remain in the loading cavities of the gel.
-
12.
Incubation over night at −20 °C is recommended. The sample can also be incubated for several days at −20 °C.
-
13.
Protein pellets can be very small or transparent. If the protein pellet is difficult to recognize, do not remove all liquid to ensure that the pellet is not removed with the supernatant.
-
14.
Avoid formation of foam by gently pipetting the sample up and down. When the protein pellet does not dissolve add another 10 μL of 1% (m/v) RapiGest and multiply the required volumes of the following steps by two.
-
15.
Avoid transferring decomposed RapiGest . When RapiGest is transferred to the fresh sample tube, repeat centrifugation and transfer to a fresh tube (Subheading 3.2.1, steps 16 and 17).
-
16.
Due to their size, cross-linked peptides elute in early fractions during size exclusion chromatography. Combine fractions that show low intensity (usually early and late fractions).
-
17.
Adjust the volume for dissolving dried peptides. Note that the required injection volume depends on the sensitivity of the mass spectrometer and needs to be adjusted to obtain sufficient signal.
-
18.
Depending on the used software, different file formats are supported.
-
19.
In most cases, inspection of the top scoring spectra of an identified cross-link is sufficient for manual validation.
-
20.
Cross-links are validated confidentially when series of y- or b-ions of at least 4 successive amino acids of both peptides are observed and the most intense peaks of the spectrum are assigned.
-
21.
The isotope effect of deuterium might lead to earlier elution times for cross-linked peptides containing the deuterated linker. For detection of the peak pair, expand the elution time window toward earlier elution times for deuterated cross-links and toward later elution times for the non-labelled analogue.
-
22.
Ratios of approx. 1 indicate no change in intensity of the individual protein interaction in the two states. Ratios >1 correspond to increased intensity and <1 to decreased intensity of the protein interactions in the different states.
References
Chen ZA, Rappsilber J (2018) Protein dynamics in solution by quantitative crosslinking/mass spectrometry. Trends Biochem Sci 43(11):908–920
Tinnefeld V, Sickmann A, Ahrends R (2014) Catch me if you can: challenges and applications of cross-linking approaches. Eur J Mass Spectrom (Chichester, Eng) 20(1):99–116
Staros JV, Wright RW, Swingle DM (1986) Enhancement by N-hydroxysulfosuccinimide of water-soluble carbodiimide-mediated coupling reactions. Anal Biochem 156(1):220–222
Bitan G, Teplow DB (2004) Rapid photochemical cross-linking—a new tool for studies of metastable, amyloidogenic protein assemblies. Acc Chem Res 37(6):357–364
Bragg PD, Hou C (1975) Subunit composition, function, and spatial arrangement in the Ca2+−and Mg2+−activated adenosine triphosphatases of Escherichia coli and Salmonella typhimurium. Arch Biochem Biophys 167(1):311–321
Zhang Q, Crosland E, Fabris D (2008) Nested Arg-specific bifunctional crosslinkers for MS-based structural analysis of proteins and protein assemblies. Anal Chim Acta 627(1):117–128
Wu CW, Yarbrough LR (1976) N-(1-pyrene)maleimide: a fluorescent cross-linking reagent. Biochemistry 15(13):2863–2868
Kovalenko OV, Yang XH, Hemler ME (2007) A novel cysteine cross-linking method reveals a direct association between claudin-1 and tetraspanin CD9. Mol Cell Proteomics 6(11):1855–1867
Kim Y, Ho SO, Gassman NR et al (2008) Efficient site-specific labeling of proteins via cysteines. Bioconjug Chem 19(3):786–791
Nagao R, Suzuki T, Okumura A et al (2010) Topological analysis of the extrinsic PsbO, PsbP and PsbQ proteins in a green algal PSII complex by cross-linking with a water-soluble carbodiimide. Plant Cell Physiol 51(5):718–727
Rampler E, Stranzl T, Orban-Nemeth Z et al (2015) Comprehensive cross-linking mass spectrometry reveals parallel orientation and flexible conformations of plant HOP2-MND1. J Proteome Res 14(12):5048–5062
Suchanek M, Radzikowska A, Thiele C (2005) Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells. Nat Methods 2(4):261–267
Zhang H, Tang X, Munske GR et al (2009) Identification of protein-protein interactions and topologies in living cells with chemical cross-linking and mass spectrometry. Mol Cell Proteomics 8(3):409–420
Mattson G, Conklin E, Desai S et al (1993) A practical approach to crosslinking. Mol Biol Rep 17(3):167–183
Iacobucci C, Piotrowski C, Aebersold R et al (2019) First community-wide, comparative cross-linking mass spectrometry study. Anal Chem 91(11):6953–6961
Kalkhof S, Sinz A (2008) Chances and pitfalls of chemical cross-linking with amine-reactive N-hydroxysuccinimide esters. Anal Bioanal Chem 392(1–2):305–312
Schilling B, Row RH, Gibson BW et al (2003) MS2Assign, automated assignment and nomenclature of tandem mass spectra of chemically crosslinked peptides. J Am Soc Mass Spectrom 14(8):834–850
Trester-Zedlitz M, Kamada K, Burley SK et al (2003) A modular cross-linking approach for exploring protein interactions. J Am Chem Soc 125(9):2416–2425
Fujii N, Jacobsen RB, Wood NL et al (2004) A novel protein crosslinking reagent for the determination of moderate resolution protein structures by mass spectrometry (MS3-D). Bioorg Med Chem Lett 14(2):427–429
Hurst GB, Lankford TK, Kennel SJ (2004) Mass spectrometric detection of affinity purified crosslinked peptides. J Am Soc Mass Spectrom 15(6):832–839
Chowdhury SM, Du X, Tolic N et al (2009) Identification of cross-linked peptides after click-based enrichment using sequential collision-induced dissociation and electron transfer dissociation tandem mass spectrometry. Anal Chem 81(13):5524–5532
Leitner A, Reischl R, Walzthoeni T et al (2012) Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol Cell Proteomics 11(3):M111.014126
Fritzsche R, Ihling CH, Gotze M et al (2012) Optimizing the enrichment of cross-linked products for mass spectrometric protein analysis. Rapid Commun Mass Spectrom 26(6):653–658
Leitner A, Walzthoeni T, Aebersold R (2014) Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat Protoc 9(1):120–137
Singh P, Panchaud A, Goodlett DR (2010) Chemical cross-linking and mass spectrometry as a low-resolution protein structure determination technique. Anal Chem 82(7):2636–2642
Trnka MJ, Baker PR, Robinson PJ et al (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol Cell Proteomics 13(2):420–434
Trnka MJ, Burlingame AL (2010) Topographic studies of the GroEL-GroES chaperonin complex by chemical cross-linking using diformyl ethynylbenzene: the power of high resolution electron transfer dissociation for determination of both peptide sequences and their attachment sites. Mol Cell Proteomics 9(10):2306–2317
Rinner O, Seebacher J, Walzthoeni T et al (2008) Identification of cross-linked peptides from large sequence databases. Nat Methods 5(4):315–318
Liu F, Rijkers DT, Post H et al (2015) Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat Methods 12(12):1179–1184
Hoopmann MR, Zelter A, Johnson RS et al (2015) Kojak: efficient analysis of chemically cross-linked protein complexes. J Proteome Res 14(5):2190–2198
Gotze M, Pettelkau J, Schaks S et al (2012) StavroX—a software for analyzing crosslinked products in protein interaction studies. J Am Soc Mass Spectrom 23(1):76–87
Gotze M, Pettelkau J, Fritzsche R et al (2015) Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J Am Soc Mass Spectrom 26(1):83–97
Mendes ML, Fischer L, Chen ZA et al (2019) An integrated workflow for crosslinking mass spectrometry. Mol Syst Biol 15(9):e8994
Yang B, Wu YJ, Zhu M et al (2012) Identification of cross-linked peptides from complex samples. Nat Methods 9(9):904–906
Graham M, Combe C, Kolbowski L et al (2019) xiView: a common platform for the downstream analysis of crosslinking mass spectrometry data. bioRxiv:561829. https://doi.org/10.1101/561829
Combe CW, Fischer L, Rappsilber J (2015) xiNET: cross-link network maps with residue resolution. Mol Cell Proteomics 14(4):1137–1147
Zheng C, Weisbrod CR, Chavez JD et al (2013) XLink-DB: database and software tools for storing and visualizing protein interaction topology data. J Proteome Res 12(4):1989–1995
Grimm M, Zimniak T, Kahraman A et al (2015) xVis: a web server for the schematic visualization and interpretation of crosslink-derived spatial restraints. Nucleic Acids Res 43(W1):W362–W369
Riffle M, Jaschob D, Zelter A et al (2016) ProXL (protein cross-linking database): a platform for analysis, visualization, and sharing of protein cross-linking mass spectrometry data. J Proteome Res 15(8):2863–2870
Courcelles M, Coulombe-Huntington J, Cossette E et al (2017) CLMSVault: a software suite for protein cross-linking mass-spectrometry data analysis and visualization. J Proteome Res 16(7):2645–2652
Kahraman A, Malmstrom L, Aebersold R (2011) Xwalk: computing and visualizing distances in cross-linking experiments. Bioinformatics 27(15):2163–2164
Kosinski J, von Appen A, Ori A, Karius K et al (2015) Xlink analyzer: software for analysis and visualization of cross-linking data in the context of three-dimensional structures. J Struct Biol 189(3):177–183
Leitner A, Faini M, Stengel F et al (2016) Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem Sci 41(1):20–32
Barysz H, Kim JH, Chen ZA et al (2015) Three-dimensional topology of the SMC2/SMC4 subcomplex from chicken condensin I revealed by cross-linking and molecular modelling. Open Biol 5(2):150005
Chavez JD, Eng JK, Schweppe DK et al (2016) A general method for targeted quantitative cross-linking mass spectrometry. PLoS One 11(12):e0167547
Muller F, Kolbowski L, Bernhardt OM et al (2019) Data-independent acquisition improves quantitative cross-linking mass spectrometry. Mol Cell Proteomics 18(4):786–795
Muller F, Graziadei A, Rappsilber J (2019) Quantitative photo-crosslinking mass spectrometry revealing protein structure response to environmental changes. Anal Chem 91(14):9041–9048
Chavez JD, Schweppe DK, Eng JK et al (2015) Quantitative interactome analysis reveals a chemoresistant edgotype. Nat Commun 6:7928
Chavez JD, Schweppe DK, Eng JK et al (2016) In vivo conformational dynamics of Hsp90 and its interactors. Cell Chem Biol 23(6):716–726
Yu C, Huszagh A, Viner R et al (2016) Develo** a multiplexed quantitative cross-linking mass spectrometry platform for comparative structural analysis of protein complexes. Anal Chem 88(20):10,301–10,308
Ross PL, Huang YN, Marchese JN et al (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3(12):1154–1169
Muller DR, Schindler P, Towbin H et al (2001) Isotope-tagged cross-linking reagents. A new tool in mass spectrometric protein interaction analysis. Anal Chem 73(9):1927–1934
Schmidt C, Robinson CV (2014) A comparative cross-linking strategy to probe conformational changes in protein complexes. Nat Protoc 9(9):2224–2236
Fischer L, Chen ZA, Rappsilber J (2013) Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J Proteome 88:120–128
Walzthoeni T, Joachimiak LA, Rosenberger G et al (2015) xTract: software for characterizing conformational changes of protein complexes by quantitative cross-linking mass spectrometry. Nat Methods 12(12):1185–1190
Bruderer R, Bernhardt OM, Gandhi T et al (2015) Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol Cell Proteomics 14(5):1400–1410
Jurado LA, Chockalingam PS, Jarrett HW (1999) Apocalmodulin. Physiol Rev 79(3):661–682
Bahler M, Rhoads A (2002) Calmodulin signaling via the IQ motif. FEBS Lett 513(1):107–113
Bender J, Schmidt C (2020) The CroCo cross-link converter: a user-centred tool to convert results from crosslinking mass spectrometry experiments. Bioinformatics 36:1296–1297
Acknowledgments
We acknowledge funding from the Federal Ministry for Education and Research (BMBF, ZIK programme, 03Z22HN22), the European Regional Development Funds (EFRE, ZS/2016/04/78115), and the MLU Halle-Wittenberg.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this protocol

Cite this protocol
Barth, M., Schmidt, C. (2021). Quantitative Cross-Linking of Proteins and Protein Complexes. In: Marcus, K., Eisenacher, M., Sitek, B. (eds) Quantitative Methods in Proteomics. Methods in Molecular Biology, vol 2228. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1024-4_26
Download citation
DOI: https://doi.org/10.1007/978-1-0716-1024-4_26
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-1023-7
Online ISBN: 978-1-0716-1024-4
eBook Packages: Springer Protocols