
Abstract
Microsporidia represent an evolutionary outlier in the tree of life and occupy the extreme edge of the eukaryotic domain with some of their biological features. Many of these unicellular fungi-like organisms have reduced their genomic content to potentially the lowest limit. With some of the most compacted eukaryotic genomes, microsporidia are excellent model organisms to study reductive evolution and its functional consequences. While the growing number of sequenced microsporidian genomes have elucidated genome composition and organization, a recent increase in complementary post-genomic studies has started to shed light on the impacts of genome reduction in these unique pathogens. This chapter will discuss the biological framework enabling genome minimization and will use one of the most ancient and essential macromolecular complexes, the ribosome, to illustrate the effects of extreme genome reduction on a structural, molecular, and cellular level. We outline how reductive evolution in microsporidia has shaped DNA organization, the composition and function of the ribosome, and the complexity of the ribosome biogenesis process. Studying compacted mechanisms, processes, or macromolecular machines in microsporidia illuminates their unique lifestyle and provides valuable insights for comparative eukaryotic structural biology.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Microsporidia
- Genome reduction
- Comparative evolutionary structural biology
- Ribosome structure and function
- Ribosome biogenesis
- Reductive evolution
1.1 Causes and Effects of Reductive Evolution
There is a common bias to describe evolution as a march toward decreased entropy and increased complexity. After all, the regular ordering of atoms to create larger and more complex systems is an intrinsic feature of life on earth. While this suggests that a gradual increase in organism complexity is inevitable, complexity is balanced in many environmental niches by selective pressures that favor rapid and efficient reproduction, leading to the elimination of superfluous traits. This process of ablation is known as “reductive evolution” and can result in simpler organisms deriving from more complex ancestors.
Reductive evolution typically results from efficient nutrient usage. Macroscopic examples of this include the loss or atrophy of eyes in a wide variety of cave fish or the somewhat ironic loss of a digestive tract in tapeworms (Castro 1996; Morris et al. 2012). At the microscopic level, reductive evolution usually involves eliminating extraneous metabolic processes. Many freshwater chrysophytes, for example, have switched from autotrophy/mixotrophy to heterotrophy in order to combat limited carbon availability and have lost photosynthetic pathways and large swaths of their genomes along the way (Olefeld et al. 2018; Majda et al. 2021).
Gene loss is the most common example of reductive evolution and is often made feasible by co-occurring organisms. One interesting case of this is the loss of the catalase-peroxidase protein, KatG, in the marine cyanobacteria Prochlorococcus spp. (Scanlan et al. 2009). KatG serves an important role in protecting many cyanobacteria from hydrogen peroxide (Perelman et al. 2003), which builds up in oceans due to photooxidation of dissolved organic carbon (Cooper et al. 1988). In a few hours of direct sunlight, enough hydrogen peroxide can be produced to kill off Prochlorococcus cultures (Morris et al. 2011), indicating that they have a very high sensitivity for the molecule. It is surprising then that Prochlorococcus spp. would have lost katG. Instead, co-occurring cyanobacteria have retained katG and scavenge surrounding hydrogen peroxide from marine environments (Petasne and Zika 1997). The loss of katG is therefore only possible because organisms in the natural community provide a protective function.
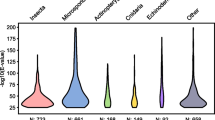
Interactions between Prochlorococcus spp. and other marine cyanobacteria inspired the “Black Queen Hypothesis,” which posits that natural selection for genomic streamlining breeds dependencies on co-occurring organisms (Morris et al. 2012). This contrasts with the “Red Queen Hypothesis,” inspired by Lewis Carroll’s Through the Looking-Glass, which postulates that competition breeds coevolution (Van Valen 1973). As a corollary to the Black Queen Hypothesis, the more dependent an organism is on other organisms, the more thoroughly a genome will be streamlined. We might therefore hypothesize that genome reduction scales with metabolic dependence on other organisms, i.e., the average genome size of evolutionarily related phototrophs > heterotrophs, and facultative parasites > obligate parasites, which seems to be the case (de Castro et al. 2009; Merhej et al. 2009; Clark et al. 2010; Majda et al. 2021) (Fig. 1.1).

Microsporidia have the smallest known eukaryotic genomes. Logarithmic plot of the number of annotated protein coding genes as a function of the respective organism’s genome size. All entries present in NCBI (https://www.ncbi.nlm.nih.gov/) were included, but the data were broadly filtered to remove untenable outliers, partial sequences, and nucleomorphs. Because of the broad filtering, some partially sequenced or annotated entries are still present. The plot was generated using source code from https://github.com/smsaladi/genome_size_vs_protein_count. Eukaryotes are colored in different shades of red, with microsporidia in black. Prokaryotes and viruses are represented in shades of green
Parasites are some of the greatest beneficiaries of reductive evolution, and nowhere is this more conspicuous than in microsporidia. As obligate intracellular parasites, microsporidia have dramatically reduced many elements of their genomes. In the sections below, we describe the various factors facilitating genome reduction and outline elements of the genome that are absent in microsporidia. We then delve more deeply into the effects of genome ablation at the protein and RNA level by comparing aspects of ribosome structure, function, and maturation in microsporidia to other eukaryotes.
1.2 The Price of a Large Genome
The cost of genome replication is threefold and requires payment in time, nutrients, and space. All three costs increase with genome size, although there is some variation between prokaryotes and eukaryotes. In this section, we discuss the impact of each of these factors on genome replication and describe how they contribute to reduction in microsporidian genome sizes.
1.2.1 Time
Time is required both to collect materials for DNA synthesis and to physically duplicate the genome. The amount of time required for genome replication depends largely on the catalytic rate of the DNA polymerase. In E. coli, DNA polymerase III copies around 1000 nucleotides/second (Kelman and O’Donnell 1995; Naufer et al. 2017). On the other hand, the equivalent yeast polymerase, Pol ϵ, has a maximal catalytic rate of only 350 nt/s (Ganai et al. 2015). Yeast replication is further decreased to 50 nt/s by proofreading, lagging strand synthesis, etc. Fortunately, eukaryotes are able to offset slower catalytic rates and considerably larger genomes by segregating genetic material into different chromosomes and amplifying from multiple origins of replication. Consequently, yeast and E. coli grown in ideal conditions have replication times commensurate to their genome sizes: 90–120 min for 12 Mbp in yeast (Salari and Salari 2017), versus 40 min for 4.6 Mbp in E. coli (Fossum et al. 2007). Interestingly, replication rates for many cancerous human cells are on the order of only 20 h (Pereira et al. 2017), despite having genomes 250 times larger than yeast. This shows that various factors contribute to dramatically decrease the necessary time for eukaryotic replication, but that total replication time typically increases with increasing genome sizes. Thus, it is beneficial for intracellular parasites like microsporidia to reduce their genome size in order to decrease doubling times. Unfortunately, very little is currently known about microsporidian polymerases, or even whether their chromosomes harbor multiple origins of replication. One study on the microsporidia Nematocida parisii determined that their population doubles in around 140 min (Balla et al. 2016). Although there are a variety of confounding factors, such as growth occurring in infected nematodes rather than in an optimized broth, the replication rate of the 4.3 Mbp N. parisii genome is considerably slower than in yeast (about 1/4 the rate). This suggests that the catalytic rate of the polymerase is slower and/or that N. parisii has fewer chromosomes and origins of replication per Mbp than yeast.
1.2.2 Nutrients
Nitrogen and phosphorous are key elements in DNA and are considered the limiting nutrients for growth in most ecosystems (Ågren et al. 2012; Elser 2012). The biosynthesis of DNA is thus an extremely resource-intensive investment. In fact, comprehensive estimates for the ATP requirements for DNA replication suggest that it costs as much as 500 high-energy bonds/bp in diploid eukaryotes (Lynch and Marinov 2015). While this estimate includes indirect costs such as the production of nucleosomes to stabilize the DNA, most expenses scale linearly with genome size. The larger the genome, the more NTPs are required, and the less high-energy bonds are available for alternative functions like protein production or cell defense. Many organisms therefore pass through a cell-cycle checkpoint, called START, which acts as a nutrient-sensing step to assess available resources prior to replication (Foster et al. 2010). Cells lacking requisite nutrients enter a quiescent state until conditions are more favorable for DNA biosynthesis.
Nutrient limitations are even more restrictive for obligate intracellular parasites. Indeed, microsporidia are almost completely reliant on their hosts and are metabolically inactive in nutrient-poor, extracellular environments (Weiss and Becnel 2014). The hijacking of host systems allows them to bypass much of the innate cost of DNA replication, and simply importing nucleotides instead of synthesizing their own reduces the ATP requirements per base pair by nearly 50% (Lynch and Marinov 2015). Intriguingly, microsporidia have opted to eliminate the majority of enzymes required for nucleotide biosynthesis (Dean et al. 2016) and have instead expanded families dedicated to nucleotide import (Cuomo et al. 2012). This indicates that microsporidia have increased import proteins but greatly decreased biosynthetic pathways, facilitating a net decrease in overall genome size (Dean et al. 2016). Similar trends are identifiable in microsporidia for many other central eukaryotic pathways, such as glycolysis or fatty acid metabolism (Wiredu Boakye et al. 2017).
1.2.3 Space
Although space is perhaps the least conspicuous cost for DNA, many studies have noted and discussed the intricate relationship between genome size and cell size in eukaryotes (Gregory 2001; Cavalier-Smith 2005). The crux of this argument lies in the relatively invariant karyoplasmic ratio, i.e., the ratio of the nuclear volume to cytoplasm is important for cell function, and is generally conserved (Huxley 1925; Trombetta 1942; Cavalier-Smith 2005). The nuclear size is in turn proportional to the total volume of the chromatin (Cavalier-Smith 2005). Although the underlying causes of this effect are still being determined (Cantwell and Nurse 2019; Blommaert 2020), a decrease in genome size will generally lead to a decreased nuclear size, catalyzing a decrease in cell size. The reverse also holds true, where a decrease in cell size will herald a decrease in genome size. This relationship was cleanly demonstrated in a eukaryotic phytoplankton by Malerba et al. (2020). In this study, 72 different Dunaliella tertiolecta lineages with cell volumes spanning two orders of magnitude were placed under selective pressures favoring smaller cells. After 100 generations, lineages that were initially much larger displayed an up to 11% decrease in genome size, while smaller lineages were unaffected. This suggests that (1) selective pressures favoring smaller cells indirectly select for smaller genomes and (2) lineages with larger genomes contain a set of superfluous genes that can be lost, while smaller lineages are already operating at closer to the minimal genome (Malerba et al. 2020).
For intracellular parasites like microsporidia, the space available within their hosts directly restricts the number of spores produced. Cells infected with microsporidia are often saturated with spores (Weiss and Becnel 2014; Grigsby et al. 2020), suggesting the host cell walls limit the number of spores created per infection. In fact, the spatial costs of DNA are twofold, as not only does DNA indirectly determine the size of the spores or meronts, but it also takes up valuable real estate within the cell. It is therefore extremely beneficial for microsporidia to minimize genome size, and it is unsurprising that they are some of the physically smallest eukaryotes. As a consequence of cell-wall limitations to genome size, microsporidian species that exit via exocytosis may have less stringent spatial costs than lytic species. Mature spores are constantly being shed in exocytosed species, increasing the effective available space compared to lytic species. Currently, only one pair of species can be used as an example: Nematocida displodere is primarily released via cell lysis, while N. parisii can be exocytosed in vesicles (Luallen et al. 2016). Although the genome of N. displodere is, in fact, smaller than the genome of N. parisii (Luallen et al. 2016), more data are required to determine whether the “spore release method” contributes to genome size variation between related microsporidians.
1.3 Paths of Reductive Evolution in Microsporidia
Microsporidia are characterized by many unique and interesting features, such as a lack of innate mobility (Weiss and Becnel 2014) and a fishing-line like infection apparatus (Han et al. 2017). Despite their innovations, microsporidia are perhaps most frequently referenced for their exquisitely small genomes (Keeling and Slamovits 2004; Corradi et al. 2010; Corradi and Slamovits 2011) and minimized macromolecular complexes (Melnikov et al. 2018a; Barandun et al. 2019; Ehrenbolger et al. 2020). Microsporidian genomes are indeed very small and have the honors of claiming both the smallest known eukaryotic genome (Corradi et al. 2010) and one of the highest known eukaryotic gene densities (Fig. 1.2) (Keeling and Slamovits 2004; Keeling 2007). The genome of Encephalitozoon intestinalis, for example, is only 2.3 Mbp (Corradi et al. 2010). That is only half the size of the E. coli genome (4.6 Mbp) and 1/65,000 the size of Paris japonica (150 Gbp), a flowering perennial with the largest confirmed eukaryotic genome (Pellicer et al. 2010).

Microsporidia have one of the most gene-dense eukaryotic genomes. Gene density across different kingdoms was calculated by dividing the number of annotated protein coding genes by the genome size of the respective organism in kilobase pairs. All entries present in NCBI (https://www.ncbi.nlm.nih.gov/) were included, but the data were broadly filtered to remove untenable outliers, partial sequences, and nucleomorphs. Because of the broad filtering, some partially sequenced or annotated entries are still present. Eukaryotes are colored in different shades of red, with microsporidia in black. Prokaryotes and viruses are represented in shades of green
Early studies on microsporidia noted the absence or modification of several cellular structures characteristic of eukaryotes. For example, microsporidia lack peroxisomes, have unstacked Golgi bodies, and have highly reduced mitochondria called mitosomes (Corradi and Keeling 2009; Vávra and Ronny Larsson 2014). These observations led to speculation that microsporidia represent an ancient and unsophisticated eukaryotic lineage. They were therefore classified as Archezoa, with the prevailing hypothesis stating that they diverged prior to endosymbiosis of the mitochondrial ancestor (Cavalier-Smith 1983). This theory was disproven when further genetic analyses demonstrated that a subset of genes found in eukaryotic mitochondria have been transferred to microsporidian chromosomes (Germot et al. 1996; Katinka et al. 2001), indicating that microsporidia diverged after endosymbiosis and are therefore simplified organisms derived from more complex ancestors. Likewise, the small genomes of microsporidia are not a representation of a primitive ancestral state but are instead the result of minimization of multifarious genomic features. In this section, we describe several features affecting genome size, such as gene loss, intron minimization/removal, reductions in gene length, deletions of redundant genes, and the shortening of intergenic regions (IGRs) (Fig. 1.3a).

Mechanisms of genome compaction in microsporidia. (a) Schematic representation of a relatively expanded (top) and compacted genome (bottom). Different genomic elements are colored, and the processes leading to their compaction (lower panel) are labeled on top. (b) The size of intergenic sequence (IGS) regions correlates with the directionality of adjacent genes, likely due to the presence of transcriptional control elements upstream of the transcriptional start site (e.g., enhancers, promotors). Gene directionality is indicated with arrows and 5′ or 3′ labels. Transcriptional control elements and their binding partners (e.g., transcription factors, RNA polymerases etc.) are shown as symbolic cartoons and colored in shades of green
1.3.1 Non-coding Regions
Microsporidian genomes are similar to those of other eukaryotes in structure and organization. Multiple linear chromosomes can be segregated into telomeres, subtelomeres containing ribosomal DNA (rDNA) and repetitive elements, and gene-rich cores (Dia et al. 2016). Variation is more localized to individual elements of the genome, like coding sequences and intergenic regions. The regions between genes are essential for efficient transcription and contain binding sites for various promoters and enhancers, which are often thousands of nucleotides away from the gene they enhance. It is intriguing then that many microsporidia have tiny IGRs, with E. intestinalis averaging only 115 bp between genes (Corradi et al. 2010). The genes themselves are an average of 1.04 kbp (Corradi et al. 2010). By taking into account the gene density (1.16 kbp/gene) (Corradi et al. 2010), we can determine that coding regions account for as much as 90% of the E. intestinalis genome. To put this in perspective, around 70% of the yeast genome codes for proteins (Dujon 1996) and only 2% of the human genome is protein coding (Piovesan et al. 2019). The low ratio of non-coding to coding sequences suggests that microsporidia have extremely streamlined IGRs. In fact, contrary to most eukaryotes, non-coding regions in microsporidia have higher sequence conservation than coding regions (Corradi et al. 2010; Corradi and Slamovits 2011; Whelan et al. 2019), indicating that the remaining bases form important molecular recognition motifs.
Most regulatory elements are found upstream of the 5′ end of a gene. Tellingly, the length of microsporidian IGRs appears to correlate with the directionality of adjacent genes (Fig. 1.3b) (Keeling and Slamovits 2004). For Encephalitozoon cuniculi, regions wedged between the termini of two genes (the 3′ ends) are about 20% shorter than regions between parallel genes (one 3′, and one 5′ end), while regions abutting divergent 5′ ends are a further 20% longer on average. This pattern is indicative of severe reductive selection operating on IGRs (Keeling and Slamovits 2004), as zero, one, or two sets of upstream transcription factors need to bind between convergent, parallel, and divergent genes, respectively.
Several other factors suggest that Encephalitozoon spp. are operating at the limit of IGR reduction. Firstly, the length of IGRs sometimes dips into negative values, i.e., genes overlap one another (Katinka et al. 2001; Akiyoshi et al. 2009; Corradi et al. 2010). Secondly, multiple studies have noted that transcripts initiate in upstream genes and read through into downstream genes (Williams et al. 2005; Corradi et al. 2008; Gill et al. 2010), suggesting that transcriptional start sites and termination sequences are often located within adjacent genes. Finally, microsporidia produce many multigene transcripts, which surprisingly encode both sense and antisense genes (Peyretaillade et al. 2009; Corradi and Slamovits 2011; Watson et al. 2015). These transcripts, known as “noncontiguous operons,” are thought to regulate protein expression levels and result from evolutionary pressure to minimize genome size (Sáenz-Lahoya et al. 2019). These three examples provide evidence that microsporidia trim and eliminate IGRs wherever possible and have adapted more spatially efficient mechanisms to regulate protein expression levels.
Microsporidian parsimony is not only directed toward IGRs but also impacts other non-coding regions like introns. Splicing machinery and introns appear to have been convergently eliminated in at least three microsporidian genera: Edhazardia, Nematocida, and Enterocytozoon (Keeling et al. 2010; Desjardins et al. 2015). Even when introns are retained, they are reduced in both number and length (Lee et al. 2010; Campbell et al. 2013). In E. cuniculi, for example, a total of 36 introns have been identified, ranging from only 23 to 76 bases in length (Lee et al. 2010). The splicing efficiency for many of these introns is very low, often around 10–25%, and many putative introns display no active splicing (Grisdale et al. 2013; Campbell et al. 2013; Desjardins et al. 2015). For comparison, the yeast genome contains at least 300 introns ranging from around 100 to 1000 bases (S**ola et al. 1999; ** the “eukaryotic” or “e” proteins of the ribosome. It is somewhat surprising then that the drastic microsporidian reduction in ESs is not accompanied by a concomitant loss in the number of ribosomal proteins (Fig. 1.5).
To better understand the proteinaceous changes to microsporidian ribosomes, we have collected sequences from genomes available on MicrosporidiaDB (Aurrecoechea et al. 2011) and compared their conservation in Fig. 1.8. Caution is advised while drawing conclusion from these data, as many microsporidian genomes are derived from incomplete assemblies and microsporidian proteins are rapidly evolving. It is therefore very likely that some of the proteins marked as absent were simply not identified via our methods. That said, microsporidia have retained most of the ribosomal proteins found in yeast, and only a few of the 80 yeast proteins are potentially absent in many microsporidian species. Remaining proteins have a 38% average sequence identity to yeast homologs and are often considerably shorter (Fig. 1.8a and b). Some proteins have lost loops or linkers, while others have been truncated at the N- or C-terminus. Additionally, low levels of sequence identity can be used to demarcate proteins that have structurally diverged from yeast (Fig. 1.8c).

Microsporidian ribosomal protein phylogeny, identity, and structure relative to their yeast homologs. (a) Ribosomal protein phylogeny generated by using protein sequences conserved in all listed microsporidian species. Connected to the microsporidian phylogeny (black) is a simplified tree for other non-microsporidian species, based on (James et al. 2006, 2013; Haag et al. 2014). For the microsporidian phylogenetic tree, the protein sequences were obtained by performing translated nucleotide blast (tblastn) searches with an E-value cutoff of 0.05, using the S. cerevisiae sequences or verified microsporidian hits as query and MicrosporidiaDB (Aurrecoechea et al. 2011) as database. For P. locustae and V. necatrix, protein sequences were obtained from (Barandun et al. 2019; Ehrenbolger et al. 2020) or local genome databases. For the non-microsporidian species, sequences were obtained from https://www.ncbi.nlm.nih.gov/. Proteins were aligned using MUSCLE 3.8.31 (Edgar 2004) and trimmed using trimAl (Capella-Gutiérrez et al. 2009) with the –gappyout option. The trimmed alignments were then concatenated using FASconcat 1.11 (Kück and Meusemann 2010). The phylogenetic tree was constructed with RAxML 8.2.12 using the model PROTGAMMAILGF, determined with ProtTest 3.4.2, and 1000 bootstrap replicates. The sequence identity heatmap was constructed using MUSCLE 3.8.31 (Edgar 2004) and Clustal-Omega (Sievers et al. 2011). The S. cerevisiae sequences were set as reference, except for eL28 and msL1, where H. sapiens and V. necatrix were used. The different shades of blue describe the percentage identity of the protein sequence compared to the reference. The row for S. cerevisiae contains viability data, color coded for lethal (dark yellow), slow-growing (yellow), and normal-growing (cream) ribosomal gene knockouts (Giaever et al. 2002; Gao et al. 2015). A black dot is used to mark genes that are duplicated in the yeast genome. Only single gene knockouts were performed in the referenced study. The sequence of eS31* was modified by removing the ubiquitin moiety to create the mature protein. (b) Difference in length between the V. necatrix or P. locustae and S. cerevisiae ribosomal proteins. (c) Comparison of the region around ES4 between the S. cerevisiae (left, PDB 4 V88), V. necatrix (middle, PDB 6RM3), and P. locusate ribosome (PDB 6ZU5). Selected ribosomal proteins are colored and labeled with name and N- and C-termini in shades of red. The lost eL38 and the gained msL1 are shown in shades of green. (d) The same view is shown as in (c) with selected proteins colored solid and the ribosome structure transparent
Genome-wide knockout screens have been performed in yeast, which allows us to identify essential ribosomal proteins (Giaever et al. 2002; Gao et al. 2015) (Fig. 1.8a). These studies further noted knockouts that led to slow-growth defects. It is important to mention, however, that yeast have duplicated the majority of ribosomal genes. Some deleterious effects may have therefore been ameliorated by the presence of paralogs during single-gene deletion studies. Nevertheless, comparisons between gene conservation and essentiality reveal several interesting results. Firstly, as might be expected, many of the essential genes in yeast were not duplicated. Secondly, essential genes are still extant in almost all microsporidia. Instances of their loss, such as uL16 in Enterocytozoon hepatopenaei, are more likely a result of incomplete genome assemblies or low sequence conservation. This is evinced by the isolation of purported losses. Only in the case of uL23 are essential genes unidentifiable in a related cluster of microsporidia (Trachipleistophora hominis and Pseudoloma neurophilia). Numerous studies have demonstrated the essentiality of uL23 for the formation of the polypeptide exit tunnel (Kaur and Stuart 2011; Polymenis 2020). We therefore find it more probable that its absence is a matter of incomplete genome assemblies; however, a genuine absence would undoubtedly provide useful insights into evolutionary strategies developed by microsporidia to minimize the ribosome exit tunnel. Thirdly, all of the yeast proteins unidentifiable in most microsporidia are nonessential (see eL28, eL38, eL41, P1, and P2), as are some of the frequently missing proteins (eS12, eS25, and eL29). The nonessential eL38 is present in all earlier branching eukaryotes and is absent in all but two microsporidian species (M. daphinae, Amphiamblys sp.), suggesting a relatively recent loss of this ribosomal protein (Barandun et al. 2019). These findings demonstrate that microsporidia have typically retained essential proteins and eliminated nonessential ones.
The nonessential protein eL41 is the only yeast subunit absent in all sequenced microsporidia (Fig. 1.8a) (Barandun et al. 2019; Ehrenbolger et al. 2020). It is remarkably short in other eukaryotes, only ~25 amino acids, and forms a small bridge between the LSU and the SSU (Tamm et al. 2019). Deletions of eL41 are easily tolerated, with knockout yeast strains displaying growth rates similar to wild-type strains (Giaever et al. 2002). More in-depth analyses have revealed that eL41 plays a role in translational efficiency (Dresios et al. 2003; Meskauskas et al. 2003). Ribosomes lacking eL41 had both lower translational fidelity and slower rates of peptidyltransferase activity. This suggests that the removal of eL41 in microsporidia may be another factor contributing to their markedly high rate of missense mutations (Melnikov et al. 2018b). The deletion of eL41 may also result in a slower translation rate, although no information is currently available on the kinetics of microsporidian ribosomes.
The ribosomal stalk proteins, which also have a purported role in translational efficiency (Wawiórka et al. 2017), are reduced in most microsporidia. A typical eukaryotic ribosomal stalk is composed of uL10, two subunits of P1, and two subunits of P2. All five protomers contain a highly conserved, C-terminal SDDDMGFGLFD motif, preceded by a long and flexible linker (Choi et al. 2015). This organization and motif is found in organisms as diverged as humans and the archaeon Pyrococcus horikoshii (Ito et al. 2014). During active translation, the C-termini of the pentamer bind to and recruit the essential elongation factor EF1α, which delivers charged aminoacyl-tRNA to the ribosome. It is proposed that the five redundant motifs aid in the rapid and efficient recruitment of the correct aminoacyl-tRNA, by greatly increasing the local concentrations of EF1α (Wawiórka et al. 2017). Additionally, this kinetic model of decoding suggests that ribosomal pausing leads to the acceptance of near-cognate anticodons, resulting in missense mutations. It is therefore interesting that the majority of microsporidia do not to encode P1, and some may have lost P2 (Fig. 1.8), implying a single EF1α-binding motif is present. Previous work has demonstrated that P1 and P2 are nonessential in eukaryotes only because uL10 retains an EF1α binding domain (Santos and Ballesta 1995; Remacha et al. 1995). On the other hand, the prokaryotic equivalents to P1/P2 are required for translation (Huang et al. 2010), as prokaryotic L10 lacks the binding motif. Remarkably, the uL10 homologs for microsporidian clades have lost the linker and the SDDDMGFGLFD motif (data not shown). Some microsporidia therefore have no identified proteins that can recruit EF1α to ribosomes. This finding may indicate that the translation rate and fidelity are much lower in microsporidia. Alternatively, microsporidia might have developed novel proteins or binding motifs to recruit EF1α. This possibility is of particular interest, as the C-terminal motif utilized by eukaryotes and archaea is a common target for potent toxins like ricin (Choi et al. 2015; Fan et al. 2016). A unique motif would represent an attractive target for therapeutics or pesticides.
1.5.3 Retained and Gained Ribosomal Proteins
Most microsporidian proteins have relatively low sequence identity to yeast proteins (Fig. 1.8). This is not entirely unexpected, as even proteins from two closely related Nematocida species share only ~70% of their amino acid sequence (Balla and Troemel 2013). A noticeable outlier in this divergence is eS31, an essential protein located in the beak of the SSU. Interestingly, eS31 is always produced as a fusion with a ubiquitin moiety. The ubiquitin acts as a chaperone protein to assist in the production and folding of eS31 and is cleaved off before eS31 is incorporated into ribosomes (Martín-Villanueva et al. 2019). The high sequence identity for eS31 derives from this ubiquitin moiety, as a realignment without ubiquitin results in much lower values (see eS31 vs eS31* in Fig. 1.8). Another highly conserved protein is eL15, which is present in all sequenced microsporidia. Little is known about eL15’s function other than that it is essential; however, it is a structural protein that is mostly buried and is therefore likely to have many conserved intermolecular interactions. Additionally, eL15 seems to mediate concentrations of other core ribosomal proteins, and its dysregulation leads to various cancers and diseases (Wlodarski et al. 2018; Ebright et al. 2020). Despite the lack of focused studies, the high conservation of eL15 in microsporidia evinces a high level of functional significance, which is not amenable to mutations in sequence or structure.
In addition to retaining most ribosomal proteins, microsporidia have also gained at least one novel subunit. The microsporidia-specific ribosomal protein (msL1) binds to V. necatrix ribosomes in a gap left by the loss of four ESs (Fig. 1.5) (Barandun et al. 2019). Although the specific role of this protein is unknown, it may be required to stabilize the ribosome in the absence of ESs. Genomic erosion in organelles, such as mitochondria, has resulted in a similarly minimized rRNA. In response, many mitochondria have acquired unique proteins used to patch unstable ribosomes (Petrov et al. 2019). It is likely that msL1 serves a similar patching function in microsporidia where rRNA reduction led to structural instability.
1.5.4 Conserving Energy by Utilizing Ribosome Hibernation Factors
Translational costs are high, and an estimated 30 ATPs are required for the biosynthesis and attachment of each amino acid (Wagner 2007). Such costs are unsustainable in nutrient-poor conditions. Organisms therefore express proteins known as hibernation factors, which bind to and inhibit ribosomes when nutrients are scarce (Prossliner et al. 2018). These factors allow cells to sequester intact ribosomes instead of degrading them (Brown et al. 2018; Trösch and Willmund 2019). The ability to inactivate ribosomes and recover them post-quiescence is of vital importance to microsporidia, as they spend a significant portion of their lifecycle as metabolically inactive spores (Weiss and Becnel 2014).
Microsporidia encode multiple hibernation factors, including the late-annotated short open reading frame 2 (Lso2), and microsporidian dormancy factors (MDF) 1 and 2 (Barandun et al. 2019; Ehrenbolger et al. 2020). All three of these proteins block active sites of the ribosome (Fig. 1.6) and are incompatible with active translation. In yeast, Lso2 is important for recovery of ribosomes post-starvation (Wang et al. 2018), and roughly 10% of ribosomes isolated from starved yeast are bound by Lso2 (Wells et al. 2020). Microsporidian ribosomes isolated from spores, on the other hand, displayed an approximately 92% occupancy rate, indicating that the vast majority of ribosomes in spores are in an inactivated state (Ehrenbolger et al. 2020). MDF1 and MDF2 have not been biochemically characterized; however, their high occupancy in spores and mechanisms of binding indicate that they are likely hibernation factors (Barandun et al. 2019). While MDF1 is broadly conserved in eukaryotes, MDF2 may be species-specific. Orthologs have thus far only been identified in V. necatrix, Nosema ceranae, and Nosema apis. The high occupancy of these factors bound to spore-stage ribosomes, and the fact that microsporidia have potentially evolved species-specific hibernation factors, demonstrates that sequestration of ribosomes during the spore stage is crucial. Although hibernation factors are not specifically associated with reductive evolution, they provide an additional example of the mechanisms by which microsporidia conserve energy.
1.6 Microsporidian Ribosome Assembly
In eukaryotes, ribosome biogenesis is a multidimensional process requiring the action of all three RNA polymerases (Pol) and a complex repertoire of over 300 assembly factors and snoRNAs (Woolford and Baserga 2013; Ebersberger et al. 2014; Klinge and Woolford 2019). The pathway starts in the nucleolus, a subcompartment of the nucleus, where the transcription of a precursor ribosomal RNA (pre-rRNA) initiates a co-transcriptional maturation pathway. In yeast, the precursor contains the rRNAs of both the small subunit (18S) and the large subunit (5.8S, 25S). These rRNAs are flanked by four transcribed spacer regions, two external and two internal (ETS, ITS; Fig. 1.9a). The third rRNA of the large subunit (5S) is transcribed from a different locus and is not part of this long precursor RNA. Assembly factors associate in a co-transcriptional manner with the rRNA precursor, including the transcribed spacers, to assist in the folding and enzymatic processing of the pre-rRNA and to incorporate ribosomal proteins. Several co-transcriptional endonucleolytic cleavage events are required to process the spacers and release the partially matured pre-ribosomal particles. Maturation then continues in the nucleus, where the pre-mature rRNA ends (e.g., 5′ ETS or ITS2) are further processed and degraded. After a controlled export through the nuclear pore complex, the last ribosomal maturation steps and quality control events occur in the cytoplasm.

Compaction of the microsporidian rDNA locus to a prokaryotic-like organization. Schematic representation of a single rDNA locus, below a diagram indicating the genomic distribution of all rDNA loci, from (a) S. cerevisiae, (b) microsporidia (nucleotide sizes from V. necatrix), and (c) E. coli. The genes and known spacer sizes are indicated and drawn to scale for comparative purposes
The transcribed spacers are not present in the mature ribosome, but are essential elements required to recruit ribosome assembly factors. The level of spacer processing is also used to demarcate the maturation stage of this complex particle (Klinge and Woolford 2019). In addition, eukaryotic ribosomal expansion segments, which are part of the mature ribosome, are also involved in recruiting specific assembly factors. Genome compaction in microsporidia has not only removed rRNA elements, such as eukaryotic ESs, but also drastically affected the transcribed spacers of the ribosomal precursor (e.g., removal of ITS2; Fig. 1.9b). While the pre-ribosomal and ribosomal RNA have been minimized, the number of ribosomal proteins associated with the mature microsporidian ribosome has been less affected (see Sect. 1.5.2) (Barandun et al. 2019; Ehrenbolger et al. 2020). This raises the question of whether ribosome assembly factors and the maturation pathway have been similarly reduced overall, or if specific assembly factor categories have been more impacted by genome reduction than others. Have microsporidia lost ribosome assembly factors with a role in maturing eukaryotic-specific RNA or protein elements? The following section discusses the impact of reductive evolution on the organization of rDNA loci and the maturation of pre-rRNA in microsporidia.
1.6.1 Impact of Genome Compaction on Number and Localization of the rDNA Loci
In most organisms, the ribosomal RNAs are transcribed from one or more polycistronic ribosomal DNA loci. The number of rDNA loci increases considerably from prokaryotes to eukaryotes: from a single rDNA locus in slow-growing bacteria (e.g., Mycobacterium tuberculosis) to ~150–200 copies in yeast (Petes 1979) to more than 10,000 in some plants (Kobayashi 2014). In S. cerevisiae, the primary model organism to study eukaryotic ribosome assembly, all rDNA loci are clustered head to tail on a single chromosome (Petes 1979) (Fig. 1.9a). Within eukaryotes, the size of one pre-rRNA coding locus varies substantially. These size variations are mostly due to differences in the lengths of external and internal spacer elements or eukaryotic-specific ribosomal expansion segments. Eukaryotic rDNA sizes range from the minimal microsporidian version with approximately 4.5 kbp (calculated from the V. necatrix sequences), which has lost many regulatory spacers and ESs, to ~9.1 kbp in yeast or ~ 43 kbp in humans, which contain long ETSs and extensive intergenic spacer regions.
In microsporidia, the rDNA organization and localization within the genome differ between species. While other eukaryotes contain large numbers of clustered rDNA repeats, microsporidia are left with fewer and often not clustered rDNA genes. Twenty-two rDNA copies have been reported for E. cuniculi, located on both telomeric ends of its 11 chromosomes (Brugère et al. 2000; Katinka et al. 2001; Dia et al. 2016). Forty-six partial and polymorph rDNA loci have been found in N. ceraneae (Cornman et al. 2009), and similar to the rDNA loci in N. bombycis, they appear to be distributed over all chromosomes (Liu et al. 2008). While the individual loci are scattered throughout different chromosomes in many microsporidian species, in N. apis, the rDNA genes cluster as repeats head to tail (Gatehouse and Malone 1998), which is more similar to the classical arrangement observed in other eukaryotic organisms.
In most eukaryotes, the 5S encoding gene is dispersed throughout the genome and is not adjacent to the other three rRNAs. One exception to this observation is S. cerevisiae, where the 5S rRNA gene clusters in the intergenic spaces between rDNA repeats (Fig. 1.9a). Both arrangements have been observed in microsporidia. Similar to yeast, in N. bombycis, the 5S gene is located next to the rDNA locus (Huang et al. 2004). Other species, such as E. cuniculi and E. intestinalis, have dispersed the 5S throughout the genome. In these two microsporidia, three copies for the 5S have been detected (Katinka et al. 2001; Corradi et al. 2010), in contrast to the 22 rDNA loci. While the rDNA locus is transcribed by RNA Pol I, the 5S rRNA is transcribed by RNA Pol III (Ciganda and Williams 2011). The microsporidian transcription machinery includes elements for RNA pol I, II, and III (Katinka et al. 2001), indicating that the use of separate polymerases for 5S and rDNA transcription may be retained in microsporidia.
The comparatively small number of rDNA repeats in microsporidia may be a result of their diminutive cell size, simple genomes, and low proteomic complexity. Fewer and shorter genes might require a reduced number of ribosomes, which in turn can be synthesized from fewer rDNA repeats. Indeed, a strong positive correlation between genome size and the number of rDNA repeats in eukaryotes has been noted (Prokopowich et al. 2003). Although this correlation exists, in general, only a fraction of all rDNA repeats are transcriptionally active. The actual rRNA synthesis rate is more so determined by the rate of RNA polymerase recruitment. A yeast strain with only 42 rDNA repeats, compared to the original 142 repeats, grows as well as wild type because two times more RNA polymerases are recruited to the rDNA locus (French et al. 2003). In addition to a potentially reduced need for ribosomes and increased RNA polymerase recruitment to a single locus, the simplified microsporidian rDNA gene organization might allow for a more streamlined ribosome maturation. Fewer pre-rRNA processing steps might be required than in other eukaryotes, due to missing pre-rRNA elements such as internal transcribed spacer 2 (ITS2).
1.6.2 Loss and Minimization of Transcribed Spacers
In many microsporidian species, genome compaction and gene fusion led to a reduction in the total number of ribosomal RNAs from four to three, which represents a reversal of the evolutionary trend seen in eukaryotic ribosomes. The eukaryotic 5.8S rRNA sequence and the 5′ end of the prokaryotic large subunit gene are homologous (Jacq 1981). In typical eukaryotes, ITS2 separates the 5.8S from the remainder of the large subunit rRNA gene (Fig. 1.9b). Early branching microsporidia like M. daphnia and Chytridiopsis typographi still contain highly reduced versions of ITS2 and thereby preserve the traditional eukaryote-specific separation of the 5.8S from the LSU gene (Corsaro et al. 2019). In all later-branching microsporidia, ITS2 has been removed (Vossbrinck and Woese 1986). The reductive evolution in these organisms led to a complete loss of ITS2 and fusion of the 5.8S rRNA with the LSU rRNA (23S), which has created a unique eukaryotic rDNA locus (Fig. 1.9b) with prokaryotic features (Fig. 1.9c). The remaining ITS has been reduced to a surprisingly short sequence in some microsporidia. While N. bombycis (Huang et al. 2004) contains an ITS of ~179 nt, other microsporidians, such as V. necatrix or N. apis (Gatehouse and Malone 1998), compacted this element to only ~33/34 nt. The intergenic spacer regions are important signal sequences for co-transcriptional endonucleolytic processing of the pre-rRNA fragment. Together with an apparent reduction of the 5′ and 3′ ETS regions and the removal of ITS2, the shortening of the ITS has significant implications for the ribosome maturation process, which is tightly controlled by ribosome assembly factors binding to these regions.
1.6.3 Impact of rDNA Compaction on Ribosome Biogenesis Factors
In 2014, Ebersberger et al. performed an evolutionary analysis of 255 yeast protein factors involved in ribosome biogenesis and included four microsporidian species in their analyses (Ebersberger et al. 2014). From these initial factors, 244 were proposed to be present in the last common ancestor shared with the microsporidia. Remarkably, only about half of them could be identified in microsporidia, which was highlighted as “the most remarkable gene loss” observed among the eukaryotic supertaxa (Ebersberger et al. 2014). Although extensive lists of factors involved in yeast ribosome biogenesis existed at the time, the precise functions or binding sites of most of these factors were unknown due to a lack of structural and biochemical data. During the decade since, our knowledge of fungal ribosome biogenesis has advanced to a detailed structural and functional description of the individual factors. This is mainly due to the technical progress made in cryo-EM, which provided high-resolution information and enabled the study of previously inaccessible pre-ribosomal particles from the fungi S. cerevisiae or Chaetomium thermophilum. These structures now provide an updated and comprehensive picture of fungal ribosome maturation and depict the intricate interaction network of assembly factors and ribosomal proteins bound to pre-ribosomal rRNA elements (Barandun et al. 2018; Klinge and Woolford 2019). They show how ribosome maturation proceeds in a hierarchical manner through several different conformational states to produce the final mature eukaryotic ribosome (Klinge and Woolford 2019). The emerging structural data on the fungal biogenesis process, together with recent studies on the microsporidian ribosomes (Barandun et al. 2019; Ehrenbolger et al. 2020), allows us to give a few selected examples of why expansion segment and transcribed spacer removal or shortening might have enabled assembly factor loss (or vice versa).
In yeast, the 5′ ETS is 700 nt long and is involved in the co-transcriptional recruitment of up to 27 ribosome biogenesis factors and the formation of an assembly platform for the SSU. In microsporidia, the exact size and structure of the 5′ ETS pre-rRNA fragment are not known. However, several factors that typically bind to this region have not been identified in microsporidia (Fig. 1.10). One of the first and largest multi-subunit complexes bound to the newly synthesized 5′ ETS is UtpA (Fig. 1.10b) (Hunziker et al. 2016). UtpA is a 7-subunit complex in yeast but appears to be absent or drastically reduced in microsporidia. The UtpA binding site on the 5′ ETS is shared with Utp18, a subunit of another early binding biogenesis complex, UtpB. The potential absence of Utp18 and the entire UtpA complex (Fig. 1.10a) suggests microsporidia may contain a shorter 5′ ETS sequence, which recruits a minimal small subunit assembly platform. Alternatively, assembly factors may be too divergent to be identified.

A reduced set of ribosome biogenesis factors and selected examples of assembly factor and expansion segment loss in microsporidia. (a) Presence and conservation of ribosome assembly factors in selected eukaryotes and microsporidia. The protein sequences were obtained by performing translated nucleotide blast (tblastn) or protein blast (blastp) searches with an E-value cutoff of 0.05, using the S. cerevisiae sequences and MicrosporidiaDB (Aurrecoechea et al. 2011) as database. For P. locustae and V. necatrix, protein sequences were obtained from local genome databases. For the non-microsporidian species, sequences were obtained from https://www.ncbi.nlm.nih.gov/. For phylogenetic tree calculation, see legend of Fig. 1.8. Many biogenesis factors display significant sequence similarity (e.g., WD40 domain proteins). It was therefore common for the same open reading frame to be identified as homologous to multiple different biogenesis factors. In such cases, we selected hits with the lowest E-value. It should be noted that the figure thus serves as only a guide to general trends for absent and present proteins, since annotations may be inaccurate. Proteins that were not identifiable (NI) are shown in cream. Biogenesis factors are clustered based on known or predicted binding regions within the 5’ ETS, SSU, ITS2, or LSU of the pre-rRNA. Conservation correlates between members of the same complex. Example complexes are labelled below (a) in shades of gray. (b-d) Structures of S. cerevisiae pre-ribosomal particles denoting selected maturation factors that are often absent in microsporidia, as highlighted in (a). Pre-SSU structures from PDB 5WLC (Barandun et al. 2017) (b), PDB 7AJU (Lau et al. 2021) (c), and a pre-LSU-structure from PDB 6C0F (Sanghai et al. 2018) (d) are displayed with expansion segments missing in V. necatrix, colored in shades of orange and yellow (SSU) or shades of blue and green (LSU), and selected biogenesis factors colored as in (a). These examples demonstrate a correlation between ES reduction and the loss of biogenesis factors that typically bind to those ESs
During pre-rRNA maturation, several eukaryotic expansion segments of the ribosomal RNA are bound and remodeled by assembly factors. In general, the absence of an assembly factor correlates with removal of its binding site in other organisms (Fig. 1.10). One striking example includes the SSU segments es3 and es6, which are bound and stabilized by the large HEAT repeat protein Utp20 (Fig. 1.10c). Es3 and es6 are the two largest small subunit expansion segments and have been completely lost or strongly reduced in microsporidia (Barandun et al. 2019; Ehrenbolger et al. 2020). Similarly, Utp20 appears to be absent in all microsporidian species. This suggests the primary role of Utp20 in chaperoning the maturation of these two expansion segments is no longer required in microsporidia. Similarly, the loss of h41 correlates with the loss of Utp30, an assembly factor binding to this rRNA element in pre-ribosomal particles (Fig. 1.10b). In the large subunit, ES7 is bound by two assembly factors, Rrp1 and Nsa1. Again, both the ES7 and the two assembly factors seem to be eliminated from microsporidian genomes.
A key step in large subunit maturation in S. cerevisiae involves processing of the ITS2 prior to nuclear export. Absence of ITS2, the spacer separating the two LSU rRNAs, explains the absence of many ribosome assembly factors binding this RNA region, such as Cic1, Rlp7, or the Las1 complex (Woolford and Baserga 2013). ITS1 processing in yeast is catalyzed by the essential ribozyme-protein complex RNAse MRP. While microsporidia still contain a highly reduced version of RNAse MRP (Zhu et al. 2006), ITS1 has been ablated to only 33 nt. It is unclear if this short ITS region can fold into a structure recognized by the minimized RNAse MRP, or if a simpler mechanism is used.
Apart from the mature ribosome structure, genomic data, and bioinformatics, very little is known about ribosome assembly in microsporidia. By studying the process in these minimal organisms, we can learn more about the still relatively unknown role of expansion segments during the assembly process in other eukaryotic organisms. The compaction of rRNAs together with the removal of transcribed spacer regions appears to have significantly affected the assembly process in microsporidia. A more thorough analysis of how expansion segment removal correlates with assembly factor loss will be required to understand the process in microsporidia and relate loss and compaction to a potential functional role in other eukaryotes.
1.7 Conclusion and Future Perspectives
Genome reduction and size appear to correlate with the degree of metabolic dependence on other organisms. Consequently, an obligate intracellular lifestyle provides a plausible explanation for the loss of redundant metabolic pathways and the invention of novel and more energetically efficient mechanisms of host exploitation. The drastic impact of genome compaction in microsporidia, however, has not only reduced the complexity of metabolic pathways but also affected intergenic regions, minimized gene sizes, and removed regulatory elements and features considered to be essential in eukaryotic organisms.
Genome erosion has significantly altered the microsporidian ribosomal DNA locus. By removing eukaryote-specific elements, such as ITS2 and nearly all expansion segments, the rDNA gene arrangement regressed to a prokaryote-like organization. The recent structural characterization of the microsporidian ribosome has illustrated the impact of genome reduction on the composition and assembly of this essential and ancient particle. It provided the surprising information that despite the loss of their rRNA binding site, almost all eukaryote-specific ribosomal proteins, albeit shortened, are still retained in the structure. Could limited access to primary metabolites precipitate a more compact ribosome? Are nucleotides more “rare” than amino acids, and could this be one reason why the rRNA is much more compacted than ribosomal proteins? Does the extensive rRNA loss affect the fidelity of the ribosome? Further studies are required to delineate the functional implications of ribosome compaction on protein synthesis and to reveal the suitability of ribosome-targeting antibiotics as translation inhibitors in microsporidia.
Microsporidia are of great interest in the fields of infection biology and comparative structural biology. They act as a reservoir for many unique and peculiar structures and have developed the most minimized versions of eukaryotic macromolecular complexes. Additional biochemical and structural studies in microsporidia not only will illuminate their own lifecycle but will also shed light on optional elements in many highly conserved cellular processes.
References
Ågren GI, Wetterstedt JÅM, Billberger MFK (2012) Nutrient limitation on terrestrial plant growth - modeling the interaction between nitrogen and phosphorus. New Phytol 194:953–960. https://doi.org/10.1111/j.1469-8137.2012.04116.x
Akiyoshi DE, Morrison HG, Lei S et al (2009) Genomic survey of the non-cultivatable opportunistic human pathogen, Enterocytozoon bieneusi. PLoS Pathog 5:1000261. https://doi.org/10.1371/journal.ppat.1000261
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Armache JP, Anger AM, Márquez V et al (2013) Promiscuous behaviour of archaeal ribosomal proteins: implications for eukaryotic ribosome evolution. Nucleic Acids Res 41:1284–1293. https://doi.org/10.1093/nar/gks1259
Arnesen T, Van Damme P, Polevoda B et al (2009) Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci U S A 106:8157–8162. https://doi.org/10.1073/pnas.0901931106
Aurrecoechea C, Barreto A, Brestelli J et al (2011) AmoebaDB and MicrosporidiaDB: functional genomic resources for Amoebozoa and microsporidia species. Nucleic Acids Res 39:D612–D619. https://doi.org/10.1093/nar/gkq1006
Balla KM, Troemel ER (2013) Caenorhabditis elegans as a model for intracellular pathogen infection. Cell Microbiol 15:1313–1322. https://doi.org/10.1111/cmi.12152
Balla KM, Luallen RJ, Bakowski MA, Troemel ER (2016) Cell-to-cell spread of microsporidia causes Caenorhabditis elegans organs to form syncytia. Nat Microbiol 1:16144. https://doi.org/10.1038/nmicrobiol.2016.144
Ban N, Beckmann R, Cate JHD et al (2014) A new system for naming ribosomal proteins. Curr Opin Struct Biol 24:165–169
Barandun J, Chaker-Margot M, Hunziker M et al (2017) The complete structure of the small-subunit processome. Nat Struct Mol Biol 24:944–953. https://doi.org/10.1038/nsmb.3472
Barandun J, Hunziker M, Klinge S (2018) Assembly and structure of the SSU processome — a nucleolar precursor of the small ribosomal subunit. Curr Opin Struct Biol 49:1–9. https://doi.org/10.1016/j.sbi.2018.01.008
Barandun J, Hunziker M, Vossbrinck CR, Klinge S (2019) Evolutionary compaction and adaptation visualized by the structure of the dormant microsporidian ribosome. Nat Microbiol 4:1798–1804. https://doi.org/10.1038/s41564-019-0514-6
Bennett GM, Moran NA (2013) Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol Evol 5:1675–1688. https://doi.org/10.1093/gbe/evt118
Ben-Shem A, Garreau de Loubresse N, Melnikov S et al (2011) The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334:1524–1529. https://doi.org/10.1126/science.1212642
Bhattacharya S, Takada S, Jacobson RH (2007) Structural analysis and dimerization potential of the human TAF5 subunit of TFIID. Proc Natl Acad Sci U S A 104:1189–1194. https://doi.org/10.1073/pnas.0610297104
Blommaert J (2020) Genome size evolution: towards new model systems for old questions. Proc Biol Sci 287(1933):20201441. https://doi.org/10.1098/rspb.2020.1441
Bremer H, Dennis PP (2008) Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus 3(1). https://doi.org/10.1128/ecosal.5.2.3
Brown CA, Murray AW, Verstrepen KJ (2010) Rapid expansion and functional divergence of Subtelomeric gene families in yeasts. Curr Biol 20:895–903. https://doi.org/10.1016/j.cub.2010.04.027
Brown A, Baird MR, Yip MC et al (2018) Structures of translationally inactive mammalian ribosomes. elife 7:1–18. https://doi.org/10.7554/eLife.40486
Brugère JF, Cornillot E, Méténier G et al (2000) Encephalitozoon cuniculi (Microspora) genome: physical map and evidence for telomere-associated rDNA units on all chromosomes. Nucleic Acids Res 28:2026–2033. https://doi.org/10.1093/nar/28.10.2026
Campbell SE, Williams TA, Yousuf A et al (2013) The genome of Spraguea lophii and the basis of host-microsporidian interactions. PLoS Genet 9:1003676. https://doi.org/10.1371/journal.pgen.1003676
Cantwell H, Nurse P (2019) Unravelling nuclear size control. Curr Genet 65:1281–1285
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. https://doi.org/10.1093/bioinformatics/btp348
Castro GA (1996) Helminths: structure, classification, growth, and development. University of Texas Medical Branch at Galveston, Galveston
Cavalier-Smith T (1983) A 6-Klngdom classification and a unified phylogeny. In: Schenk HEA, Schwemmler W (eds) Intracellular space as oligogenetic ecosystem. Proceedings. De Gruyter, Berlin, Boston, pp 1027–1034
Cavalier-Smith T (2005) Economy, speed and size matter: evolutionary forces driving nuclear genome miniaturization and expansion. In: Annals of botany. Oxford University Press, Oxford, pp 147–175
Choi AKH, Wong ECK, Lee KM, Wong KB (2015) Structures of eukaryotic ribosomal stalk proteins and its complex with trichosanthin, and their implications in recruiting ribosome-inactivating proteins to the ribosomes. Toxins (Basel) 7:638–647
Ciganda M, Williams N (2011) Eukaryotic 5S rRNA biogenesis. Wiley Interdiscip Rev RNA 2:523–533. https://doi.org/10.1002/wrna.74
Clark EL, Karley AJ, Hubbard SF (2010) Insect endosymbionts: manipulators of insect herbivore trophic interactions? Protoplasma 244:25–51
Cooper WJ, Zika RG, Petasne RG, Plane JMC (1988) Photochemical formation of H2O2 in natural waters exposed to sunlight. Environ Sci Technol 22:1156–1160. https://doi.org/10.1021/es00175a004
Cormier A, Chebbi MA, Giraud I et al (2021) Comparative genomics of strictly vertically transmitted, feminizing microsporidia endosymbionts of amphipod crustaceans. Genome Biol Evol 13:evaa245. https://doi.org/10.1093/gbe/evaa245
Cornman RS, Chen YP, Schatz MC et al (2009) Genomic analyses of the microsporidian Nosema ceranae, an emergent pathogen of honey bees. PLoS Pathog 5(6):e1000466. https://doi.org/10.1371/journal.ppat.1000466
Corradi N, Keeling PJ (2009) Microsporidia: a journey through radical taxonomical revisions. Fungal Biol Rev 23:1–8
Corradi N, Slamovits CH (2011) The intriguing nature of microsporidian genomes. Brief Funct Genomics 10:115–124. https://doi.org/10.1093/bfgp/elq032
Corradi N, Gangaeva A, Keeling PJ (2008) Comparative profiling of overlap** transcription in the compacted genomes of microsporidia Antonospora locustae and Encephalitozoon cuniculi. Genomics 91:388–393. https://doi.org/10.1016/j.ygeno.2007.12.006
Corradi N, Pombert JF, Farinelli L et al (2010) The complete sequence of the smallest known nuclear genome from the microsporidian Encephalitozoon intestinalis. Nat Commun 1:77. https://doi.org/10.1038/ncomms1082
Corsaro D, Wylezich C, Venditti D et al (2019) Filling gaps in the microsporidian tree: rDNA phylogeny of Chytridiopsis typographi (microsporidia: Chytridiopsida). Parasitol Res 118:169–180. https://doi.org/10.1007/s00436-018-6130-1
Cuomo CA, Desjardins CA, Bakowski MA et al (2012) Microsporidian genome analysis reveals evolutionary strategies for obligate intracellular growth. Genome Res 22:2478–2488. https://doi.org/10.1101/gr.142802.112
De Albuquerque NRM, Ebert D, Haag KL (2020) Transposable element abundance correlates with mode of transmission in microsporidian parasites. Mob DNA 11:1–8. https://doi.org/10.1186/s13100-020-00218-8
de Castro F, Gaedke U, Boenigk J (2009) Reverse evolution: driving forces behind the loss of acquired photosynthetic traits. PLoS One 4:e8465. https://doi.org/10.1371/journal.pone.0008465
Dean EJ, Davis JC, Davis RW, Petrov DA (2008) Pervasive and persistent redundancy among duplicated genes in yeast. PLoS Genet 4:1000113. https://doi.org/10.1371/journal.pgen.1000113
Dean P, Hirt RP, Embley TM (2016) Microsporidia: why make nucleotides if you can steal them? PLoS Pathog 12:1–13. https://doi.org/10.1371/journal.ppat.1005870
Derilus D, Rahman MZ, Pinero F, Massey SE (2020) Synergism between the black queen effect and the proteomic constraint on genome size reduction in the photosynthetic picoeukaryotes. Sci Rep 10:1–13. https://doi.org/10.1038/s41598-020-65476-1
Desjardins CA, Sanscrainte ND, Goldberg JM et al (2015) Contrasting host-pathogen interactions and genome evolution in two generalist and specialist microsporidian pathogens of mosquitoes. Nat Commun 6:1–12. https://doi.org/10.1038/ncomms8121
Dia N, Lavie L, Faye N et al (2016) Subtelomere organization in the genome of the microsporidian Encephalitozoon cuniculi: patterns of repeated sequences and physicochemical signatures. BMC Genomics 17:1–20. https://doi.org/10.1186/s12864-015-1920-7
Dresios J, Panopoulos P, Suzuki K, Synetos D (2003) A dispensable yeast ribosomal protein optimizes peptidyltransferase activity and affects translocation. J Biol Chem 278:3314–3322. https://doi.org/10.1074/jbc.M207533200
Dujon B (1996) The yeast genome project: what did we learn? Trends Genet 12:263–270
Ebersberger I, Simm S, Leisegang MS et al (2014) The evolution of the ribosome biogenesis pathway from a yeast perspective. Nucleic Acids Res 42:1509–1523. https://doi.org/10.1093/nar/gkt1137
Ebright RY, Lee S, Wittner BS et al (2020) Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science (80-) 367:1468–1473. https://doi.org/10.1126/science.aay0939
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Ehrenbolger K, Jespersen N, Sharma H et al (2020) Differences in structure and hibernation mechanism highlight diversification of the microsporidian ribosome. PLoS Biol 18:e3000958. https://doi.org/10.1371/journal.pbio.3000958
Elser JJ (2012) Phosphorus: a limiting nutrient for humanity? Curr Opin Biotechnol 23:833–838
Fan X, Zhu Y, Wang C et al (2016) Structural insights into the interaction of the ribosomal P stalk protein P2 with a type II ribosome-inactivating protein ricin. Sci Rep 6:1–10. https://doi.org/10.1038/srep37803
Fischer K, Chavchich M, Huestis R et al (2003) Ten families of variant genes encoded in subtelomeric regions of multiple chromosomes of Plasmodium chabaudi, a malaria species that undergoes antigenic variation in the laboratory mouse. Mol Microbiol 48:1209–1223. https://doi.org/10.1046/j.1365-2958.2003.03491.x
Forte GMA, Pool MR, Stirling CJ (2011) N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS Biol 9:1001073. https://doi.org/10.1371/journal.pbio.1001073
Fossum S, Crooke E, Skarstad K (2007) Organization of sister origins and replisomes during multifork DNA replication in Escherichia coli. EMBO J 26:4514–4522. https://doi.org/10.1038/sj.emboj.7601871
Foster DA, Yellen P, Xu L, Saqcena M (2010) Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 1:1124–1131
French SL, Osheim YN, Cioci F et al (2003) In exponentially growing Saccharomyces cerevisiae cells, rRNA synthesis is determined by the summed RNA polymerase I loading rate rather than by the number of active genes. Mol Cell Biol 23:1558–1568. https://doi.org/10.1128/mcb.23.5.1558-1568.2003
Fujii K, Susanto TT, Saurabh S, Barna M (2018) Decoding the function of expansion segments in ribosomes. Mol Cell 72:1013–1020.e6. https://doi.org/10.1016/j.molcel.2018.11.023
Galindo LJ, Torruella G, Moreira D et al (2018) Evolutionary genomics of metchnikovella incurvata (metchnikovellidae): an early branching microsporidium. Genome Biol Evol 10:2736–2748. https://doi.org/10.1093/gbe/evy205
Ganai RA, Osterman P, Johansson E (2015) Yeast DNA polymerase ε catalytic core and holoenzyme have comparable catalytic rates. J Biol Chem 290:3825–3835. https://doi.org/10.1074/jbc.M114.615278
Gao F, Luo H, Zhang CT, Zhang R (2015) Gene essentiality analysis based on DEG 10, an updated database of essential genes. Methods Mol Biol 1279:219–233. https://doi.org/10.1007/978-1-4939-2398-4_14
Gatehouse HS, Malone LA (1998) The ribosomal RNA gene region of Nosema apis (Microspora): DNA sequence for small and large subunit rRNA genes and evidence of a large tandem repeat unit size. J Invertebr Pathol 71:97–105. https://doi.org/10.1006/jipa.1997.4737
Gerlitz G, Darhin E, Giorgio G et al (2005) Novel functional features of the Lis-H domain: role in protein dimerization, half-life and cellular localization. Cell Cycle 4:1632–1640. https://doi.org/10.4161/cc.4.11.2151
Germot A, Philippe H, Le Guyader H (1996) Presence of a mitochondrial-type 70-kDa heat shock protein in Trichomonas vaginalis suggests a very early mitochondrial endosymbiosis in eukaryotes. Proc Natl Acad Sci U S A 93:14614–14617. https://doi.org/10.1073/pnas.93.25.14614
Giaever G, Chu AM, Ni L et al (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391. https://doi.org/10.1038/nature00935
Gill EE, Lee RCH, Corradi N et al (2010) Splicing and transcription differ between spore and intracellular life stages in the parasitic microsporidia. Mol Biol Evol 27:1579–1584. https://doi.org/10.1093/molbev/msq050
Gogala M, Becker T, Beatrix B et al (2014) Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 506:107–110. https://doi.org/10.1038/nature12950
Gregory TR (2001) Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biol Rev Camb Philos Soc 76:65–101. https://doi.org/10.1017/s1464793100005595
Grigsby A, Kelly BJ, Sanscrainte ND et al (2020) Propagation of the microsporidian parasite Edhazardia aedis in aedes aegypti mosquitoes. J Vis Exp 2020:1–10. https://doi.org/10.3791/61574
Grisdale CJ, Bowers LC, Didier ES, Fast NM (2013) Transcriptome analysis of the parasite Encephalitozoon cuniculi: an in-depth examination of pre-mRNA splicing in a reduced eukaryote. BMC Genomics 14:207. https://doi.org/10.1186/1471-2164-14-207
Haag KL, James TY, Pombert J-F et al (2014) Evolution of a morphological novelty occurred before genome compaction in a lineage of extreme parasites. Proc Natl Acad Sci 111:15480–15485. https://doi.org/10.1073/pnas.1410442111
Haag KL, Pombert J-F, Sun Y et al (2020) Microsporidia with vertical transmission were likely shaped by nonadaptive processes. Genome Biol Evol 12:3599–3614. https://doi.org/10.1093/gbe/evz270
Han K, Li ZF, Peng R et al (2013) Extraordinary expansion of a Sorangium cellulosum genome from an alkaline milieu. Sci Rep 3:1–7. https://doi.org/10.1038/srep02101
Han B, Polonais V, Sugi T et al (2017) The role of microsporidian polar tube protein 4 (PTP4) in host cell infection. PLoS Pathog 13:e1006341. https://doi.org/10.1371/journal.ppat.1006341
Heinz E, Williams TA, Nakjang S et al (2012) The genome of the obligate intracellular parasite Trachipleistophora hominis: new insights into microsporidian genome dynamics and reductive evolution. PLoS Pathog 8:e1002979. https://doi.org/10.1371/journal.ppat.1002979
Huang WF, Tsai SJ, Lo CF et al (2004) The novel organization and complete sequence of the ribosomal RNA gene of Nosema bombycis. Fungal Genet Biol 41:473–481. https://doi.org/10.1016/j.fgb.2003.12.005
Huang C, Mandava CS, Sanyal S (2010) The ribosomal stalk plays a key role in IF2-mediated association of the ribosomal subunits. J Mol Biol 399:145–153. https://doi.org/10.1016/j.jmb.2010.04.009
Hua-Van A, Le Rouzic A, Boutin TS et al (2011) The struggle for life of the genome’s selfish architects. Biol Direct 6:19. https://doi.org/10.1186/1745-6150-6-19
Hunziker M, Barandun J, Petfalski E et al (2016) UtpA and UtpB chaperone nascent pre-ribosomal RNA and U3 snoRNA to initiate eukaryotic ribosome assembly. Nat Commun 7:1–10. https://doi.org/10.1038/ncomms12090
Huxley JS (1925) The cell in development and heredity. Nature 115:669–671. https://doi.org/10.1038/115669a0
Ito K, Honda T, Suzuki T et al (2014) Molecular insights into the interaction of the ribosomal stalk protein with elongation factor 1α. Nucleic Acids Res 42:14042–14052. https://doi.org/10.1093/nar/gku1248
Jacq B (1981) Sequence homologies between eukaryotic 5.8S rRNA and the 5′ end of prokaryotic 23S rRNA: evidences for a common evolutionary origin. Nucleic Acids Res 9:2913–2932. https://doi.org/10.1093/nar/9.12.2913
James TY, Kauff F, Schoch CL et al (2006) Reconstructing the early evolution of fungi using a six-gene phylogeny. Nature 443:818–822. https://doi.org/10.1038/nature05110
James TY, Pelin A, Bonen L et al (2013) Shared signatures of parasitism and phylogenomics unite cryptomycota and microsporidia. Curr Biol 23:1548–1553. https://doi.org/10.1016/j.cub.2013.06.057
Katinka MDD, Duprat S, Cornillott E et al (2001) Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 414:450–453. https://doi.org/10.1038/35106579
Kaur J, Stuart RA (2011) Truncation of the Mrp20 protein reveals new ribosome-assembly subcomplex in mitochondria. EMBO Rep 12:950–955. https://doi.org/10.1038/embor.2011.133
Keeling PJ (2007) Ostreococcus tauri: seeing through the genes to the genome. Trends Genet 23:151–154
Keeling PJ, Slamovits CH (2004) Simplicity and complexity of microsporidian genomes. Eukaryot Cell 3:1363–1369
Keeling PJ, Corradi N, Morrison HG et al (2010) The reduced genome of the parasitic microsporidian Enterocytozoon bieneusi lacks genes for core carbon metabolism. Genome Biol Evol 2:304–309. https://doi.org/10.1093/gbe/evq022
Kelman Z, O’Donnell M (1995) DNA polymerase III holoenzyme: structure and function of a chromosomal replicating machine. Annu Rev Biochem 64:171–200
Klinge S, Woolford JL (2019) Ribosome assembly coming into focus. Nat Rev Mol Cell Biol 20:116–131. https://doi.org/10.1038/s41580-018-0078-y
Knorr AG, Schmidt C, Tesina P et al (2019) Ribosome–NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nat Struct Mol Biol 26:35–39. https://doi.org/10.1038/s41594-018-0165-y
Kobayashi T (2014) Ribosomal RNA gene repeats, their stability and cellular senescence. Proc Japan Acad Ser B 90:119–129. https://doi.org/10.2183/pjab.90.119
Kück P, Meusemann K (2010) FASconCAT: convenient handling of data matrices. Mol Phylogenet Evol 56:1115–1118. https://doi.org/10.1016/j.ympev.2010.04.024
Kung C, Martinac B, Sukharev S (2010) Mechanosensitive channels in microbes. Annu Rev Microbiol 64:313–329
Kurze C, Dosselli R, Grassl J et al (2016) Differential proteomics reveals novel insights into Nosema–honey bee interactions. Insect Biochem Mol Biol 79:42–49. https://doi.org/10.1016/j.ibmb.2016.10.005
Lau B, Cheng J, Flemming D et al (2021) Structure of the maturing 90S pre-ribosome in association with the RNA exosome. Mol Cell 81:293–303.e4. https://doi.org/10.1016/j.molcel.2020.11.009
Lee RCH, Gill EE, Roy SW, Fast NM (2010) Constrained intron structures in a microsporidian. Mol Biol Evol 27:1979–1982. https://doi.org/10.1093/molbev/msq087
Ling J, O’Donoghue P, Söll D (2015) Genetic code flexibility in microorganisms: novel mechanisms and impact on physiology. Nat Rev Microbiol 13:707–721
Liu H, Pan G, Song S et al (2008) Multiple rDNA units distributed on all chromosomes of Nosema bombycis. J Invertebr Pathol 99:235–238. https://doi.org/10.1016/j.jip.2008.06.012
Luallen RJ, Reinke AW, Tong L et al (2016) Discovery of a natural microsporidian pathogen with a broad tissue tropism in Caenorhabditis elegans. PLoS Pathog 12:e1005724. https://doi.org/10.1371/journal.ppat.1005724
Luo H, Friedman R, Tang J, Hughes AL (2011) Genome reduction by deletion of paralogs in the marine cyanobacterium Prochlorococcus. Mol Biol Evol 28:2751–2760. https://doi.org/10.1093/molbev/msr081
Luo J, He Q, Xu J-Z et al (2021) Microsporidia infection upregulates host energy metabolism but maintains ATP homeostasis. J Invertebr Pathol 186:107596. https://doi.org/10.1016/j.jip.2021.107596
Lynch M, Marinov GK (2015) The bioenergetic costs of a gene. Proc Natl Acad Sci U S A 112:15690–15695. https://doi.org/10.1073/pnas.1514974112
Maitra A, Dill KA (2015) Bacterial growth laws reflect the evolutionary importance of energy efficiency. Proc Natl Acad Sci U S A 112:406–411. https://doi.org/10.1073/pnas.1421138111
Majda S, Beisser D, Boenigk J (2021) Nutrient-driven genome evolution revealed by comparative genomics of chrysomonad flagellates. Commun Biol 4:1–11. https://doi.org/10.1038/s42003-021-01781-3
Malerba ME, Ghedini G, Marshall DJ (2020) Genome size affects fitness in the eukaryotic alga Dunaliella tertiolecta. Curr Biol 30:3450–3456.e3. https://doi.org/10.1016/j.cub.2020.06.033
Martín-Villanueva S, Fernández-Pevida A, Kressler D, de la Cruz J (2019) The ubiquitin moiety of Ubi1 is required for productive expression of ribosomal protein eL40 in Saccharomyces cerevisiae. Cell 8:850. https://doi.org/10.3390/cells8080850
Melnikov SV, Manakongtreecheep K, Rivera KD et al (2018a) Muller’s ratchet and ribosome degeneration in the obligate intracellular parasites microsporidia. Int J Mol Sci 19:4125. https://doi.org/10.3390/ijms19124125
Melnikov SV, Rivera KD, Ostapenko D et al (2018b) Error-prone protein synthesis in parasites with the smallest eukaryotic genome. Proc Natl Acad Sci U S A 115:E6245–E6253. https://doi.org/10.1073/pnas.1803208115
Merhej V, Royer-Carenzi M, Pontarotti P, Raoult D (2009) Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol Direct 4:13. https://doi.org/10.1186/1745-6150-4-13
Meskauskas A, Harger JW, Jacobs KLM, Dinman JD (2003) Decreased peptidyltransferase activity correlates with increased programmed −1 ribosomal frameshifting and viral maintenance defects in the yeast Saccharomyces cerevisiae. RNA 9:982–992. https://doi.org/10.1261/rna.2165803
Miranda I, Silva-Dias A, Rocha R et al (2013) Candida albicans CUG mistranslation is a mechanism to create cell surface variation. MBio 4:e00285-13. https://doi.org/10.1128/mBio.00285-13
Miranda-Saavedra D, Stark MJR, Packer JC et al (2007) The complement of protein kinases of the microsporidium Encephalitozoon cuniculi in relation to those of Saccharomyces cerevisiae and Schizosaccharomyces pombe. BMC Genomics 8:1–21. https://doi.org/10.1186/1471-2164-8-309
Moran NA, Bennett GM (2014) The tiniest tiny genomes. Annu Rev Microbiol 68:195–215
Morris JJ, Johnson ZI, Szul MJ et al (2011) Dependence of the cyanobacterium Prochlorococcus on hydrogen peroxide scavenging microbes for growth at the ocean’s surface. PLoS One 6:e16805. https://doi.org/10.1371/journal.pone.0016805
Morris JJ, Lenski RE, Zinser ER (2012) The black queen hypothesis: evolution of dependencies through adaptive gene loss. MBio 3:e00036-12. https://doi.org/10.1128/mBio.00036-12
Muraki K, Murnane JP (2017) The DNA damage response at dysfunctional telomeres, and at interstitial and subtelomeric DNA double-strand breaks. Genes Genet Syst 92:135–152. https://doi.org/10.1266/ggs.17-00014
Muszewska A, Steczkiewicz K, Stepniewska-Dziubinska M, Ginalski K (2019) Transposable elements contribute to fungal genes and impact fungal lifestyle. Sci Rep 9:1–10. https://doi.org/10.1038/s41598-019-40965-0
Nakjang S, Williams TA, Heinz E et al (2013) Reduction and expansion in microsporidian genome evolution: new insights from comparative genomics. Genome Biol Evol 5:2285–2303. https://doi.org/10.1093/gbe/evt184
Natchiar SK, Myasnikov AG, Kratzat H et al (2017) Visualization of chemical modifications in the human 80S ribosome structure. Nature 551:472–477. https://doi.org/10.1038/nature24482
Naufer MN, Murison DA, Rouzina I et al (2017) Single-molecule mechanochemical characterization of E. coli pol III core catalytic activity. Protein Sci 26:1413–1426. https://doi.org/10.1002/pro.3152
Noeske J, Wasserman MR, Terry DS et al (2015) High-resolution structure of the Escherichia coli ribosome. Nat Struct Mol Biol 22:336–341. https://doi.org/10.1038/nsmb.2994
Olefeld JL, Majda S, Albach DC et al (2018) Genome size of chrysophytes varies with cell size and nutritional mode. Org Divers Evol 18:163–173. https://doi.org/10.1007/s13127-018-0365-7
Pan G, Xu J, Li T et al (2013) Comparative genomics of parasitic silkworm microsporidia reveal an association between genome expansion and host adaptation. BMC Genomics 14:1–14. https://doi.org/10.1186/1471-2164-14-186
Parisot N, Pelin A, Gasc C et al (2014) Microsporidian genomes harbor a diverse array of transposable elements that demonstrate an ancestry of horizontal exchange with metazoans. Genome Biol Evol 6:2289–2300. https://doi.org/10.1093/gbe/evu178
Parker MS, Balasubramaniam A, Sallee FR, Parker SL (2018) The expansion segments of 28S ribosomal RNA extensively match human messenger RNAs. Front Genet 9:66. https://doi.org/10.3389/fgene.2018.00066
Pellicer J, Fay MF, Leitch IJ (2010) The largest eukaryotic genome of them all? Bot J Linn Soc 164:10–15. https://doi.org/10.1111/j.1095-8339.2010.01072.x
Pereira PMR, Berisha N, Bhupathiraju NVSDK et al (2017) Cancer cell spheroids are a better screen for the photodynamic efficiency of glycosylated photosensitizers. PLoS One 12:e0177737. https://doi.org/10.1371/journal.pone.0177737
Perelman A, Uzan A, Hacohen D, Schwarz R (2003) Oxidative stress in Synechococcus sp. strain PCC 7942: various mechanisms for H2O2 detoxification with different physiological roles. J Bacteriol 185:3654–3660. https://doi.org/10.1128/JB.185.12.3654-3660.2003
Petasne RG, Zika RG (1997) Hydrogen peroxide lifetimes in South Florida coastal and offshore waters. Mar Chem 56:215–225. https://doi.org/10.1016/S0304-4203(96)00072-2
Petes TD (1979) Yeast ribosomal DNA genes are located on chromosome XII. Proc Natl Acad Sci U S A 76:410–414. https://doi.org/10.1073/pnas.76.1.410
Petrov AS, Wood EC, Bernier CR et al (2019) Structural patching fosters divergence of mitochondrial ribosomes. Mol Biol Evol 36:207–219. https://doi.org/10.1093/molbev/msy221
Peyretaillade E, Gonçalves O, Terrat S et al (2009) Identification of transcriptional signals in Encephalitozoon cuniculi widespread among microsporidia phylum: support for accurate structural genome annotation. BMC Genomics 10:1–13. https://doi.org/10.1186/1471-2164-10-607
Peyretaillade E, Parisot N, Polonais V et al (2012) Annotation of microsporidian genomes using transcriptional signals. Nat Commun 3:1–9. https://doi.org/10.1038/ncomms2156
Piir K, Paier A, Liiv A et al (2011) Ribosome degradation in growing bacteria. EMBO Rep 12:458–462. https://doi.org/10.1038/embor.2011.47
Piovesan A, Antonaros F, Vitale L et al (2019) Human protein-coding genes and gene feature statistics in 2019. BMC Res Notes 12:1–5. https://doi.org/10.1186/s13104-019-4343-8
Polymenis M (2020) Ribosomal proteins: mutant phenotypes by the numbers and associated gene expression changes. Open Biol 10(8):200114
Pombert JF, Selman M, Burki F et al (2012) Gain and loss of multiple functionally related, horizontally transferred genes in the reduced genomes of two microsporidian parasites. Proc Natl Acad Sci U S A 109:12638–12643. https://doi.org/10.1073/pnas.1205020109
Prokopowich CD, Gregory TR, Crease TJ (2003) The correlation between rDNA copy number and genome size in eukaryotes. Genome 46:48–50. https://doi.org/10.1139/g02-103
Prossliner T, Skovbo Winther K, Sørensen MA, Gerdes K (2018) Ribosome hibernation. Annu Rev Genet 52:321–348. https://doi.org/10.1146/annurev-genet-120215-035130
Quast C, Pruesse E, Yilmaz P et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:590–596. https://doi.org/10.1093/nar/gks1219
Ramesh M, Woolford JL (2016) Eukaryote-specific rRNA expansion segments function in ribosome biogenesis. RNA 22:1153–1162. https://doi.org/10.1261/rna.056705.116
Rathore OS, Faustino A, Prudêncio P et al (2016) Absence of N-terminal acetyltransferase diversification during evolution of eukaryotic organisms. Sci Rep 6:1–13. https://doi.org/10.1038/srep21304
Ree R, Varland S, Arnesen T (2018) Spotlight on protein N-terminal acetylation. Exp Mol Med 50:90
Reinke AW, Balla KM, Bennett EJ, Troemel ER (2017) Identification of microsporidia host-exposed proteins reveals a repertoire of rapidly evolving proteins. Nat Commun 8:1–11. https://doi.org/10.1038/ncomms14023
Remacha M, Jimenez-Diaz A, Bermejo B et al (1995) Ribosomal acidic phosphoproteins P1 and P2 are not required for cell viability but regulate the pattern of protein expression in Saccharomyces cerevisiae. Mol Cell Biol 15:4754–4762. https://doi.org/10.1128/mcb.15.9.4754
Romier C, James N, Birck C et al (2007) Crystal structure, biochemical and genetic characterization of yeast and E. cuniculi TAFII5 N-terminal domain: implications for TFIID assembly. J Mol Biol 368:1292–1306. https://doi.org/10.1016/j.jmb.2007.02.039
Sáenz-Lahoya S, Bitarte N, García B et al (2019) Noncontiguous operon is a genetic organization for coordinating bacterial gene expression. Proc Natl Acad Sci U S A 116:1733–1738. https://doi.org/10.1073/pnas.1812746116
Salari R, Salari R (2017) Investigation of the best Saccharomyces cerevisiae growth condition. Electron Physician 9:3592–3597. https://doi.org/10.19082/3592
Sanghai ZA, Miller L, Molloy KR et al (2018) Modular assembly of the nucleolar pre-60S ribosomal subunit. Nature 556:126–129. https://doi.org/10.1038/nature26156
Santos C, Ballesta JPG (1995) The highly conserved protein P0 carboxyl end is essential for ribosome activity only in the absence of proteins P1 and P2. J Biol Chem 270:20608–20614. https://doi.org/10.1074/jbc.270.35.20608
Scanlan DJ, Ostrowski M, Mazard S et al (2009) Ecological genomics of marine Picocyanobacteria. Microbiol Mol Biol Rev 73:249–299. https://doi.org/10.1128/mmbr.00035-08
Senderskiy IV, Timofeev SA, Seliverstova EV et al (2014) Secretion of Antonospora (Paranosema) locustae proteins into infected cells suggests an active role of microsporidia in the control of host programs and metabolic processes. PLoS One 9:93585. https://doi.org/10.1371/journal.pone.0093585
Shajani Z, Sykes MT, Williamson JR (2011) Assembly of bacterial ribosomes. Annu Rev Biochem 80:501–526. https://doi.org/10.1146/annurev-biochem-062608-160432
Sievers F, Wilm A, Dineen D et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol 7:539–539. https://doi.org/10.1038/msb.2011.75
S**ola M, Grate L, Haussler D, Manuel A (1999) Genome-wide bioinformatic and molecular analysis of introns in Saccharomyces cerevisiae. RNA 5:221–234. https://doi.org/10.1017/S1355838299981682
Tamm T, Kisly I, Remme J (2019) Functional interactions of ribosomal intersubunit bridges in saccharomyces cerevisiae. Genetics 213:1329–1339. https://doi.org/10.1534/genetics.119.302777
Tempest DW, Neijssel OM (1984) The status of YATP and maintenance energy as biologically interpretable phenomena. Annu Rev Microbiol 38:459–513. https://doi.org/10.1146/annurev.mi.38.100184.002331
Trombetta VV (1942) The cytonuclear ratio. Bot Rev 8:317–336. https://doi.org/10.1007/BF02882227
Trösch R, Willmund F (2019) The conserved theme of ribosome hibernation: from bacteria to chloroplasts of plants. Biol Chem 400:879–893. https://doi.org/10.1515/hsz-2018-0436
Undeen AH, Vander Meer RK (1999) Microsporidian Intrasporal sugars and their role in germination. J Invertebr Pathol 73:294–302. https://doi.org/10.1006/jipa.1998.4834
Van Valen L (1973) A new evolutionary law. Evol Theory 1:1–30
Vávra J, Ronny Larsson JI (2014) Structure of the microsporidia. In: The microsporidia and microsporidiosis. ASM Press, Washington, DC, pp 7–84
Vossbrinck CR, Woese CR (1986) Eukaryotic ribosomes that lack a 5.8S RNA. Nature 320:287–288. https://doi.org/10.1038/320287a0
Vossbrinck CR, Maddox JV, Friedman S et al (1987) Ribosomal RNA sequence suggests microsporidia are extremely ancient eukaryotes. Nature 326:411–414. https://doi.org/10.1038/326411a0
Wagner A (2007) Energy costs constrain the evolution of gene expression. J Exp Zool Part B Mol Dev Evol 308:322–324. https://doi.org/10.1002/jez.b.21152
Wang YJ, Vaidyanathan PP, Rojas-Duran MF et al (2018) Lso2 is a conserved ribosome-bound protein required for translational recovery in yeast. PLoS Biol 16:e2005903. https://doi.org/10.1371/journal.pbio.2005903
Wang H, Dienemann C, Stützer A et al (2020) Structure of the transcription coactivator SAGA. Nature 577:717–720. https://doi.org/10.1038/s41586-020-1933-5
Watson AK, Williams TA, Williams BAP et al (2015) Transcriptomic profiling of host-parasite interactions in the microsporidian Trachipleistophora hominis. BMC Genomics 16:1–20. https://doi.org/10.1186/s12864-015-1989-z
Wawiórka L, Molestak E, Szajwaj M et al (2017) Multiplication of ribosomal P-stalk proteins contributes to the Fidelity of translation. Mol Cell Biol 37(17):e00060-17. https://doi.org/10.1128/mcb.00060-17
Weiss LM, Becnel JJ (2014) Microsporidia: pathogens of opportunity. John Wiley & Sons, Ames
Wells JN, Buschauer R, Mackens-Kiani T et al (2020) Structure and function of yeast Lso2 and human CCDC124 bound to hibernating ribosomes. PLoS Biol 18:e3000780. https://doi.org/10.1371/journal.pbio.3000780
Whelan TA, Lee NT, Lee RCH, Fast NM (2019) Microsporidian introns retained against a background of genome reduction: characterization of an unusual set of introns. Genome Biol Evol 11:263–269. https://doi.org/10.1093/gbe/evy260
Williams BAP, Slamovits CH, Patron NJ et al (2005) A high frequency of overlap** gene expression in compacted eukaryotic genomes. Proc Natl Acad Sci U S A 102:10936–10941. https://doi.org/10.1073/pnas.0501321102
Williams BAP, Lee RCH, Becnel JJ et al (2008) Genome sequence surveys of Brachiola algerae and Edhazardia aedis reveal microsporidia with low gene densities. BMC Genomics 9:200. https://doi.org/10.1186/1471-2164-9-200
Wiredu Boakye D, Jaroenlak P, Prachumwat A et al (2017) Decay of the glycolytic pathway and adaptation to intranuclear parasitism within Enterocytozoonidae microsporidia. Environ Microbiol 19:2077–2089. https://doi.org/10.1111/1462-2920.13734
Wlodarski MW, Da Costa L, O’donohue MF et al (2018) Recurring mutations in RPL15 are linked to hydrops fetalis and treatment independence in diamond-blackfan anemia. Haematologica 103:949–958. https://doi.org/10.3324/haematol.2017.177980
Woese CR, Fox GE (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A 74:5088–5090. https://doi.org/10.1073/pnas.74.11.5088
Woolford JL, Baserga SJ (2013) Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195:643–681. https://doi.org/10.1534/genetics.113.153197
**a X (2020) RNA-Seq approach for accurate characterization of splicing efficiency of yeast introns. Methods 176:25–33. https://doi.org/10.1016/j.ymeth.2019.03.019
Zaher HS, Green R (2009) Quality control by the ribosome following peptide bond formation. Nature 457:161–166. https://doi.org/10.1038/nature07582
Zhu Y, Stribinskis V, Ramos KS, Li Y (2006) Sequence analysis of RNase MRP RNA reveals its origination from eukaryotic RNase P RNA. RNA 12:699–706. https://doi.org/10.1261/rna.2284906
Acknowledgments
We thank all members of the Barandun laboratory and Mirjam Hunziker for discussions and critical reading of this chapter.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Ethics declarations
Conflict of Interest
The authors declare that there is no conflict of interest.
Funding
This work was funded by the Swedish Research Council (2019–02011), the European Research Council (ERC Starting Grant PolTube 948655), the SciLifeLab National Fellows program, and the Laboratory for Molecular Infection Medicine Sweden (MIMS).
Ethical Approval
The chapter is a review of previously published accounts. As such, no animal or human studies were performed.
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Jespersen, N., Monrroy, L., Barandun, J. (2022). Impact of Genome Reduction in Microsporidia. In: Weiss, L.M., Reinke, A.W. (eds) Microsporidia. Experientia Supplementum, vol 114. Springer, Cham. https://doi.org/10.1007/978-3-030-93306-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-93306-7_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-93305-0
Online ISBN: 978-3-030-93306-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)