
Abstract
There is a growing interest in first-principles investigations of materials with broken translational symmetry, for example, impurities, interfaces, and alloys. In particular, the total energy and the electronic structure calculations of systems with an arbitrary distribution of atoms on an underlying lattice will give information essential to understanding their stability and properties. At the same time two schemes most frequently used in ab initio total energy calculations for completely random alloys, the ConnollyWilliams (CW) method1 and the methods based on the single-site approximation, as the coherent potential approximation (CPA)2, have limited applicability and reliability3. Another way of approaching the solution to the electronic structure problem for disordered systems is given by the supercell technique. In this case the three-dimensional periodicity is restored although its effect on the final result can be neglected. Then conventional band structure methods can be used. However, this approach has both fundamental and technical limitations. For instance, from the basic point of view all details connected with the smearing of the electronic spectrum and the Fermi surface in alloys are lost. From the technical point of view the computational effort increases as N 3 with increasing number of atoms N in the supercell.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Preview
Unable to display preview. Download preview PDF.
Similar content being viewed by others
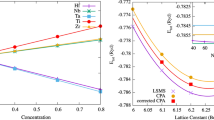

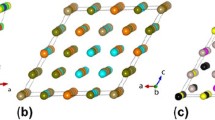
References
J.W.D. Connolly and A.R. Williams, Density-functional theory applied to phase transformations in transition metals, Phys. Rev. B 27:5169 (1983).
J.S. Faulkner, The modern theory of alloys, Prog. Mater. Sci. 27:1 (1982).
I.A. Abrikosov, A.V. Ruban, B. Johansson, and H.L. Skriver, Total energy calculations of random alloys: Connolly-Wiliams and CPA methods, in: ”Stability of Materials”, Series E: Applied Sciences, A. Gonis, P.E.A. Turchi, and J. Kudrnovsky, ed., Kluwer Academic Publishers, the Netherlands (1996).
D.M.C. Nicholson, C.M. Stocks, Y. Wang, W.A. Shelton, Z. Szotek, and W.M. Temmer-man, Stationary nature of the density-functional free energy: Application to accelerated multiple-scattering calculations, Phys. Rev. B 50:14686 (1994).
Y. Wang, C.M. Stocks, W.A. Shelton, D.M.C. Nicholson, Z. Szotek, and W. M. Temmerman, Order-N multiple scattering approach to electronic structure calculations, Phys. Rev. Lett. 75:2867 (1995).
I.A. Abrikosov, A.M.N. Niklasson, S.I. Simak, B. Johansson, A.V. Ruban, and H.L. Skriver, Order-N Green’s function technique for local environment effects in alloys, Phys. Rev. Lett. 76:4203 (1996).
D.D. Johnson and F.J. Pinski, Including charge correlations in calculations of the energetics and electronic structure for random substitutional alloys, Phys. Rev. B 48:11553 (1993).
H. L. Skriver and N. M. Rosengaard, Self-consistent Green’s function technique for surfaces and interfaces, Phys. Rev. B 43:9538 (1991).
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 1997 Springer Science+Business Media New York
About this chapter
Cite this chapter
Abrikosov, I.A., Simak, S.I., Johansson, B. (1997). Total Energy Calculations of Alloys: Locally Self-Consistent Green’s Function Method. In: Gonis, A., Meike, A., Turchi, P.E.A. (eds) Properties of Complex Inorganic Solids. Springer, Boston, MA. https://doi.org/10.1007/978-1-4615-5943-6_14
Download citation
DOI: https://doi.org/10.1007/978-1-4615-5943-6_14
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4613-7723-8
Online ISBN: 978-1-4615-5943-6
eBook Packages: Springer Book Archive